Synthesis of New L-Ascorbic Ferulic Acid Hybrids

Université de Caen Basse-Normandie, U.F.R. des Sciences Pharmaceutiques, Centre d’Etudes et de Recherche sur le Médicament de Normandie, UPRES EA-3915, 5, rue Vaubénard, 14032 CAEN Cedex, France
*
Author to whom correspondence should be addressed.
Molecules 2007, 12(11), 2533-2545; https://doi.org/10.3390/12112533
Submission received: 30 October 2007
/
Revised: 15 November 2007
/
Accepted: 15 November 2007
/
Published: 17 November 2007
Abstract
:A feasibility and chemical study of the coupling conditions of L-ascorbic acid with ferulic acid derivatives are described on the basis of the known synergistic effects of mixtures of various antioxidants. Novel L-ascorbic ferulic hybrids linked at the C-3 hydroxyl group were prepared with the aim to protect the alcohol function and the enediol system.
Introduction
Oxidative stress is thought to play an important contributory role in the pathogenesis of numerous degenerative or chronic diseases, such as cancer. Besides, experimental evidence links the production of reactive oxygen species to biological damage that can potentially provide a mechanistic basis for their initiation and/or progression. For this reason we were especially interested in the use of antioxidants in this context of photoprotection and we have already developed a synthesis program that was aimed at the preparation of antioxidant trans-1,2-diarylethenes from cinnamic acids [1].
Under normal conditions, cells and tissues are protected from the attack of active oxygen species and free radicals by various enzymes such as catalase, superoxide dismutase and glutathione peroxidase [2,3,4,5].
In the experiments, corroborated in clinical studies on volunteers, L-ascorbic acid (1) protects against photoperoxidation of serum lipids, membrane lipids and collagen and increases the neosynthesis of collagens [6,7,8,9,10]. Moreover, L-ascorbic acid prevents against mutation and immunosuppression (responsible for the skin becoming cancerous), and inflammation (through reactive oxygen species) attributable to UV radiations [11,12]. Finally, L-ascorbic acid counteracts skin aging in promoting collagen formation and in stimulating elastic fiber element synthesis [10]. Therefore, the protective effect of L-ascorbic acid allows considering it as one of the most potent naturally occurring antioxidants.
These biological characteristic activities are derived from the enediol structure, with its strong electron-donating ability. L-ascorbic acid is reversibly metabolized to dehydro-L-ascorbic acid (2) via monodehydro-L-ascorbic acid in a series of oxidative processes (Scheme 1).
Scheme 1.
L-Ascorbic acid (1) and dehydro-L-ascorbic acid (2).
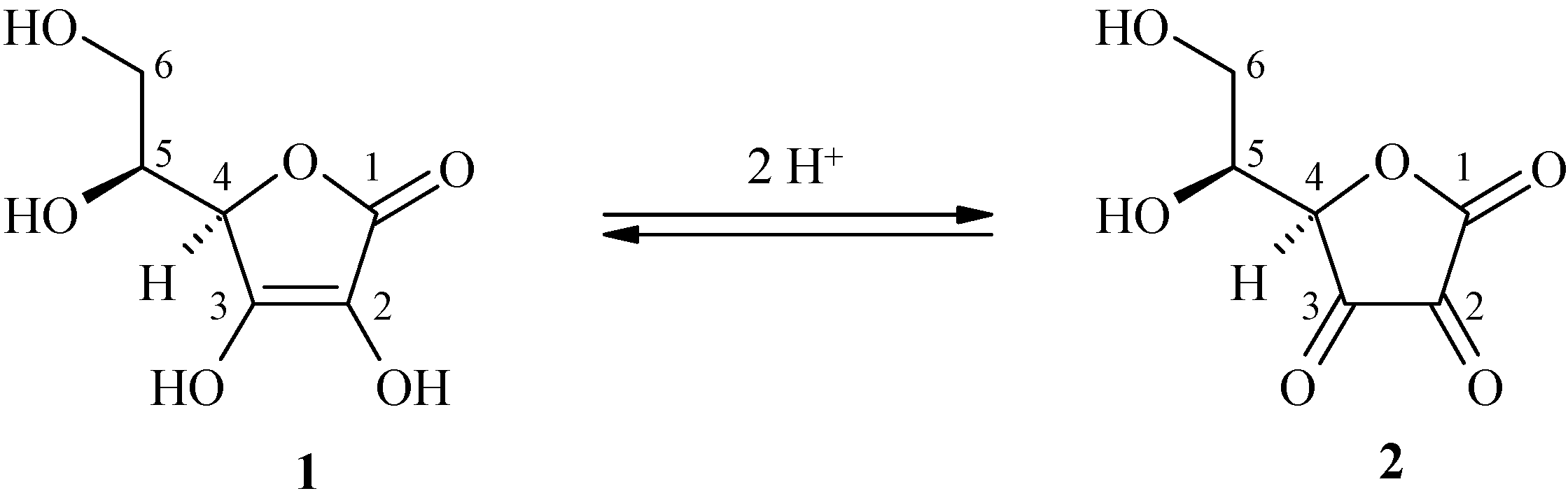
However, L-ascorbic acid loses its biological activities because of its reducing power, its low stability under aqueous conditions and its high sensitivity to oxidation. The well-known susceptibility of L-ascorbic acid to thermal and oxidative degradation has led to interest in its analogs with increased in vitro stability, while maintaining the inherent in vivo biological activity. To overcome this problem, L-ascorbic acid was chemically modified by protecting alcohol functions or the enediol system.
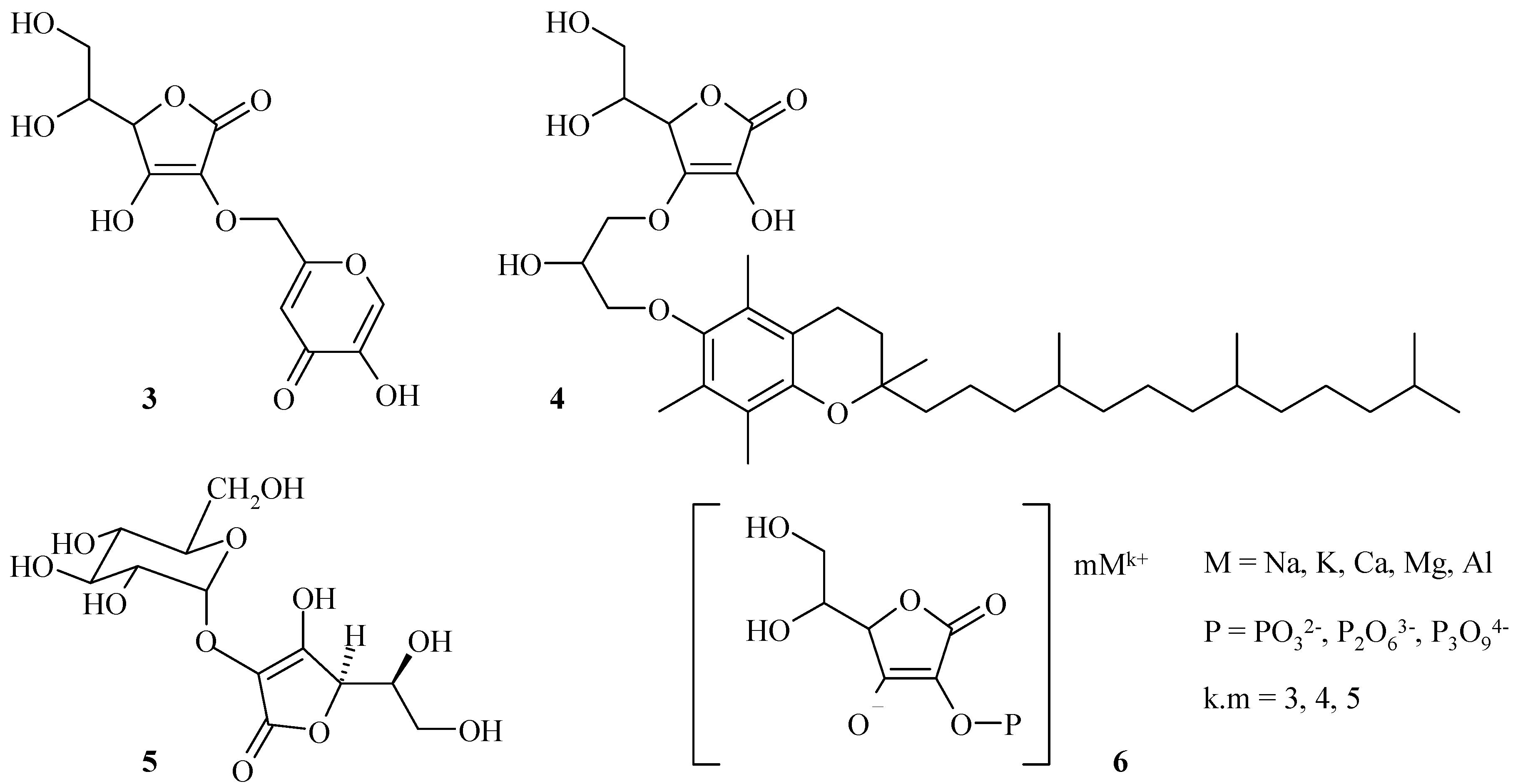
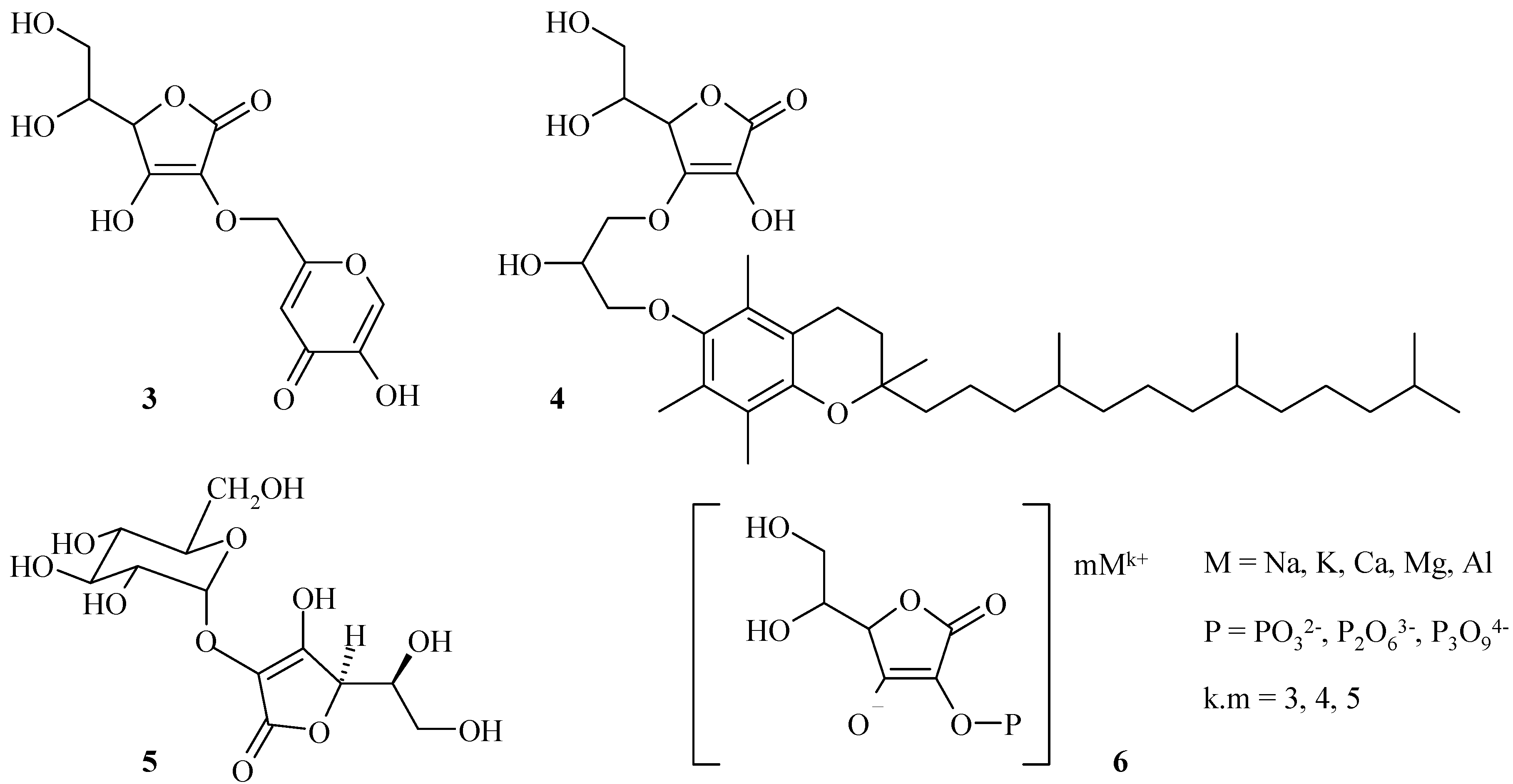
