Results and Discussion
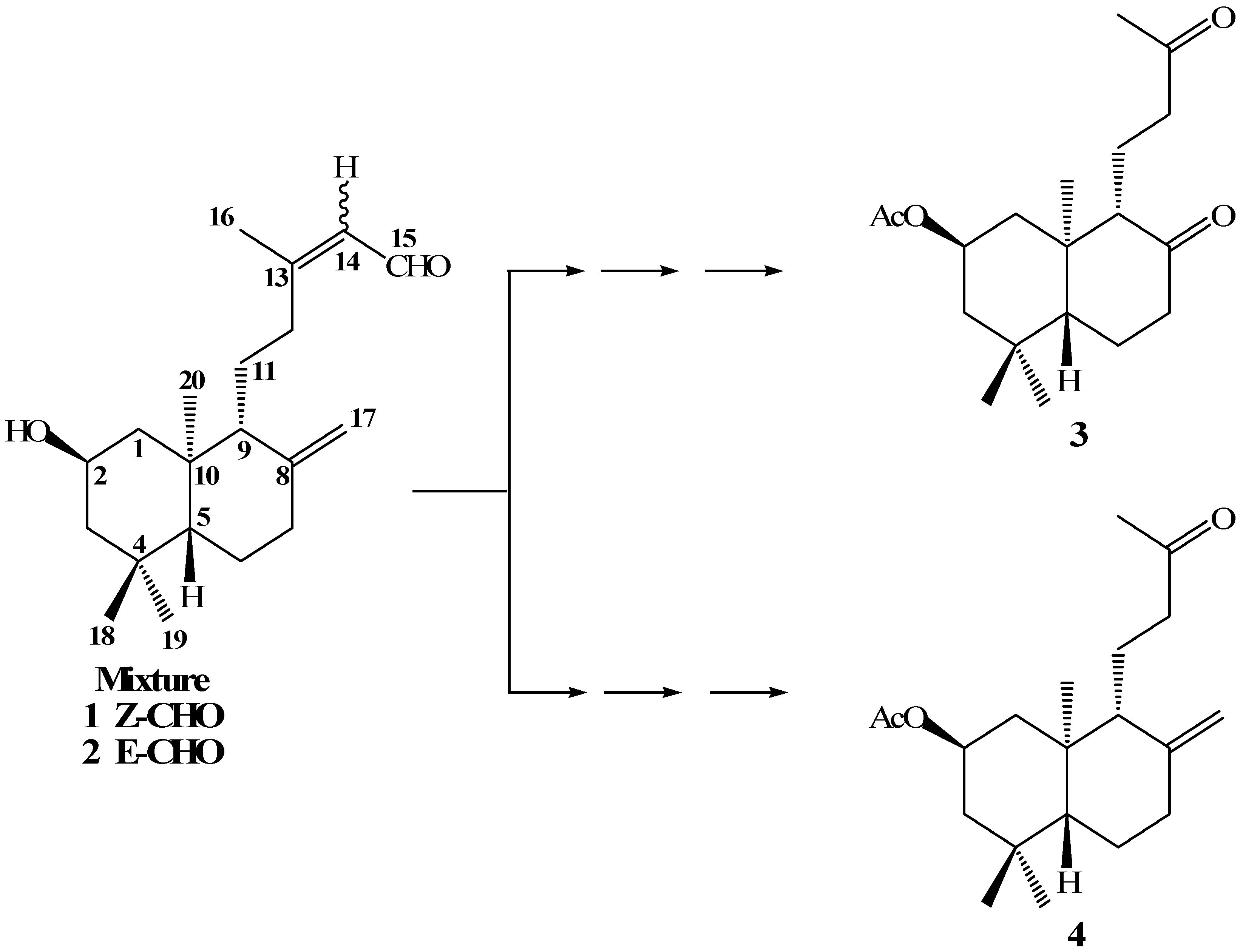
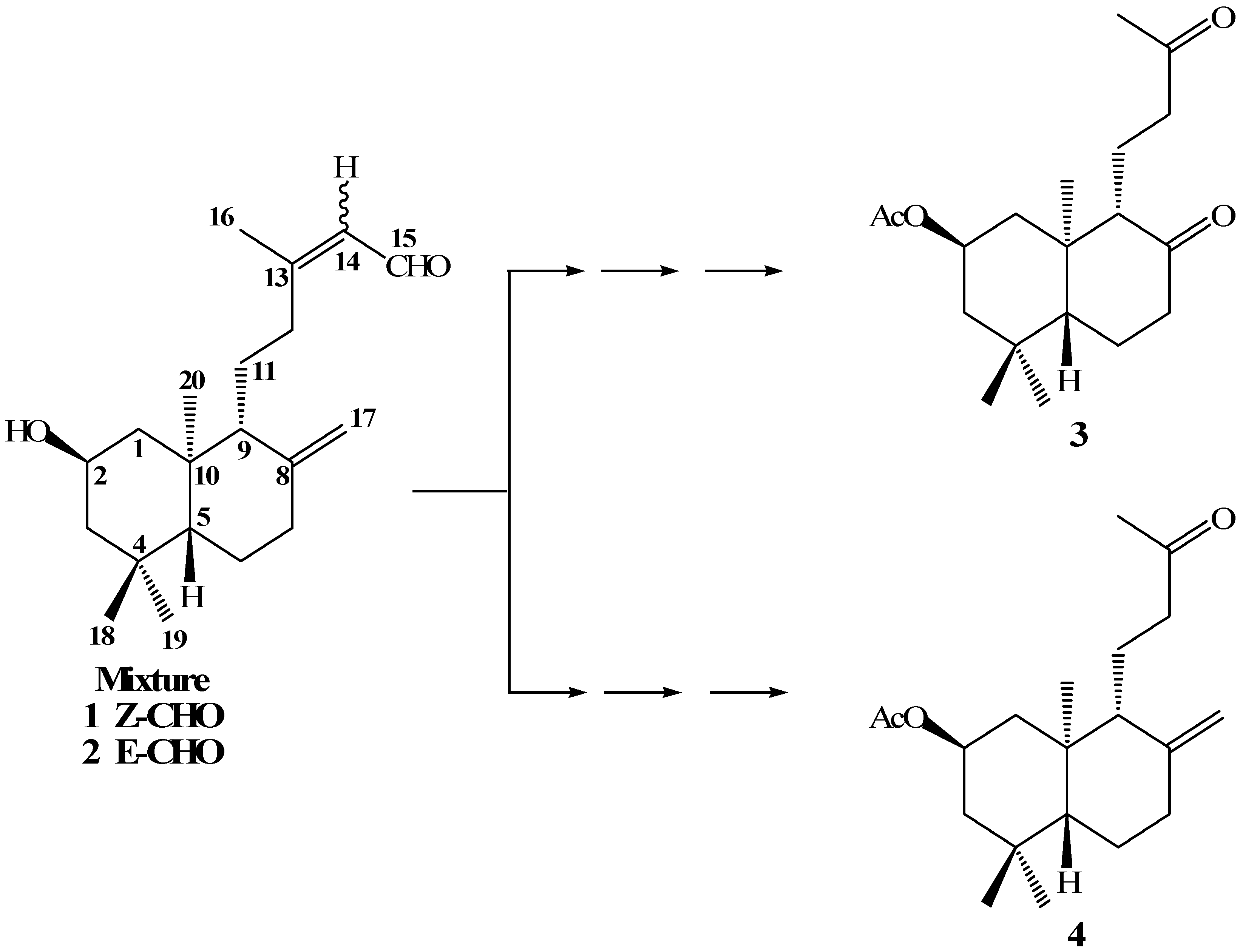
In a previous paper [
19] we reported the synthesis of compounds
3 and
4 from the mixture of
ent-labdanes
1-2 (
Scheme 1). Our main goal in the oxidative degradation of the
ent-labdane side chains was to obtain a primary alcohol at the C-12 position. This primary alcohol would then be transformed into a good leaving group, which in turn would lead to suitable terpenic fragments that could be reacted with quinone/hydroquinone moieties, in an attempt to synthesize terpenyl(hydroxy)quinones/ hydroquinones with potential anticancerogenic activities.
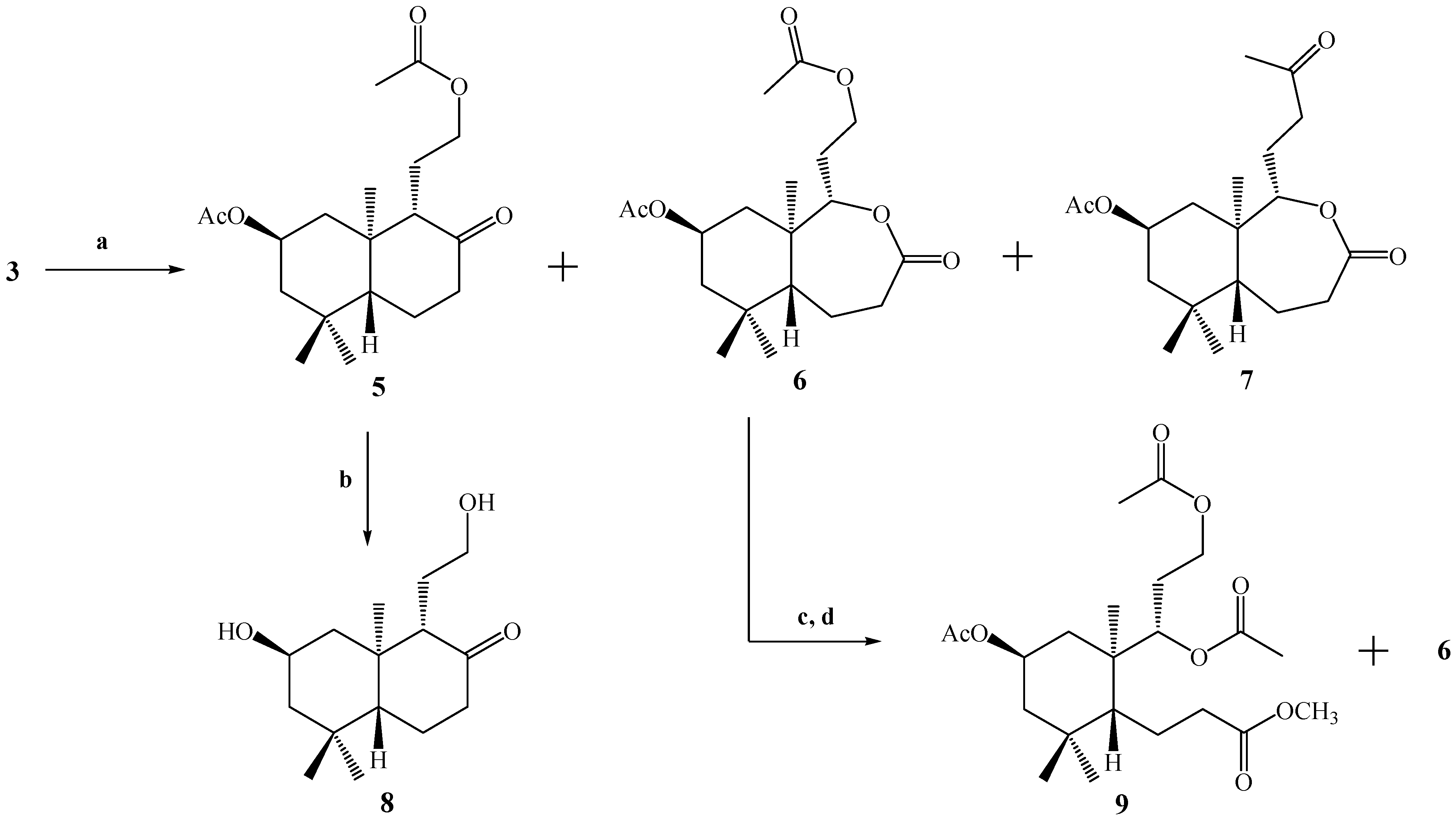
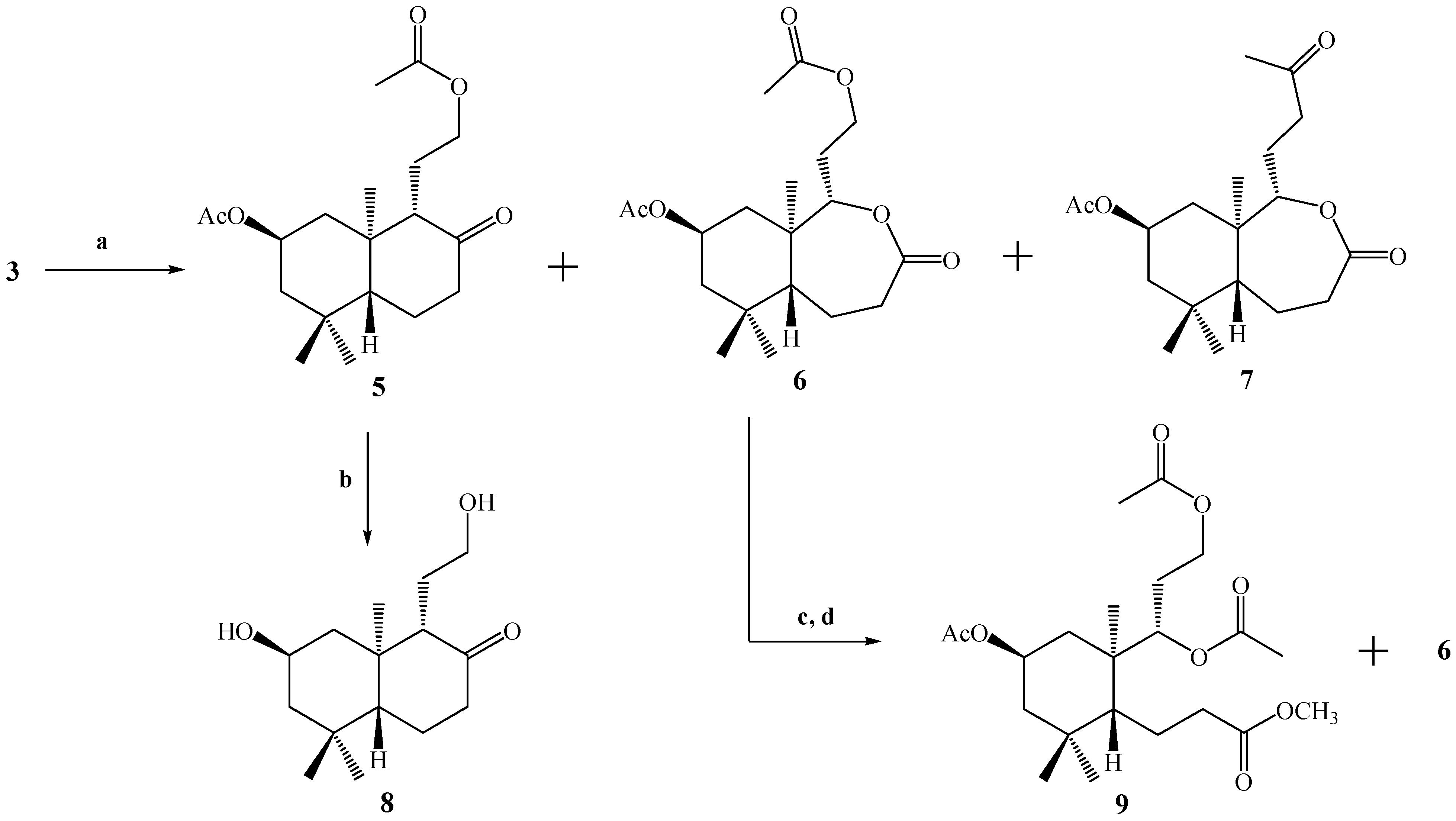
With the purpose of generating a primary alcohol at the C-12 position, compound
3 was oxidized by a Baeyer-Villiger reaction using a
m-CPBA/CH
2Cl
2 system (
Scheme 2). The reaction was not selective and three compounds were obtained: the desired compound
5 plus the lactones
6 and
7, in yields of 14%, 27.5% and 57%, respectively, in the best of the cases. The structural determination of compounds
5-7 was accomplished by IR, MS,
1H- and
13C-NMR techniques. The
1H-NMR spectrum of compound
5 shows the existence of two carbinolic hydrogens at
δ 3.87 (1H, m, H-12b) and
δ 4.09 (1H, m, H-12a) correlated (by 2D HSQC) with a carbon atom at
δ 63.8 ppm corresponding to C-12. These data also were corroborated by 2D HMBC correlations, where H-12a and H-12b showed heteronuclear
3J correlations with the acetate group carbonyl signal at
δ 171.1 ppm.
Scheme 2.
Conditions: a. m-CPBA, CH2Cl2, r.t, 24 h, C.C, 5, 14%; 6, 27.5% and 7, 57%; b. K2CO3, MeOH, reflux, 30 min. 95%; c. K2CO3, MeOH, r.t, 3 h; d. Ac2O/CH2Cl2/py, DMAP, r.t, 1.5 h, C.C; 9, 6.5% and 6 (14.2 mg).
In a similar way, in the 1H-NMR spectrum of compound 6, the presence of two carbinolic hydrogens at δ 4.11 (1H, m, H-12b) and δ 4.25 (1H, m, H-12a) correlated (by 2D HSQC) with a carbon atom at δ 61.2 ppm corresponding to C-12 was observed. These data were corroborated by 2D HMBC correlations, when H-12a and H-12b showed heteronuclear 3J correlations with the carbonyl of the acetate group at δ 170.9 ppm. In addition, the spin subsystem H-12a, H-12b, H-11a, H-11b and H-9 was assigned unequivocally by a gs-sel-1H 1D-TOCSY experiment. The presence of a lactone function in the B ring was confirmed mainly by a carbonyl (C-8) signal at δ 174.5 and 2D HMBC 3J correlations between H-9 at δ 4.16 (1H, dd, J=9.6 and 4.1 Hz) with C-8 (δ 174.5), and 2J correlations of H-7α at δ 2.70 (1H, dd, J=14.0 and 6.8 Hz) and H-7β at δ 2.53 (1H, dd, J=14.0 and 14.0 Hz) with C-8.