On the one hand, we were particularly interested in the chemical modifications of hydroxyl groups in C-2 and C-3 positions of L-ascorbic acid so as to protect the enediol structure. Among these derivatives, ascorbic acid/kojic acid and ascorbic acid/α-tocopherol hybrids 3 and 4 have been described (Scheme 2) that were more stable and have shown biological properties of both ascorbic acid and active substances [13].
In the same way, 2-O-α-D-glucopyranosylascorbic acid (5, Scheme 2) has good stability and the same biological properties as L-ascorbic acid. This compound has been widely used in cosmetics for its L-ascorbic acid-like activity [14,15]. Then, salts of ascorbyl 2-phosphoric esters, 6 (Scheme 2) have been largely studied to be less sensitive than L-ascorbic acid after dephosphorylation [16,17,18].
On the other hand, several studies such as the one by Pinnell et al. [19] suggest that mixtures of various antioxidants may have synergistic effects, yielding a greater efficiency than individual antioxidant compounds used alone. In fact, Pinnell et al. have examined ferulic acid mixed with L-ascorbic acid and α-tocopherol and have shown that this combination seemed to provide meaningful synergistic protection against oxidative stress in skin. Although the hydroxycinnamic acids, as ferulic acid, cafeic acid or p-coumaric acid, have been studied for their antioxidative potency which is related to the structure, in particular to electron delocalization of the aromatic nucleus [20], the mechanism of ferulic acid’s stabilizing effect on L-ascorbic acid and α-tocopherol is still unknown, and their chemical linkage with L-ascorbic acid has never been studied previously.
Scheme 2.
Ascorbic acid derivatives.
Therefore, we decided to study hybridization methodology conditions to synthesize new L-ascorbic acid/ferulic ester hybrids connected at the C-3 position, which is crucial for both stability and biological activity, with a view to simultaneously improving the stability and preventing diminution of activity (Scheme 3).
Scheme 3.
L-ascorbic acid / ferulic ester hybrids 20, 21 and 22.

So, this paper emphasizes a feasibility study concerning the coupling conditions via a glycidyl ether spacer of L-acid ascorbic and ferulic derivatives and we realized a chemical study in order to prepare compounds 20, 21 and 22.
At the beginning of our works, only mono- and di-esters of cinnamic acids 7 and L-ascorbic acid (Scheme 4) have been prepared [21]. Nevertheless, the two patterns, cinnamic and L-ascorbic acids, were linked at the C-2 hydroxyl group whereas the biologically active site was reported to be placed on the C-3 hydroxyl group. It is the reason why we studied the synthesis of new hybrids linked at the C-3 hydroxyl group via an ether spacer more stable than an ester group. So, we have chosen the glycidyl ether methodology previously described by Morisaki [13] which moreover can offer a greater molecular diversity.
Scheme 4.
Ascorbic acid / cinnamic acid hybrids 7.
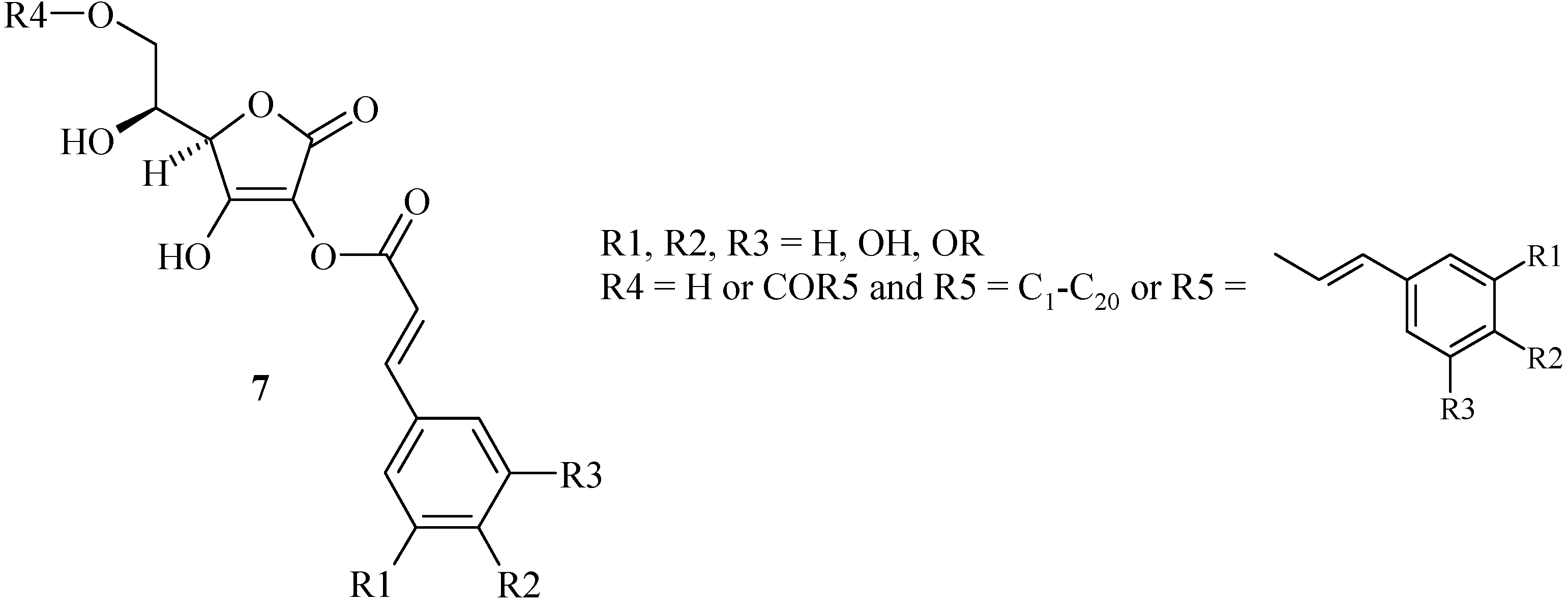
Even if L-ascorbic acid can be considered as one of the most potent naturally occurring antioxidants, its high hydrophilicity limits this latter property. These hybrids have different lipophilic properties than L-ascorbic acid (Table 1) which allows a good penetration into the stratum corneum. In fact, they could be considered as excellent vehicles of L-ascorbic acid in which the length of the alkyl chain modulates the octanol/water partition coefficient log Po/w (see hybrids 20, 21 and 22).

{kind=link}
{kind=link}
{kind=link}
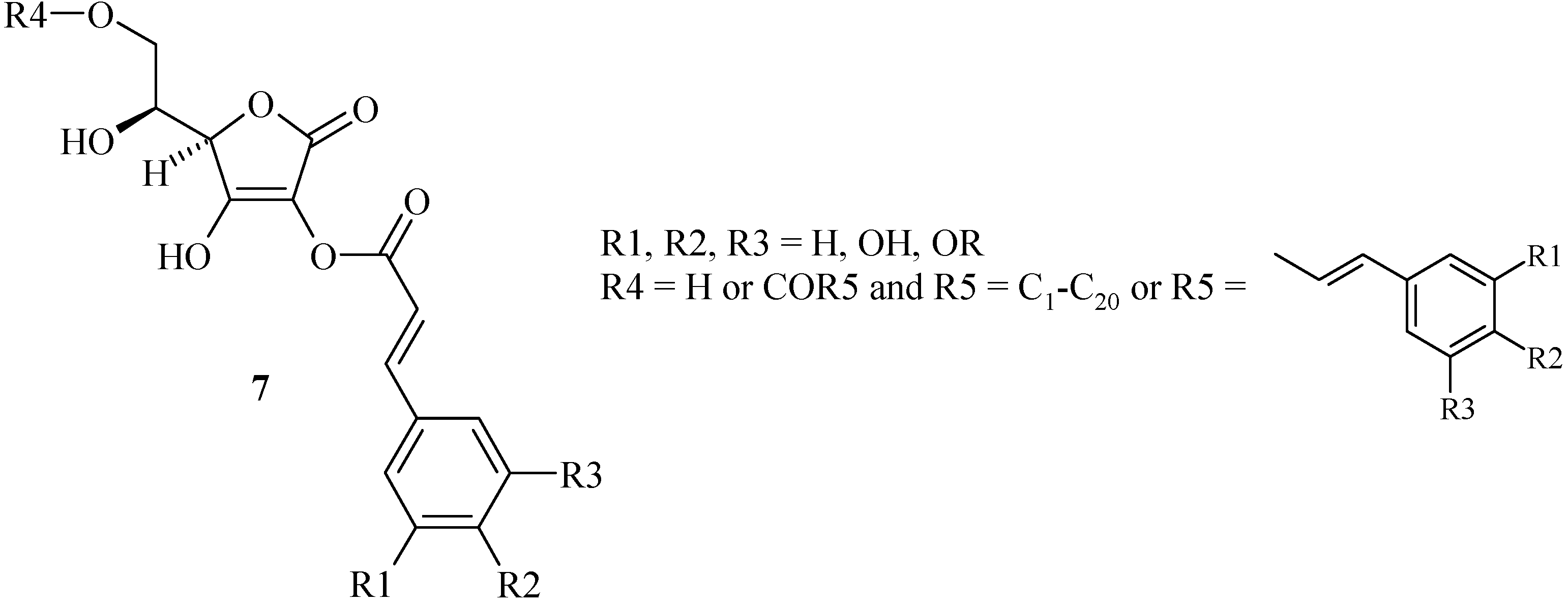{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Octanol/water partition coefficient log Po/w [22].
| Compounds | Log Po/w |
|---|---|
| L-ascorbic acid 1 | -2.34 |
| Ferulic acid 9 | 1.86 |
| Methyl ferulate 10 | 2.60 |
| Ethyl ferulate 11 | 2.94 |
| Dodecyl ferulate 13 | 6.97 |
| Hybrid 20 | -0.63 |
| Hybrid 21 | -0.23 |
| Hybrid 22 | 3.75 |
Results and Discussion
Protection of the C-5 and C-6 positions of L-ascorbic acid
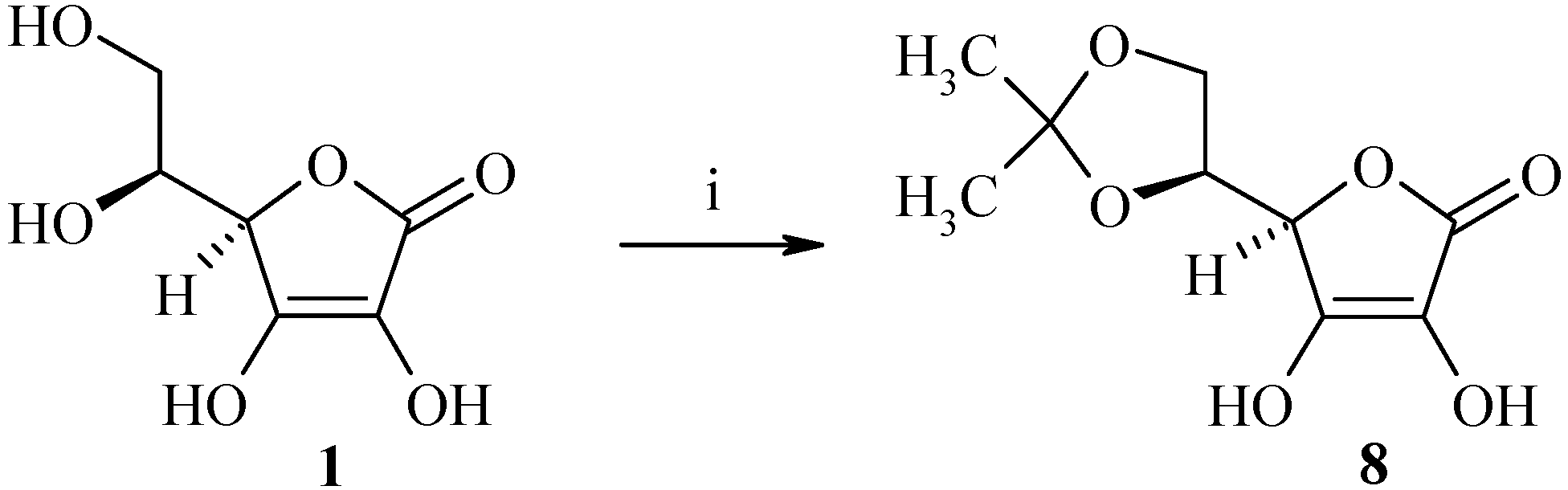
First of all, the hydroxyl groups at the C-5 and C-6 positions were protected with acetone in the presence of acetyl chloride according to Jung’s procedure [23] to afford (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8) in 74% yield (Scheme 5).
Scheme 5.
Protection of the C-5 and C-6 positions of L-ascorbic acid.
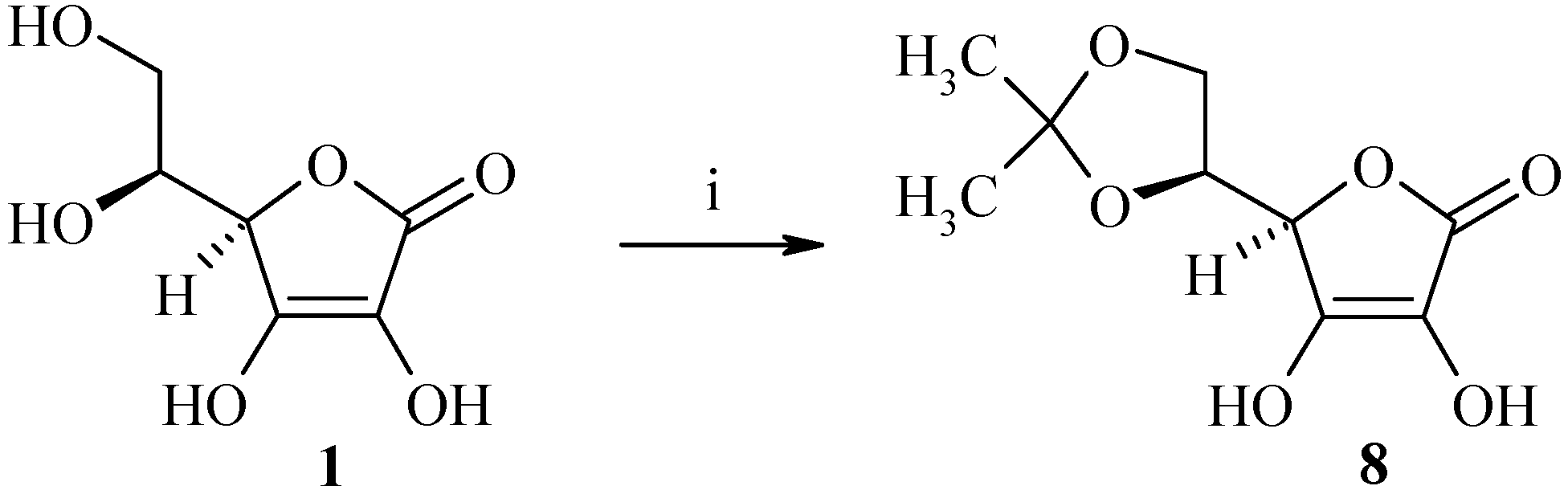
Reagents and conditions: i) CH3COCl 0.25 equiv., acetone, rt, 3h.