The structure of compound 7 was assigned mainly by the 13C-NMR spectrum. The presence of two carbonyl signals at δ 208.9 (C-13) and 175.0 (C-8) ppm confirm the presence of a ketone and a lactone function, respectively. As in the case of compound 6, in compound 7, 2D HMBC 3J correlations between H-9 at δ 4.10 (1H, dd, J=11.5 and 2.5 Hz) with C-8 (δ 175.0) were observed. On the other hand, spin subsystems between H-9 with H-11a, H-11b, H-12a and H-12b were only observed by a gs-sel-1H 1D-TOCSY experiment, when H-9 was selectively irradiated.
Hydrolysis of 5 under alkaline conditions (K2CO3/MeOH, reflux.) gave 8, bearing the desired primary alcohol in the C-12 position, in 95% yield. In the IR spectrum of 8, absorptions at 3339 and 1696 cm-1 indicated the presence of OH and C=O functions, respectively. On the other hand in the 1H spectrum of 8, carbinolic hydrogens at δ 3.32 (1H, m, H-12b) and δ 3.54 (1H, ddd, J=10.8, 5.5 and 5.5 Hz, H-12a) correlated (by 2D HSQC) with a carbon atom at δ 61.4 ppm, corresponding to C-12. These data also were corroborated by 2D HMBC correlations, where H-9 at δ 2.25 ppm (1H, d, J=9.6 Hz) showed heteronuclear 3J correlations with C-20 (δ 15.6), C-12 (δ 61.4) and 2J with C-10 (δ 43.4), C-11 (δ 24.8) and C-8 (ketone group carbonyl at δ 213.2 ppm). In addition, the H-12a, H-12b, H-11a, H-11b and H-9 spin subsystem was also unequivocally assigned by a gs-sel-1H 1D-TOCSY experiment when H-12a was selectively irradiated.
We also were interested in the hydrolysis of compound 6, but when 6 was treated with K2CO3/MeOH at room temperature, the analysis by TLC showed a very polar spot, and a routine 1H-NMR spectrum confirmed the presence of a complex mixture of compounds. Nevertheless, after acetylation of this mixture under standard conditions (Ac2O/CH2Cl2/DMAP), purification and separation by column chromatography (CC), compound 9 (6.5% yield) was obtained and starting material (compound 6) was recovered.