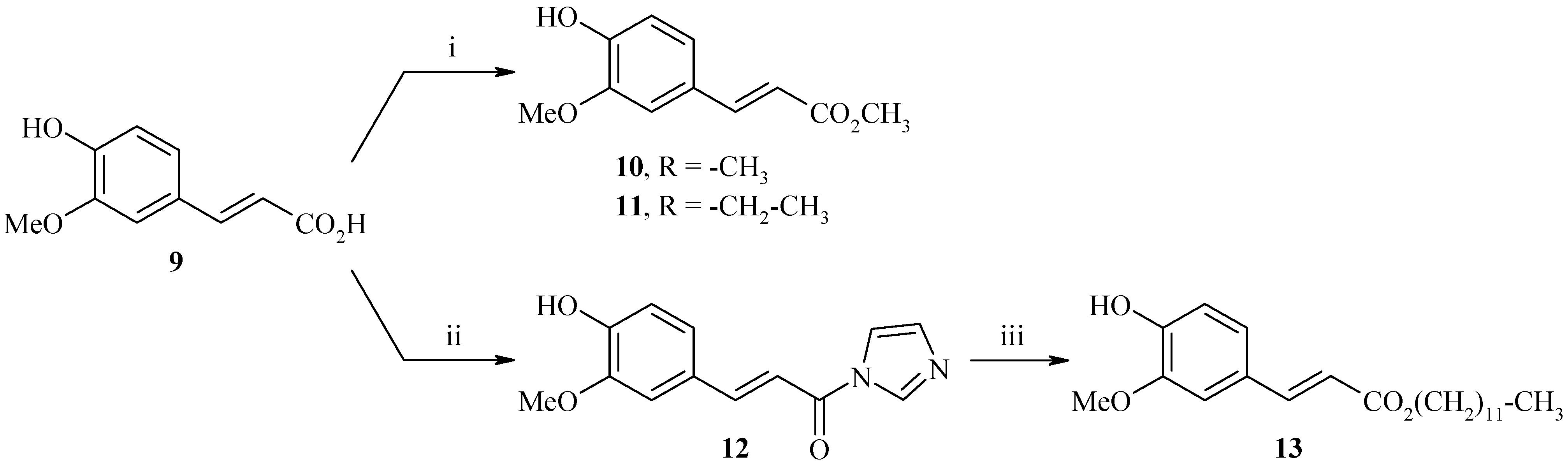
Synthesis of ethyl, methyl and dodecyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylates 10, 11 and 13
The esterification of ferulic acid (9) was carried out in the presence of methanol and sulfuric acid (95%) by a classical method to afford methyl ferulate 10 in 97% yield. The same procedure was used in the presence of ethanol to afford ethyl ferulate 11 (80%). With the previous method, we did not obtain the expected dodecyl ester (Scheme 6), so we have perfected the esterification reaction using a different methodology. Indeed, the activation of the acid function of ferulic acid with 1,1'-carbonyldiimidazole (CDI) led to (E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1H-imidazol-1-yl)prop-2-en-1-one (12) (76%), then the esterification with 1-dodecanol allowed us to obtain the dodecyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate 13 (66%).
Scheme 6.
Synthesis of ferulate derivatives 10, 11 and 13.
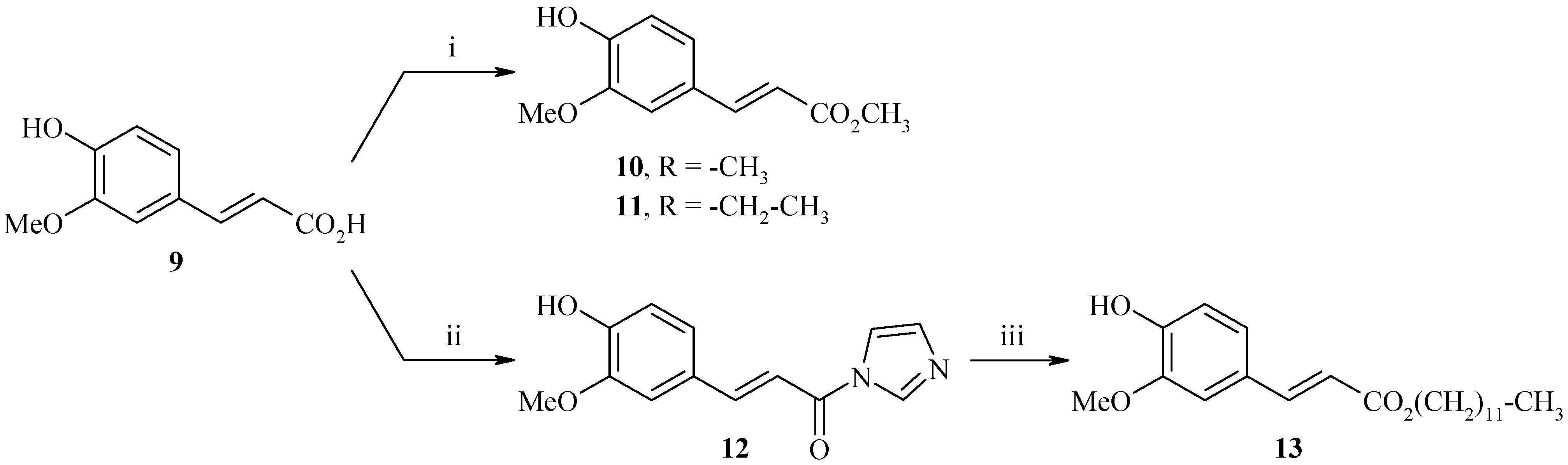
Reagents and conditions: i) MeOH or EtOH, H2SO4 (95%), reflux, 24h; ii) CDI 2 equiv., anhydrous THF, reflux, 3.5h; iii) 1-dodecanol 1 equiv., DBU 1 equiv., anhydrous THF, reflux, 48h;
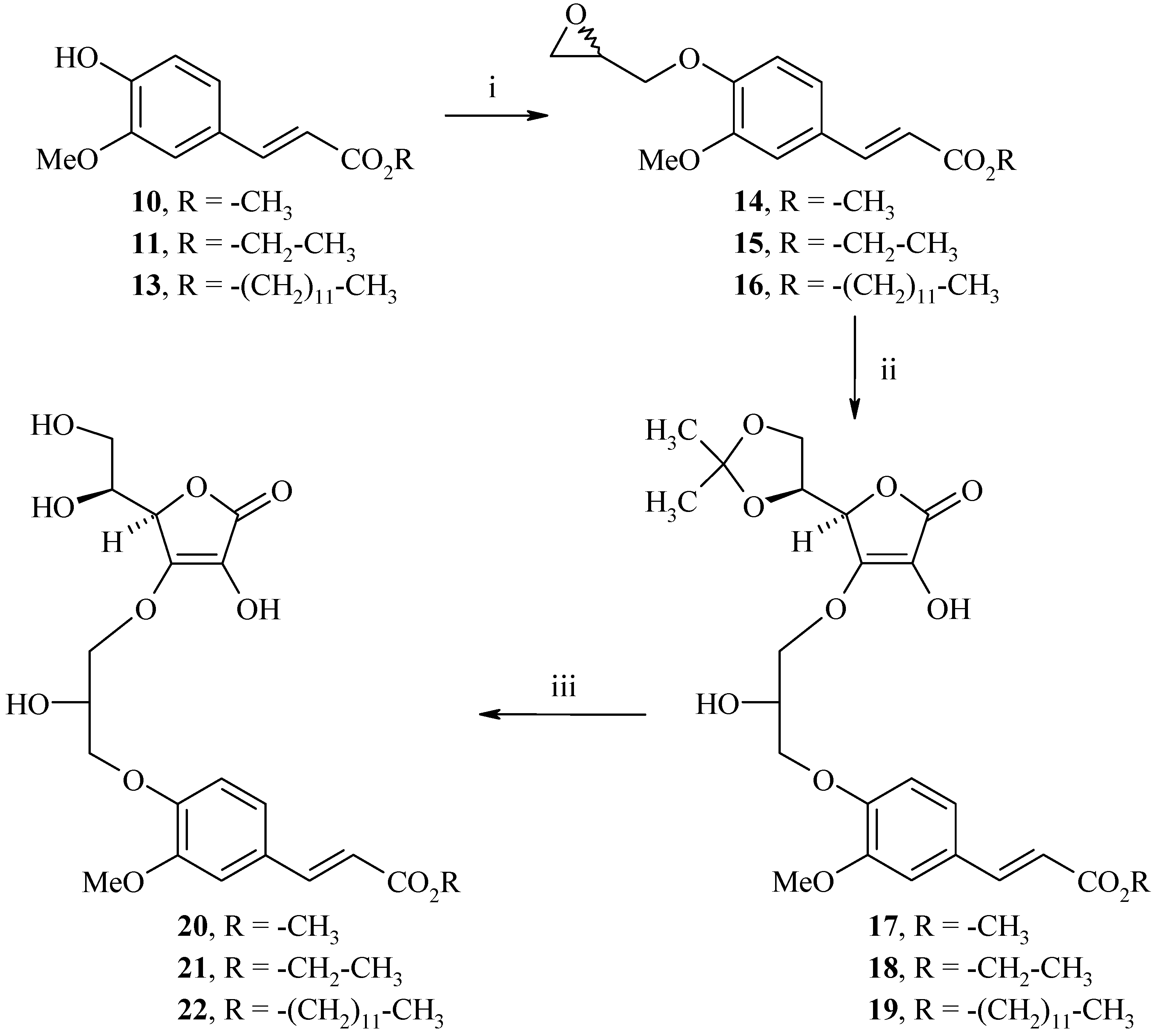
Synthesis of methyl, ethyl and dodecyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylates 20, 21 and 22
Hybrids 20, 21 and 22 were synthesized in three steps from the corresponding ferulates 10, 11 and 13, as described in Scheme 7. Alkylation with (±)-epichlorohydrin in the presence of NaOH in a water-acetone medium (1:1) gave the corresponding ethers 14, 15 and 16 in 70%, 45% and 83% yields, respectively.
Condensation of 14, 15 and 16 with the protected L-ascorbic acid derivative 8 in the presence of NaHCO3 and a catalytic amount of 4-dimethylaminopyridine (4-DMAP) in 1,4-dioxane afforded 17, 18 and 19 in 50%, 49% and 66% yields, respectively; considering that the most acidic hydrogen is the hydroxyl in the C-3 position, the condensation will take place on this position, involving the enediol system. Subsequent removal of the protecting group was achieved by acid hydrolysis to yield methyl, ethyl and dodecyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylates 20, 21 and 22 (71%, 61% and 75%).
Scheme 7.
Synthesis of hybrids 20, 21 and 22.
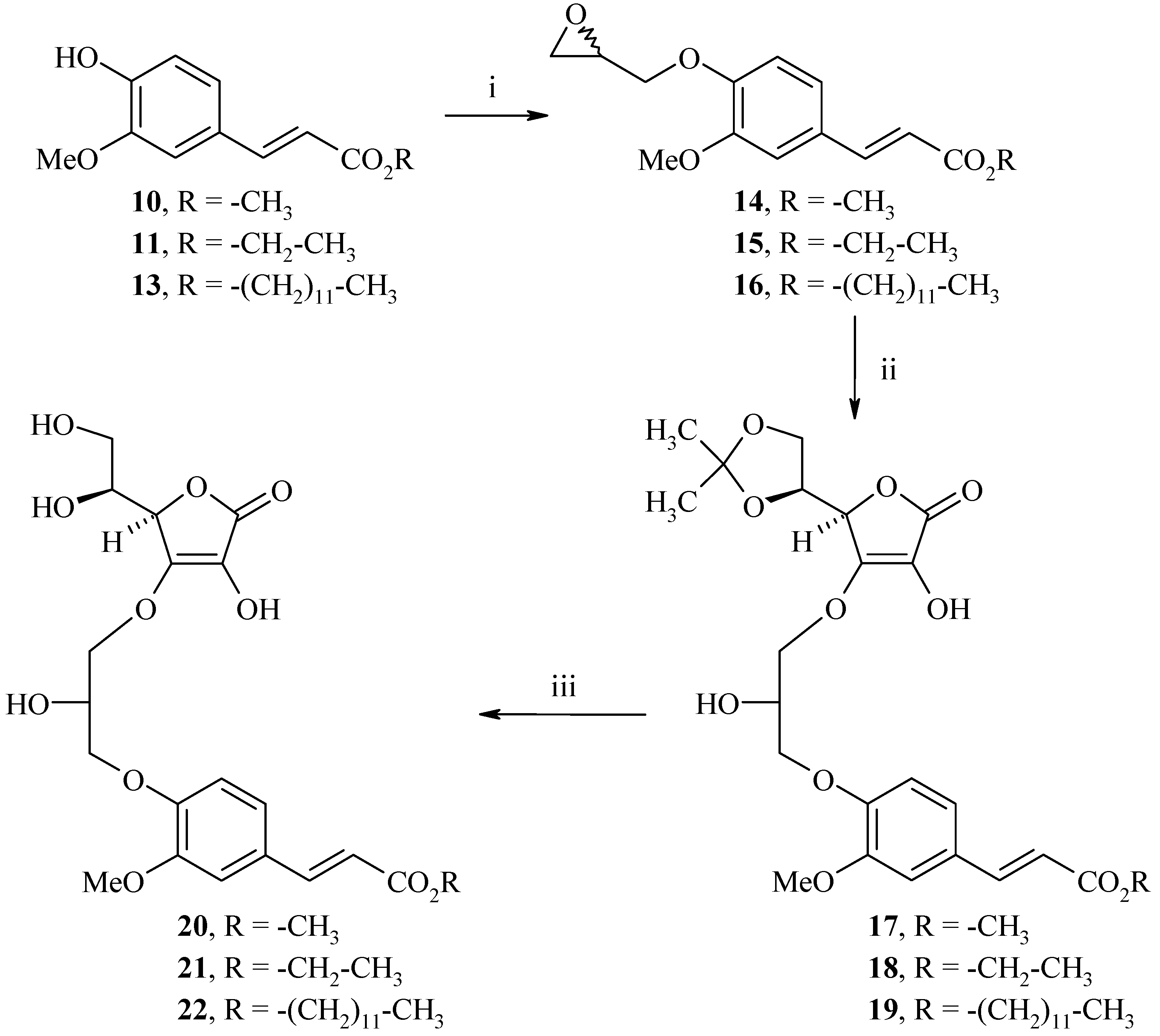
Reagents and conditions: i) (±)-Epichlorohydrin 5 equiv., NaOH 1 equiv., water/acetone:1/1, reflux, 8h; ii) 8 1.2 equiv., 4-DMAP 0.1 equiv., NaHCO3 1 equiv., 1,4-dioxane, reflux, 24h; iii) HCl 12N, 1,4-dioxane, rt, 4h.
Conclusions
We have studied the feasibility and the conditions of the hybridization methodology for the synthesis of three new L-ascorbic ferulic acid hybrids. This methodology led us to introduce a chemical spacer connected to the hydroxyl in the C-3 position of the L-ascorbic acid and the phenol of the ferulic ester. The junction could be crucial for both stability and biological activity insofar as the enediol system is protected.
The thermal stability and the inhibitory effects of the products on tyrosinase activity, and on active oxygen species and free radicals in vitro, are currently under investigation and the results will be published elsewhere.
Experimental
General
Commercial reagents were purchased from Aldrich, Acros Organics and Alfa Aesar and used without additional purification. Melting points were determined on a Kofler heating bench and are uncorrected. IR spectra were recorded on a Perkin Elmer BX FT-IR spectrophotometer. Optical rotations were obtained on a Perkin-Elmer 343 polarimeter. 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were recorded in DMSO-d6 on a JEOL Lambda 400 spectrometer. Chemical shifts are expressed in parts per million downfield from tetramethylsilane as an internal standard and coupling constants in hertz. Mass spectra were taken on a JEOL JMS GCMate spectrometer at ionising potential of 70 eV (EI) or were performed using a spectrometer LC-MS Waters alliance 2695 (ESI+). Chromatography was carried out on a column using flash silica gel 60 Merck (0.063-0.200 mm) as the stationary phase. Thin-layer chromatography was performed on 0.2 mm precoated plates of silica gel 60F-264 (Merck) and spots were visualized using an ultraviolet-light lamp.
Synthesis of (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8)
Acetyl chloride (1 mL, 14.2 mmol) was added dropwise under nitrogen to a solution of L-ascorbic acid 1 (10 g, 56.8 mmol) in acetone (40 mL) and the solution was then stirred at room temperature for 3 h. The resulting white mixture was cooled to 4°C, filtered and then washed with acetone (2 x 20 mL) to afford 9.1 g of (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8) as a white solid (yield: 74%); m.p.: 210°C; IR (cm-1): 3242 (OH), 1754 (C=O), 1663 (C=C), 1333 (C(CH3)2), 1140 (dioxolane); 1H-NMR: 11.29 (bs, 1H, OH), 8.48 (bs, 1H, OH), 4.70-4.69 (m, 1H, H4), 4.25-4.23 (m, 1H, H5), 4.08-4.06 (m, 1H, H6), 3.87-3.86 (m, 1H, H6), 1.24 (s, 6H, CH3); 13C-NMR: 173.4, 157.1, 117.8, 110.4, 76.6, 76.4, 65.1, 26.3, 25.7; MS (ESI+) m/z 219 (M++1).