The structure of compound 9 was first suggested by a simple inspection of the 1H-NMR spectrum, where three acetate group signals (3H, s, CH3CO) were observed at δ 2.02, 2.04 and 2.07 ppm, along with a singlet at δ 3.67 ppm (3H, s, CO2CH3), that was assigned to a methyl ester group. This last observation suggests an opening of the lactone ring present in B. The confirmation of the proposed structure for 9 was made by a combination of 1D and 2D NMR techniques. From 2D HSQC correlations, the signal at δ 5.09 ppm (1H, d, J=10.2 Hz) assigned to H-9 showed connection (1J) with a carbon at δ 75.0 ppm (C-9), whereas from the 2D HMBC spectrum, H-9 showed 3J correlations with the carbons at δ 18.1 ppm (C-20), 61.1 ppm (C-12) and the C=O of an acetate group and 2J correlation with the signal at δ 36.7 ppm (C-10). Nevertheless, heteronuclear 3J correlations between H-9 and C-8 were not observed, and in addition H-7a (δ 2.56 ppm, 1H, ddd, J=15.7, 10.0 and 7.0 Hz) and H-7b (δ 2.32 ppm, 1H, ddd, J=13.7, 10.0 and 7.0 Hz) showed 2J correlations with a carbon at δ 173.4 ppm (C-8) belonging to the carbonyl in the methyl ester group; all this information was consistent with the suggested origin of compound 9 (opening of the lactone ring in B)
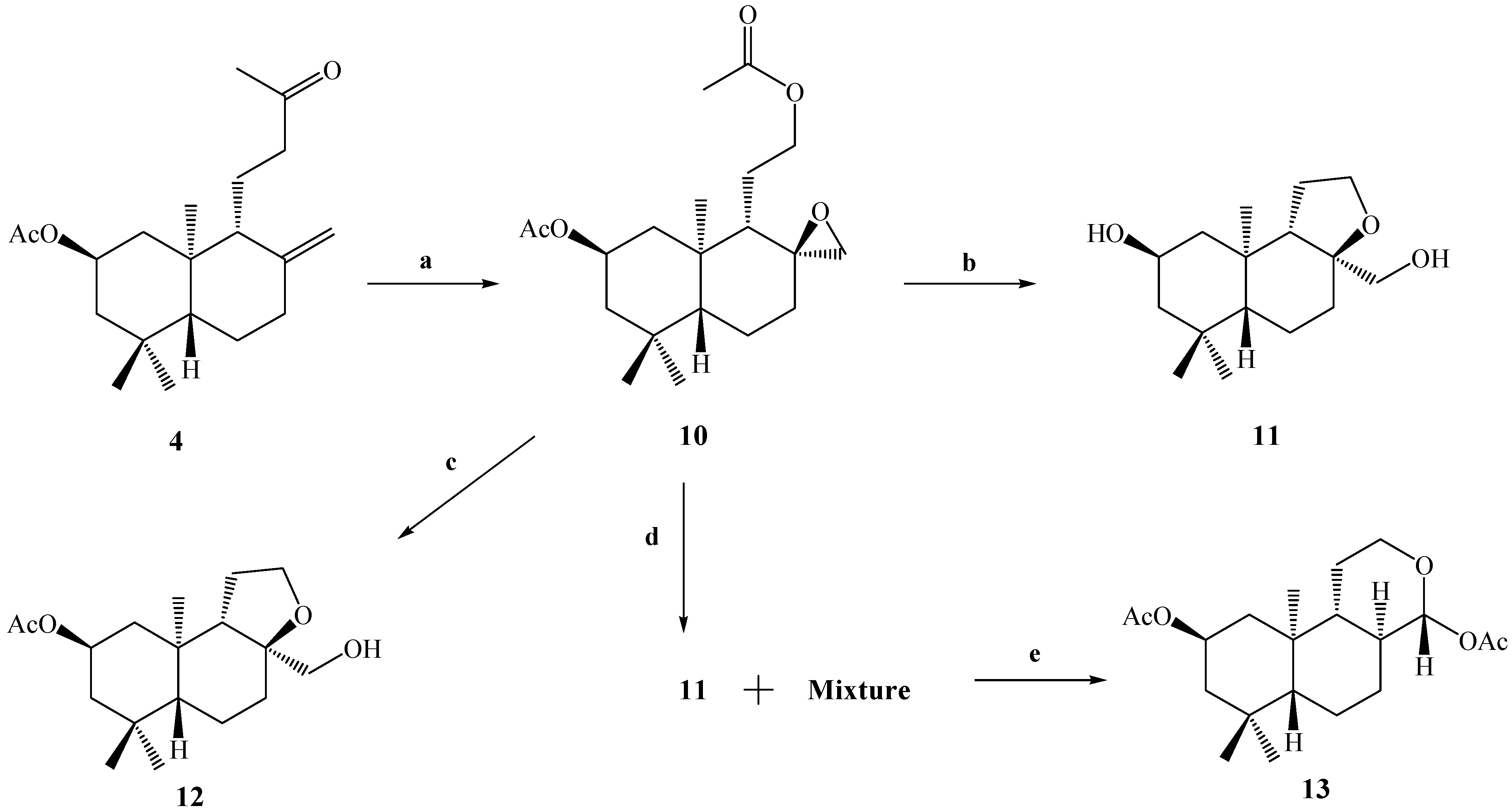
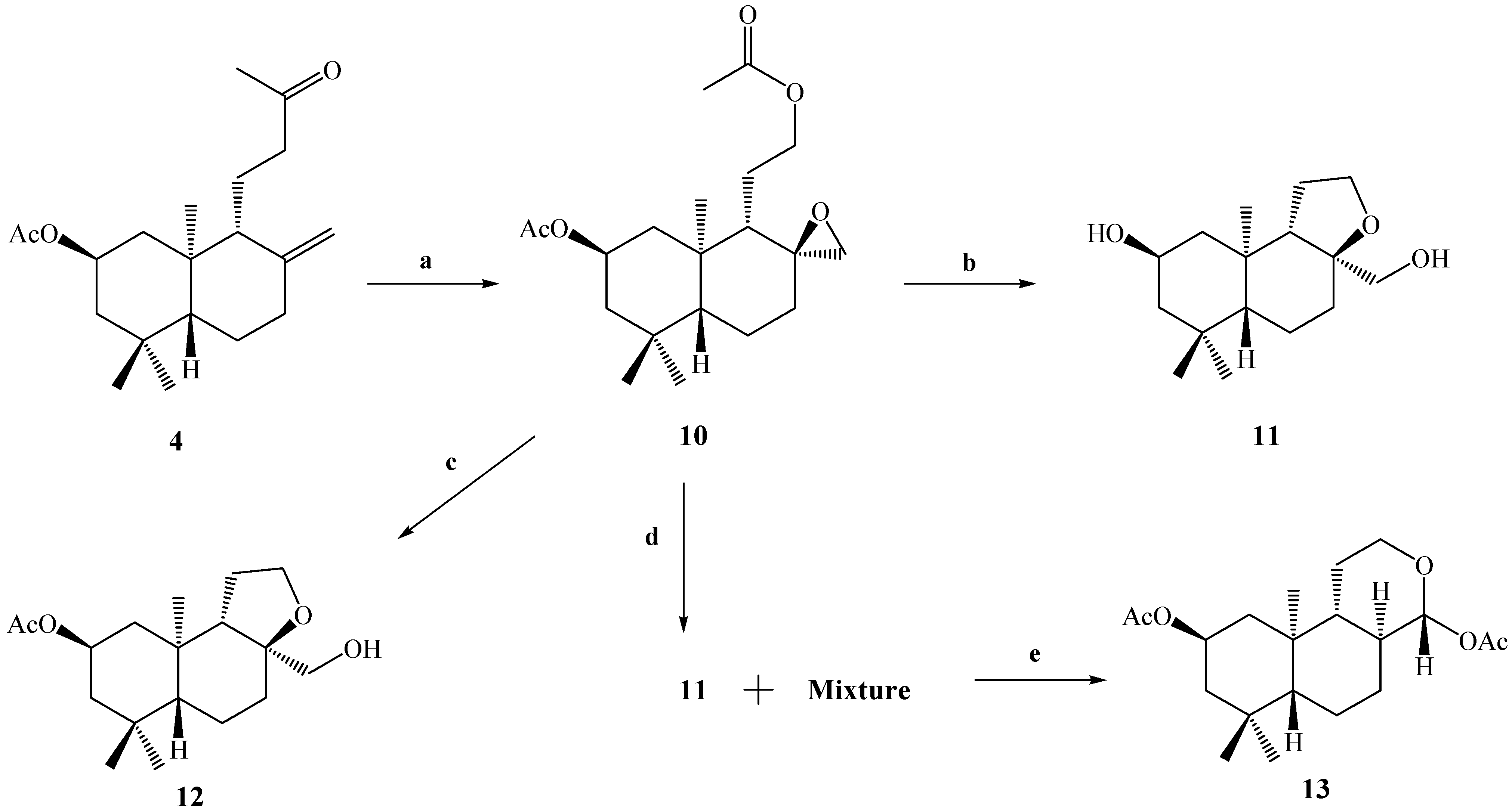
Due to the low yield of compound
8 from
3 (13.3%), we decided to carry out the Baeyer-Villiger reaction on compound
4, with the objective of performing a selective oxidation of the ketone group at C-12 and the epoxidation of the exocyclic double bond between the C-8 and C-17 carbons. The result of this reaction allowed us to obtain the desired compound
10 in 57% yield. The reaction was completely stereospecific because the compound
10 was obtained as a sole reaction product (
Scheme 3).