Synthesis of methyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (10)
Ferulic acid 9 (30.0 g, 154.0 mmol) and 5 drops of H2SO4 (95%) were refluxed in methanol for 24 h. The crude product was then evaporated to dryness and re-dissolved in dichloromethane. The organic layer was washed with saturated NaHCO3 solution (2 x 50 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane-ethylacetate: 8/2) to afford 31.2 g of methyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (10) as a white solid (yield: 97%); m.p.: 68°C; IR (cm-1): 3399 (OH), 2947 (CH3), 1718 (C=O), 1264-1160 (C-O); 1H-NMR: 9.62 (bs, 1H, OH), 7.55 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.31 (s, 1H, H2’), 7.11 (d, 3J=8.1 Hz, 1H, H6’), 6.78 (d, 3J=8.1 Hz, 1H, H5’), 6.47 (d, 3Jtrans=15.9 Hz, 1H, H3), 3.80 (s, 3H, CO2CH3), 3.68 (s, 3H, OCH3); 13C-NMR: 167.5, 149.8, 148.3, 145.5, 125.9, 123.5, 115.9, 114.6, 111.7, 53.1, 51.6; MS (ESI+) m/z 209 (M++1).
Synthesis of ethyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (11)
Ferulic acid 9 (50.0 g, 257.0 mmol) and 10 mL of H2SO4 (95%) were refluxed in ethanol for 24h. The crude product was then evaporated to dryness and re-dissolved in dichloromethane. The organic layer was washed with saturated NaHCO3 solution (2 x 50 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane-ethyl acetate: 8/2) to afford 45.8 g of ethyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (11) as a white solid (yield: 80%); m.p.: 67°C; IR (cm-1): 3180 (OH), 2952 (CH3), 1711 (C=O), 1256 (C-O); 1H-NMR: 9.58 (bs, 1H, OH), 7.53 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.31 (s, 1H, H2’), 7.10 (d, 3J=8.1 Hz, 1H, H6’), 6.77 (d, 3J=8.1 Hz, 1H, H5’), 6.45 (d, 3Jtrans=15.9 Hz, 1H, H3), 4.15 (q, J=7.0 Hz, 2H, CH2), 3.76 (s, 3H, OCH3), 1.23 (t, J=7.0 Hz, 3H, CH3); 13C-NMR: 166.9, 148.8, 148.3, 145.1, 126.9, 123.9, 116.5, 115.6, 110.7, 60.3, 56.4, 14.3; MS (ESI+) m/z 223 (M++1).
Synthesis of (E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1H-imidazol-1-yl)prop-2-en-1-one (12)
To a solution of ferulic acid (9, 5.0 g, 25.8 mmol) in freshly distilled THF (60 mL) was added 1,1'-carbonyldiimidazole (8.3 g, 51.5 mmol) and the mixture was refluxed for 3.5 h (CaCl2 guard tube). The resulting mixture was cooled down to room temperature and concentrated in vacuo. The residue was chromatographed on silica gel (ether) to afford 4.8 g of (E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1H-imidazol-1-yl)prop-2-en-1-one (12) as a white solid (yield: 76%); m.p.: 156°C; IR (cm-1): 3124 (OH), 2841 (CH3), 1772 (C=O), 1320-1243 (C-O); 1H-NMR: 8.78 (bs, 1H, Himidazole), 8.50 (bs, 1H, Himidazole), 8.03 (d, 3Jtrans=15.5 Hz, 1H, H2), 7.94 (s, 1H, H2’), 7.62 (d, 3J=8.0 Hz, 1H, H5’), 7.70 (d, 3Jtrans=15.5 Hz, 1H, H3), 7.55 (d, 3J=8.0 Hz, 1H, H6’), 7.17 (bs, 1H, Himidazole), 3.90 (s, 3H, OCH3); 13C-NMR: 163.3, 149.5, 147.8, 138.2, 129.9, 129.2, 128.2, 124.3, 115.5, 115.3, 111.8, 111.4, 57.0; MS (EI) m/z: 244 (M+ •, 100).
Synthesis of dodecyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (13)
To a solution of (E)-3-(4-hydroxy-3-methoxyphenyl)-1-(1H-imidazol-1-yl)prop-2-en-1-one (12, 4.8 g, 19.7 mmol) in freshly distilled THF (60 mL) were added DBU (3.0 g, 19.7 mmol) and 1-dodecanol (3.7 g, 19.7 mmol) and the mixture was refluxed for 48 h. The resulting mixture was cooled down to room temperature and concentrated in vacuo. The crude product was diluted with water (40 mL) and extracted with ethyl acetate (3 x 50 mL). The organic layer was dried (MgSO4), and concentrated in vacuo. The residue was chromatographed on silica gel (cyclohexane-ethyl acetate: 7/3) to afford 4.7 g of dodecyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (13) as a white solid (yield: 66%); m.p.: 46°C; IR (cm-1): 3534 (OH), 3131, 2923 (CH3), 2849, 1746 (C=O), 1269-1190 (C-O); 1H-NMR: 9.58 (bs, 1H, OH), 7.52 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.29 (s, 1H, H2’), 7.08 (d, 3J=8.1 Hz, 1H, H6’), 6.77 (d, 3J=8.1 Hz, 1H, H5’), 6.44 (d, 3Jtrans=15.9 Hz, 1H, H3), 4.09 (t, 3J=6.5 Hz, 2H, OCH2), 3.80 (s, 3H, OCH3), 1.60-1.58 (m, 2H, CH2), 1.31-1.21 (m, 18H, (CH2)9), 0.82 (t, 3J=6.4 Hz, 3H, CH3); 13C-NMR: 167.4, 147.8, 146.8, 144.6, 127.2, 123.0, 115.6, 114.6, 110.1, 64.6, 56.1, 31.9, 30.4, 29.8, 29.6, 29.5, 29.4, 29.3, 28.7, 25.9, 22.7, 15.1; MS (EI) m/z: 362 (M+ •, 95).
Synthesis of methyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (14)
To a water-acetone medium (1:1, 80 mL) were added methyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (10, 3.1 g, 15.0 mmol) and NaOH (0.6 g, 15.0 mmol). The mixture was stirred at room temperature for 10 min. (±)-Epichlorohydrin (12.8 g, 75.0 mmol) was added and the resulting mixture was refluxed for 8 h. Then, acetone was concentrated in vacuo and the aqueous layer was diluted with 70 mL of cold water and extracted with ethyl acetate (3 x 40 mL). The organic layer was dried (MgSO4), and concentrated in vacuo. Recrystallization from diethyl ether afford 2.8 g of methyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (14) as a white solid (yield: 70%); m.p.: 100°C; IR (cm-1): 2943-2925 (CH3), 2837 (CH2), 1717 (C=O), 1264-1161 (C-O); 1H-NMR: 7.58 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.37 (s, 1H, H2’), 7.22 (d, 3J=8.1 Hz, 1H, H6’), 6.98 (d, 3J=8.1 Hz, 1H, H5’), 6.57 (d, 3Jtrans=15.9 Hz, 1H, H3), 4.34-4.33 (m, 1H, Hoxirane), 3.81 (s, 3H, CO2CH3), 3.70 (s, 3H, OCH3), 3.33-3.32 (m, 2H, CH2), 2.84-2.83 (m, 1H, Hoxirane), 2.82-2.81 (m, 1H, Hoxirane); 13C-NMR: 167.9, 149.5, 147.8, 144.2, 128.8, 122.8, 117.5, 113.0, 111.3, 70.2, 55.7, 51.5, 49.8, 44.6; MS (EI) m/z: 264 (M+ •, 41), 179 (37), 15 (100).
Synthesis of ethyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (15)
The same procedure as compound 14 starting from ethyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (11, 20.0 g, 90.0 mmol) and (±)-epichlorohydrin (76.7 g, 450.0 mmol) was used to prepare 11.3 g of ethyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (15) as a white solid (yield: 45%); m.p.: 75°C; IR (cm-1): 2980 (C-H), 1703 (C=O), 1600 (C=C), 1309-1273 (C-O); 1H-NMR: 7.56 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.37 (s, 1H, H2’), 7.21 (d, 3J=8.2 Hz, 1H, H6’), 6.97 (d, 3J=8.2 Hz, 1H, H5’), 6.56 (d, 3Jtrans=15.9 Hz, 1H, H3), 4.34-4.33 (m, 1H, Hoxirane), 4,16 (q, J=7.0 Hz, 2H, CH2), 3.81 (s, 3H, OCH3), 3.33-3.32 (m, 2H, CH2), 2.84-2.83 (m, 1H, Hoxirane), 2.70-2.69 (m, 1H, Hoxirane), 1.24 (t, J=7.0 Hz, 3H, CH3); 13C-NMR: 166.9, 149.5, 147.8, 144.6, 128.3, 122.9, 116.6, 113.2, 111.3, 70.2, 60.3, 55.7, 51.5, 49.8, 44.6, 14.4; MS (ESI+) m/z 279 (M++1).
Synthesis of dodecyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (16)
The same procedure as compound 14 starting from dodecyl (2E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (13, 1.8 g, 5.0 mmol) and (±)-epichlorohydrin (2.3 g, 25.0 mmol) gave 1.7 g of dodecyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (16) as a white solid (yield: 83%); m.p.: 69°C; IR (cm-1): 2919-2849 (CH3, CH2), 1713 (C=O), 1265-1171 (C-O); 1H-NMR: 7.58 (d, 3Jtrans=16.0 Hz, 1H, H2), 7.38 (s, 1H, H2’), 7.22 (d, 3J=8.1 Hz, 1H, H6’), 6.98 (d, 3J=8.1 Hz, 1H, H5’), 6.57 (d, 3Jtrans=16.0 Hz, 1H, H3), 4.34-4.33 (m, 1H, Hoxirane), 4.12 (t, 3J=7.2 Hz, 2H, OCH2), 3.83 (s, 3H, OCH3),3.63-3.62 (m, 2H, CH2oxirane), 2.86-2.84 (m, 1H, Hoxirane), 2.71-2.69 (m, 1H, Hoxirane); 1.64-1.62 (m, 2H, CH2), 1.28-1.18 (m, 18H, (CH2)9), 0.86 (t, 3J=6.4 Hz, 3H, CH3); 13C-NMR: 167.4, 149.5, 147.8, 144.5, 128.2, 122.9, 115.6, 113.6, 111.3, 70.2, 64.6, 55.7, 49.8, 44.6, 31.9, 30.4, 29.8, 29.6, 29.5, 29.4, 29.3, 28.7, 25.9, 22.7, 14.1; MS (EI) m/z: 418 (M+ •, 37), 169 (100).
Synthesis of methyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (17)
To a solution of methyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (14, 2.2 g, 8.3 mmol) and (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8, 2.2 g, 10.0 mmol) in 1,4-dioxane (30 mL) were added NaHCO3 (0.8 g, 8.3 mmol) and 4-dimethylaminopyridine (0.1 g, 0.8 mmol). The resulting mixture was refluxed for 24 h. The organic layer was concentrated in vacuo and the crude product was diluted with water (100 mL) and extracted with ethyl acetate (3 x 40 mL). The organic layer was dried (MgSO4), and concentrated in vacuo. Recrystallization from diethyl ether afforded 2.0 g of methyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (17) as a white solid (yield: 50%); m.p.: 73°C; IR (cm-1): 3445 (OH), 2988-2950 (CH), 1760 (C=O), 1699 (C=C), 1260 (C(CH3)2); 1H-NMR: 9.03 (bs, 1H, OH), 7.58 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.35 (s, 1H, H2), 7.22 (d, 3J=8.1 Hz, 1H, H6), 6.99 (d, 3J=8.1 Hz, 1H, H5), 6.55 (d, 3Jtrans=15.9 Hz, 1H, H3), 5.43 (bs, 1H, OH), 4.78-4.77 (m, 1H, H4), 4.43-4.40 (m, 1H, H5), 4.21-4.20 (m, 1H, H6), 4.14-3.97 (m, 6H), 3.80 (s, 3H, CO2CH3), 3.70 (s, 3H, OCH3), 1.24-1.22 (m, 6H, CH3); 13C-NMR: 170.3, 167.5, 149.8, 149.2, 148.8, 148.6, 144.4, 127.9, 122.2, 119.6, 115.6, 113.1, 110.1, 74.9, 71.9, 69.9, 69.5, 68.4, 64.9, 55.6, 51.4, 25.5, 25.2; MS (EI) m/z: 480 (M+ •, 8), 280 (56), 206 (66), 194 (100).
Synthesis of ethyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (18)
The same procedure as compound 17 starting from ethyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (15, 2.2 g, 8.3 mmol) and (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8, 2.2 g, 10.0 mmol) was used to give 1.6 g of ethyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)- 3-methoxyphenyl)acrylate (18) as a white solid (yield: 49%); m.p.: 71°C; IR (cm-1): 3432 (OH), 2985 (CH), 1770 (C=O), 1698 (C=C), 1259 (C(CH3)2); 1H-NMR: 8.90 (bs, 1H, OH), 7.49 (d, 3Jtrans= 15.9 Hz, 1H, H2), 6.98 (s, 1H, H2), 6.96 (d, 3J=8.3 Hz, 1H, H6), 6.79 (d, 3J= 8.3 Hz, 1H, H5), 6.22 (d, 3Jtrans= 15.9 Hz, 1H, H3), 4.58 (bs, 1H, OH), 4.51 (m, 1H, H4), 4.30 (m, 1H, H5), 4.22 (m, 1H, H6), 4,16 (q, J=7.0 Hz, 2H, CH2), 4.10-3.86 (m, 6H), 3.78 (s, 3H, OCH3), 1.35-1.33 (m, 3H, CH3), 1.27-1.23 (m, 6H, CH3); 13C-NMR: 174.4, 172.2, 166.9, 149.3, 146.5, 144.5, 128.3, 126.7, 123.1, 116.6, 112.8, 111.4, 110.6, 77.7, 76.1, 72.0, 70.4, 70.3, 65.1, 60.3, 55.7, 26.4, 26.0, 14.4; MS (ESI+) m/z 495 (M++1).
Synthesis of dodecyl (2E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (19)
The same procedure as compound 17 starting from dodecyl (2E)-3-[3-methoxy-4-(oxiran-2-ylmethoxy)phenyl]acrylate (16, 1.7 g, 4.1 mmol) and (R)-5-((S)-2,2-dimethyl-[1,3]-dioxolan-4-yl)-3,4-dihydroxy-5H-furan-2-one (8, 1.1 g, 5.0 mmol) gave 1.7 g of dodecyl (2E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxy-propoxy)-3-methoxyphenyl)acrylate (19) as a colourless oil (yield: 66%); IR (cm-1): 3440 (OH), 2924-2854 (CH), 1769 (C=O), 1700 (C=C), 1260 (C(CH3)2); 1H-NMR: 9.03 (bs, 1H, OH), 7.56 (d, 3Jtrans=16.0 Hz, 1H, H2), 7.35 (s, 1H, H2), 7.20 (d, 3J=7.8 Hz, 1H, H6), 6.98 (d, 3J=7.8 Hz, 1H, H5), 6.55 (d, 3Jtrans=16.0 Hz, 1H, H3), 5.43 (bs, 1H, OH), 4.77-4.76 (m, 1H, H4), 4.43-4.39 (m, 2H), 4.21-4.20 (m, 1H, H6), 4.14-3.97 (m, 5H), 4.10-4.09 (m, 2H, OCH2), 3.80 (s, 3H, OCH3), 1.64-1.62 (m, 2H, CH2), 1.24 (s, 6H, CH3), 1.23-1.21 (m, 18H, (CH2)9), 0.84-0.82 (m, 3H, CH3); 13C-NMR: 174.4, 172.3, 167.4, 149.3, 146.5, 144.5, 128.2, 126.6, 123.1, 115.7, 112.8, 111.5, 110.7, 77.7, 76.1, 70.5, 72.0, 70.1, 65.1, 64.6, 53.7, 31.9, 30.4, 29.8, 29.6, 29.5, 29.4, 29.3, 28.7, 26.3, 26.1, 25.9, 22.7, 14.1; MS (EI) m/z: 635 (M+ •, 5), 436 (69), 362 (65), 194 (100).
Synthesis of methyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (20)
To a solution of methyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (17, 2.0 g, 4.2 mmol) in 1,4-dioxane (30 mL) was added HCl (12 N, 0.6 mL) and the resulting mixture was stirred at room temperature for 4 h. The organic layer was neutralized with saturated NaHCO3 solution (100 mL), and extracted with ethyl acetate (3 x 40 mL), dried (MgSO4), and concentrated in vacuo. The residue was chromatographed on silica gel (ethyl acetate) to afford 1.3 g of methyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)-acrylate (20) as a white solid (yield: 71%); m.p.: 69°C; [α = +21.0 (c 0.5, C2H5OH); IR (cm-1): 3452 (OH), 2953 (CH), 1765 (C=O), 1692 (C=C), 1259 (C-O); 1H-NMR: 8.82 (bs, 1H, OH), 7.58 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.35 (s, 1H, H2), 7.22 (d, 3J=8.1 Hz, 1H, H6), 7.00 (d, 3J=8.1 Hz, 1H, H5), 6.56 (d, 3Jtrans=15.9 Hz, 1H, H3), 5.43 (bs, 1H, OH), 4.96-4.78 (m, 2H, H4, H5), 4.43-4.41 (m, 2H, H6), 4.00-3.99 (m, 5H), 3.80 (s, 3H, CO2CH3), 3.70 (s, 3H, OCH3); 13C-NMR: 170.5, 167.0, 150.41 149.2, 144.7, 127.2, 122.9, 119.3, 118.1, 115.4, 112.9, 110.8, 74.6, 71.9, 69.7, 68.6, 67.6, 61.7, 55.7, 51.3. MS (ESI+) m/z 441 (M++1).
\Synthesis of ethyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (21)
The same procedure as compound 20 starting from ethyl 2(E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)-acrylate (18, 0.3 g, 0.5 mmol) was used to prepare 8.6 mg of ethyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)-acrylate (21) as a white solid (yield: 61%); m.p.: 68°C; [α = +21.0 (c 0.5, C2H5OH); IR (cm-1): 3419 (OH), 2936 (CH), 1760 (C=O), 1694 (C=C), 1259 (C-O); 1H-NMR: 8.83 (bs, 1H, OH), 7.56 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.34 (s, 1H, H2), 7.20 (d, 3J=8.0 Hz, 1H, H6), 6.99 (d, 3J=8.0 Hz, 1H, H5), 6.53 (d, 3Jtrans=15.9 Hz, 1H, H3), 5.44 (bs, 1H, OH), 4.97-4.38 (m, 2H, H4, H5), 4.18-4.00 (m, 5H), 3.80 (s, 3H, OCH3), 1.23 (t, J=7.0 Hz, 3H, CH3); 13C-NMR: 170.9, 166.0, 150.7, 149.6, 144.9, 127.7, 123.2, 119.8, 118.7, 116.2, 113.4, 111.2, 75.0, 72.3, 70.3, 69.0, 67.9, 62.2, 60.2, 56.1, 14.5; MS (ESI+) m/z 455 (M++1).