As in the cases of compounds 5 and 6, the presence of an acetate group at C-12 was demonstrated by the 1H-NMR spectrum of compound 10, in which the presence of two carbinolic hydrogens signals at δ 3.93 (1H, m, H-12a) and δ 4.02 (1H, m, H-12b) and two acetate groups at δ 2.00 (6H, s, CH3CO) were observed, where the first two signals were correlated (by 2D HSQC) with a carbon atom at δ 65.1 ppm corresponding to C-12. These data also were corroborated by 2D HMBC correlations, in which H-12a and H-12b showed heteronuclear 3J correlations with an acetate group carbonyl at δ 170.9 ppm and a carbon atom (C-9) at δ 50.4 ppm. In addition the H-12a, H-12b, H-11a, H-11b and H-9 spin subsystem also was unequivocally assigned by a gs-sel-1H 1D-TOCSY experiment in which H-12a and H-12b were selectively irradiated.
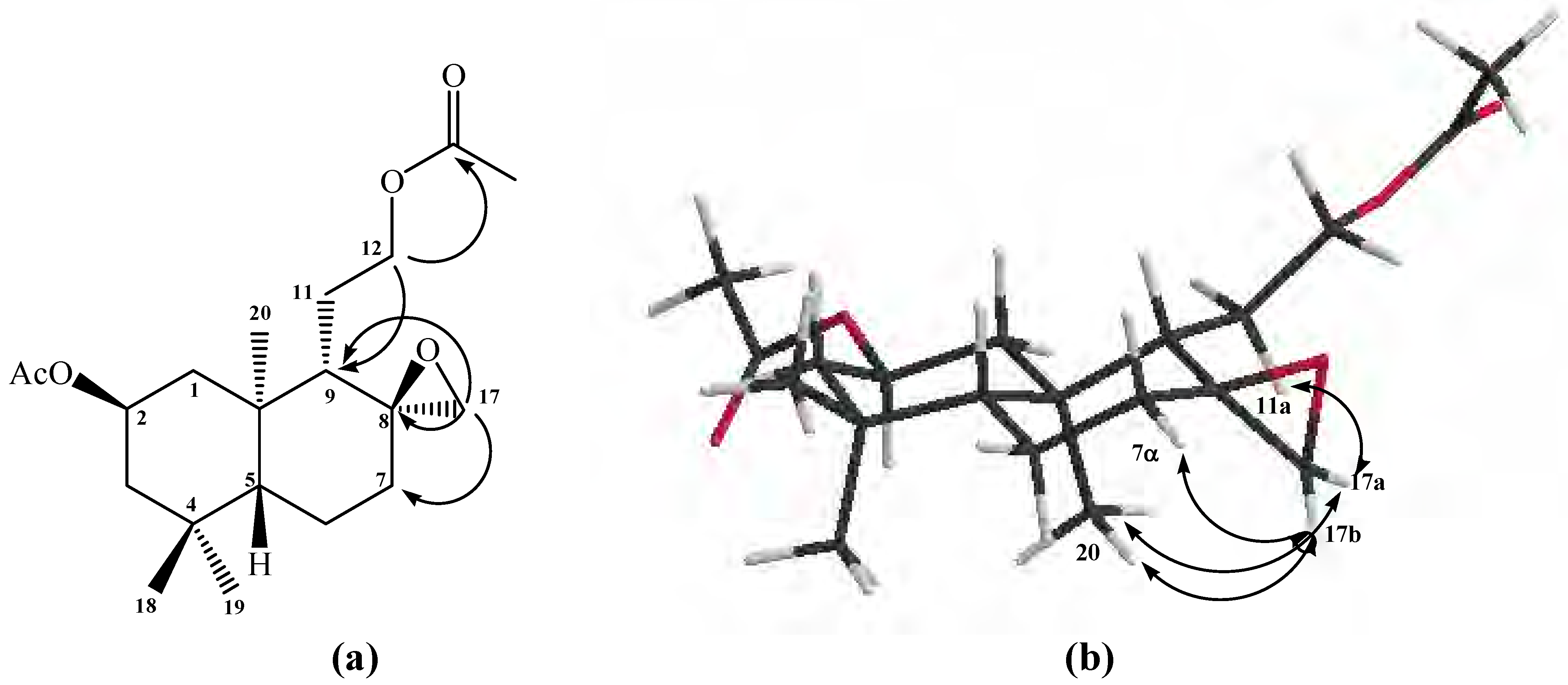
On the other hand, the presence of an oxirane ring in compound
10 was also confirmed by the following
1H-NMR data: two isolated signals at
δ 2.49 (1H, d,
J=3.9 Hz) and
δ 2.73 (1H, d,
J=3.9 Hz) were assigned to the H-17b and H-17a hydrogens, respectively. From 2D HSQC spectrum, H-17a and H-17b showed correlations with a carbon atom at
δ 50.4 ppm, assigned to C-17, whereas in the 2D HMBC spectrum, H-17a and H-17b showed
3J correlations with C-9 (
δ 50.4 ppm), with C-7 (
δ 35.9 ppm) and
2J with a quaternary carbon atom at
δ 58.2 ppm, assigned to C-8 (see
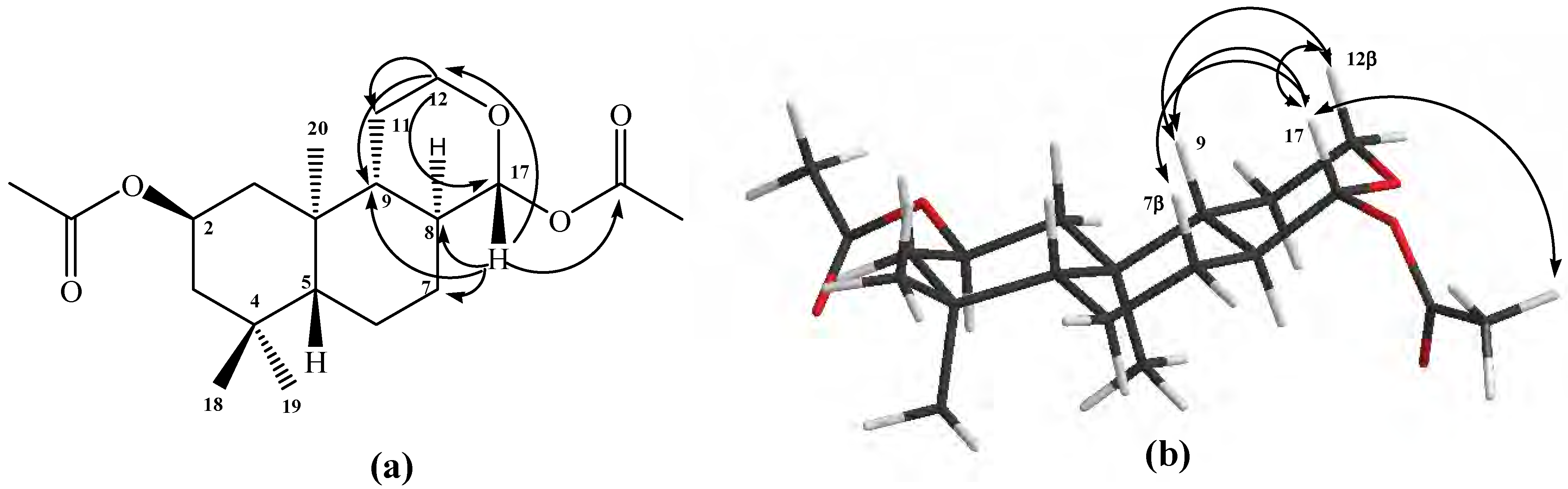
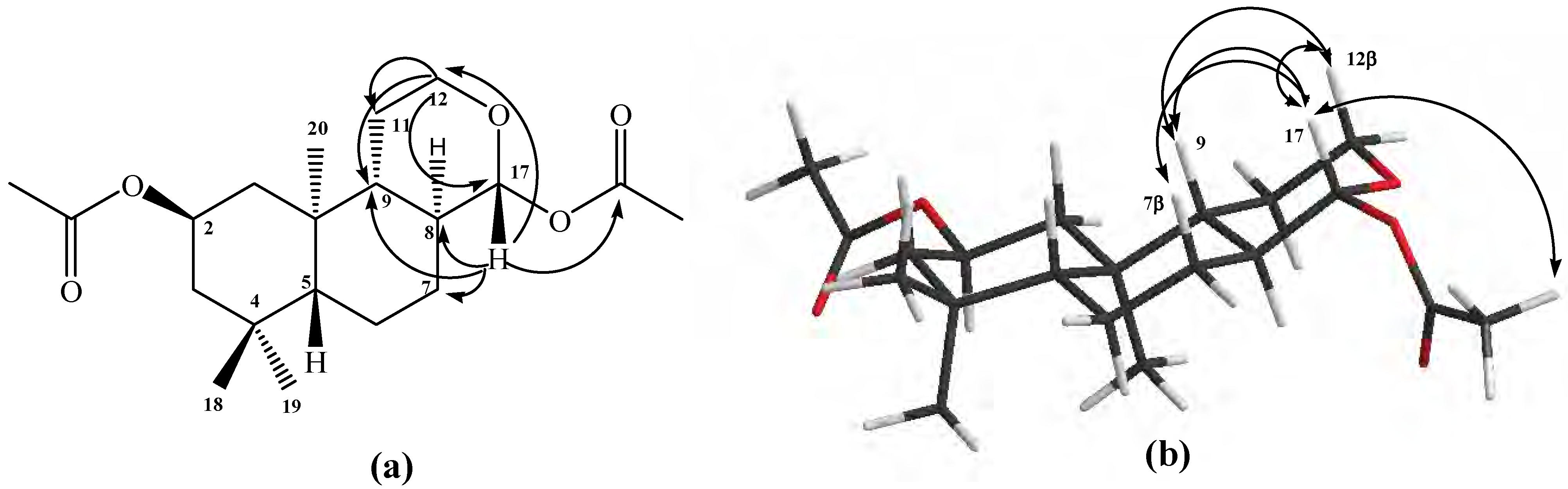
Figure 1a).
Scheme 3.
Conditions: a. m-CPBA, CH2Cl2, r.t, 8 h, 57%; b. K2CO3, MeOH, 0°C, 2 h, 95%; c. HIO4, 23.5%, THF, r.t, 2.5 h, 2.1%; d. HClO4, 6% v/v, (CH3)2CO, r.t, 24 h, C.C, 11, 52% (fraction I); e. fraction II, Ac2O/CH2Cl2/py, DMAP, r.t, 30 min. 13, 37%.
The
α-sterochemistry of the oxirane ring methylene group (C-17) was inferred mainly from gs-sel-
1H 1D-NOESY experiments: when H-17a was selectively irradiated, long range interactions (strong) with the Me-20 group (0.87 ppm) and H-11a (1.07 ppm) were observed, whereas H-17b only showed long range interactions (medium) with H-7α (1.82 ppm) and Me-20 group (see
Figure 1b).
Figure 1.
Structure of compound 10. (a) HMBC correlations. (b) NOE correlations.
Figure 1.
Structure of compound 10. (a) HMBC correlations. (b) NOE correlations.
The next goal was to perform the alkaline hydrolysis (K2CO3, MeOH, 0°C) of compound 10 with the purpose of obtaining a primary alcohol at C-12 and inducing the opening of the epoxide ring (which later, by treatment with the NaIO4/THF or Pb(OAc)4/C6H6 system, would lead to the formation of the desired compound 8). Surprisingly, under these conditions only compound 11 was obtained in 95% yield. The main spectroscopic data for the confirmation of the structure of 11 was the appearance of four carbinolic hydrogen signals at δ = 3.85 (1H, ddt, J=11.4, 11.4 and 3.8 Hz), 3.77 (2H, dd, J=8.4, and 6.5 Hz), 3.40 (1H, d, J=10.8 Hz) and 3.20 (1H, d, J=10.8 Hz), assigned to the H-2, H-12α-β, H-17a and H-17b hydrogens, respectively, which were correlated by a 2D HSQC experiment with the 13C-NMR signals at δ 64.5 (C-2), 65.5 (C-12), and 68.0 (C-17), respectively. On the other hand, in the combined 13C and 13C-DEPT-135 spectral data a fourth signal due to a quaternary carbinolic carbon was observed at δ 84.0 ppm. The downfield chemical shift of this signal suggested that this carbon could presumably be part of a third ring; in this way the molecular formula C16H28O3 was proposed for compound 11 based on the combined 1H-, 13C-, 13C-DEPT-135 spectral data. A molecular ion peak was not observed in the MS spectrum, although a peak at m/z 273 (100%), attributed to the loss of a CH2=OH fragment, was observed, suggesting the presence of a primary alcohol in the structure of 11. In addition, our unsaturation degree calculations gave a value of three: two are attributed to the A and B rings and since no signals corresponding to either C=C or C=O double bonds were observed in the 13C-NMR spectrum, the formation of a third ring C (pyran ring) fused with B, formed by the union between the carbons C-8-O-C-12, C-11, C-9 was suggested, with C-17 connected at C-8.