Synthesis of dodecyl 2(E)-3-(4-(3-((R)-2-((S)-1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)acrylate (22)
The same procedure as compound 20 starting from dodecyl (2E)-3-(4-(3-((R)-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-4-hydroxy-5-oxo-2,5-dihydrofuran-3-yloxy)-2-hydroxypropoxy)-3-methoxyphenyl)-acrylate (19, 0.4 g, 0.6 mmol) was used to prepare 0.3 g of dodecyl (2E)-3-[4-(3-[2-(1,2-dihydroxyethyl)-4-hydroxy-5-oxo-2,5-dihydro-furan-3-yloxy]-2-hydroxy-propoxy)3-methoxyphenyl]-acrylate (22) as a white solid (yield: 75%); m.p.: 67°C; [α = +21.6 (c 0.5, C2H5OH); IR (cm-1): 3387 (OH), 2953-2853 (CH), 1759 (C=O), 1695 (C=C), 1260 (C-O); 1H-NMR: 8.83 (bs, 1H, OH), 7.56 (d, 3Jtrans=15.9 Hz, 1H, H2), 7.35 (s, 1H, H2), 7.20 (d, 3J=8;1 Hz, 1H, H6), 6.99 (d, 3J=8.1 Hz, 1H, H5), 6.54 (d, 3Jtrans=15.9 Hz, 1H, H3), 5.42 (bs, 1H, OH), 4.95-4.78 (m, 2H, H4, H5), 4.46-4.45 (m, 2H, H6), 4.12-4.01 (m, 5H), 3.81 (s, 3H, OCH3), 1.61-1.60 (m, 2H, CH2), 1.22-1.19 (m, 20H, (CH2)10), 0.83 (t, 2J=5.8 Hz, 3H, CH3); 13C-NMR: 170.5, 166.6, 150.5, 150.2, 149.2, 134.9, 127.2, 122.8, 119.4, 115.6, 112.7, 110.6, 74.6, 69.7, 68.6, 67.5, 66.4, 63.8, 61.7, 55.7, 31.3, 30.4, 29.0, 28.9, 28.8, 28.7, 28.6, 28.3, 25.4, 22.1, 13.9; MS (ESI+) m/z 596 (M++1).
Acknowledgments
The authors thank the PCAS society for their financial support.
References and Notes
- Bazin, M.A.; El Kihel, L.; Lancelot, J.C.; Rault, S. Original one-pot microwave-promoted Hunsdiecker–Suzuki strategy: straightforward access to trans-1,2-diarylethenes from cinnamic acids. Tetrahedron Lett. 2007, 48, 4347–4351. [Google Scholar]
- Halliwell, B.; Chirico, S. Lipid peroxidation: its mechanism, measurement and significance. Am. J. Clin. Nutr. 1993, 57, 715S–724S. [Google Scholar]
- Loft, S.; Poulsen, H.E. Cancer risk and oxidative DNA damage in man. J. Mol. Med. 1996, 74, 297–312. [Google Scholar]
- Stahl, W.; Sies, H. Antioxidant defense: vitamins E and C and carotenoids. Diabetes 1997, 46, S14–S18. [Google Scholar]
- Bowen, P.E.; Mobarhan, S. Evidence from cancer intervention and biomarker studies and the development of biochemical markers. Am. J. Clin. Nutr. 1995, 62, 1403S–1409S. [Google Scholar]
- Rougier, A.; Nguyen, Q.L.; Boussoutra, B.; Colin, C.; Portes, P.; Cohen, C. Scientific presentations at the 1999 and 2000 meetings of AAD and EADV.
- Colven, R.M.; Pinnell, S.R. Clinics in Dermatology, Skin Aging, and Photoaging; Ledo, A., Ed.; 1996; Volume 14, pp. 227–234. [Google Scholar]
- Garcia-Mercier, C.; Richard, A.; Watier, E.; Chesne, C.; Rougier, A. Scientific presentations at the 1999 and 2000 meetings of AAD and EADV.
- Prunieras, M. Culture of skin: how and why? Int. J. Dermatol. 1974, 14, 12–22. [Google Scholar]
- Tajima, S.; Pinnell, S.R. Ascorbic acid preferentially enhances type I and III collagen gene transcription in human skin fibroblasts. J. Dermatol. Sci. 1996, 11, 250–253. [Google Scholar] [CrossRef]
- Stewart, M.S.; Cameron, G.S.; Pence, B.C. Antioxydant Nutrients Protect Against UVB-induced Oxidative Damage to DNA of Mouse Kertinocytes in Culture. J. Invest. Dermatol. 1996, 106, 1086–1089. [Google Scholar]
- Nakamura, T.; Pinnell, S.R.; Darr, D.; Kurimoto, I.; Itami, S.; Yoshikawa, K.; Streilein, J.W. Vitamin C abrogates the Deleterious Effects of UVB Radiation on Cutaneous Immunity by a Mechanism That Does Not Depend on TNF-α. J. Invest. Dermatol. 1997, 109, 20–24. [Google Scholar]
- Morisaki, K.; Ozaki, S. Design of novel hybrid vitamin C derivatives: thermal stability and biological activity. Chem. Pharm. Bull. 1996, 44, 1647–1655. [Google Scholar] [CrossRef]
- Sakai, S.; Yoneyama, M.; Miyake, T. Crystalline 2-O-α-D-glucopyranosyl-L-ascorbic acid, and its preparation and uses. US Patent 1990. [Google Scholar]
- Yamamoto, I.; Muto, N.; Murakami, K.; Suga, S.; Yamguchi, H. L-ascorbic acid α-glucoside formed by regioselective transglucosylation with rat intestinal and seed α-glucosidases: its improved stability and structure determination. Chem. Pharm. Bull. 1990, 38, 3020–3023. [Google Scholar] [CrossRef]
- Sieb, P.A.; Deyoe, C.W.; Hoseney, R.C. Method of preparation of 2-phosphate esters of ascorbic acid. US Patent US4179445, 1979. [Google Scholar]
- Austria, R.; Semenzato, A.; Bettero, A. Stability of vitamin C derivatives in solution and topical formulations. J. Pharm. Biomed. Anal. 1997, 15, 795–801. [Google Scholar] [CrossRef]
- Böttcher, A.; Gurski, H. Preparation of salts of ascorbyl 2-phosphoric esters. US Patent 2000. [Google Scholar]
- Lin, F.H.; Lin, J.Y.; Gupta, R.D.; Tournas, J.A.; Burch, J.A.; Selim, M.A.; Monteiro-Riviere, N.A.; Grichnik, J.M.; Zielinski, J.; Pinnell, S.R. Ferulic acid stabilizes a solution of vitamins C and E and doubles its photoprotection of skin. J. Invest. Dermatol. 2005, 125, 826–832. [Google Scholar] [CrossRef]
- Cuvelier, M.E.; Richard, H.; Berset, C. Comparison of the antioxidative activity of some acid-phenols: structure-activity relationship. Biosci. Biotechnol. Biochem. 1992, 56, 324–325. [Google Scholar] [CrossRef]
- Galey, J.B.; Terranova, E. Mono- and di-esters from cinnamic acid or one of its derivatives and vitamin C; process for preparing the same and their use as antioxidants in cosmetic, pharmaceutical or food compositions. EP 0 664 290 A1, 1995. [Google Scholar]
- Viswanadhan, V.N.; Ghose, A.K.; Revankar, G.R.; Robins, R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci. 1989, 29, 163–172. [Google Scholar]
- Jung, M.E.; Shaw, T.J. Total synthesis of (R)-glycerol acetonide and the anti-epileptic and hypotensive drug (-)-γ-amino-β-hydroxybutyric acid (GABOB): use of vitamin C as a chiral starting material. J. Am. Chem. Soc. 1980, 102, 6304–6311. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Voisin-Chiret, A.S.; Bazin, M.-A.; Lancelot, J.-C.; Rault, S. Synthesis of New L-Ascorbic Ferulic Acid Hybrids. Molecules 2007, 12, 2533-2545. https://doi.org/10.3390/12112533
AMA Style
Voisin-Chiret AS, Bazin M-A, Lancelot J-C, Rault S. Synthesis of New L-Ascorbic Ferulic Acid Hybrids. Molecules. 2007; 12(11):2533-2545. https://doi.org/10.3390/12112533
Chicago/Turabian StyleVoisin-Chiret, Anne Sophie, Marc-Antoine Bazin, Jean-Charles Lancelot, and Sylvain Rault. 2007. "Synthesis of New L-Ascorbic Ferulic Acid Hybrids" Molecules 12, no. 11: 2533-2545. https://doi.org/10.3390/12112533