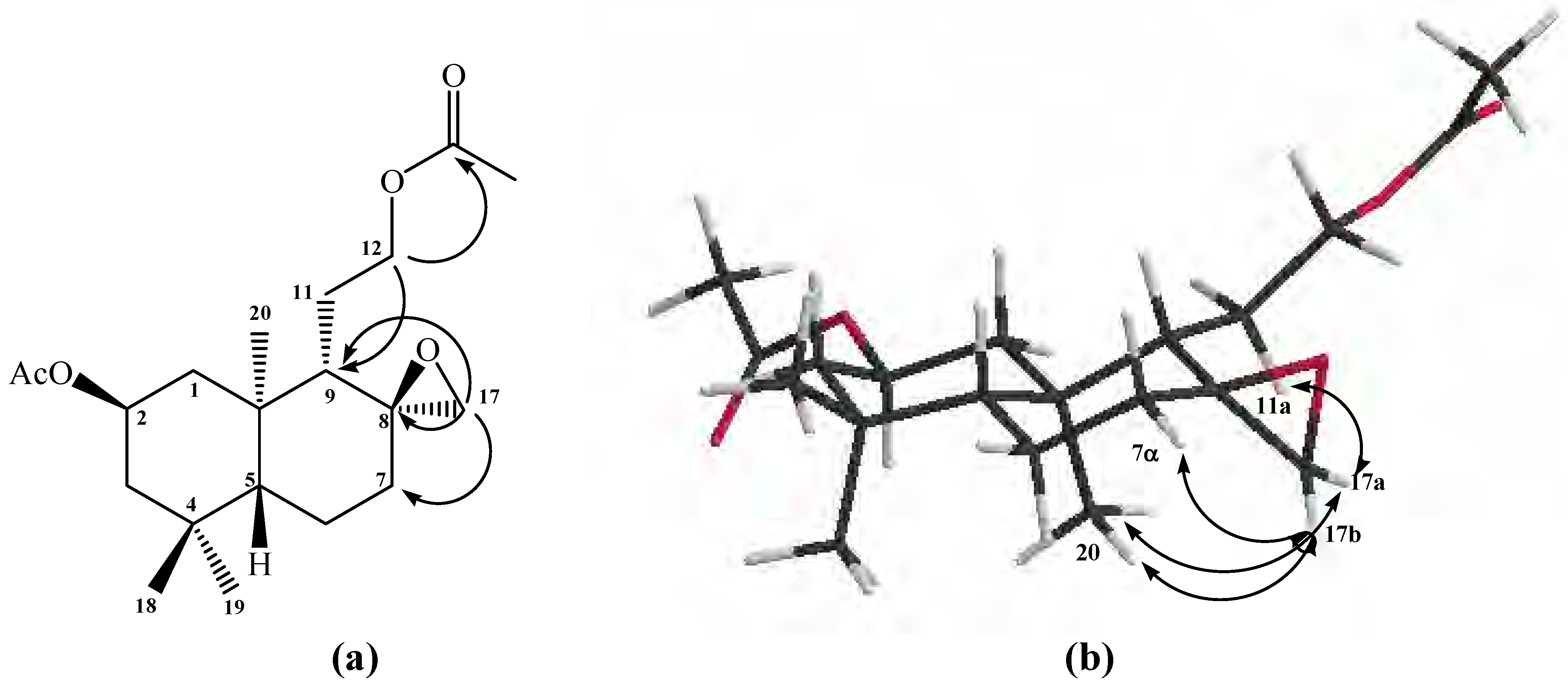
The tricyclic structure of compound
11 was confirmed mainly by the data obtained from heteronuclear 2D HSQC and HMBC experiments in which correlations between H-12α-β with C-11, C-9 and C-8 were observed, whereas H-17a showed correlations with C-7, C-9 and C-8, while H-17b only showed correlations with C-7 (see
Figure 2a). In addition, the H-12α-β, H-11α, H-11β and H-9 spin subsystem was also unequivocally assigned by a gs-sel-
1H 1D-TOCSY experiment when H-12α-β were selectively irradiated. The stereochemistry assigned to the C-8 carbon was indirectly deduced from gs-sel-
1H 1D-NOESY correlations experiments; long range interactions between H-12α with H-17b and H-17a with Me-20 were observed, therefore suggesting the stereochemistry at C-8 (see
Figure 2b).
Figure 2.
Structure of compound 11. (a) HMBC correlations. (b) NOE correlations.
Figure 2.
Structure of compound 11. (a) HMBC correlations. (b) NOE correlations.
Because under these conditions (K
2CO
3/MeOH) it was not possible to obtain the desired compound, we then focused on attempting the opening of the epoxide ring with HIO
4, according to a method previously described for other oxirane rings [
20,
21]. Under these conditions, we hoped to generate a diol function between the C-8 and C-17 carbons, which by subsequent treatment with Pb(OAc)
4 in benzene [
22], would give the desired ketone group at C-8.
When 10 was treated with aq. HIO4 (23.5%) in THF, the TLC analysis showed a complex reaction mixture, and when the crude was purified by CC, only one pure product 12 was obtained in 2.1% yield. Simple inspection of the 1H-NMR spectrum of this compound and comparison with the corresponding 1H- NMR spectrum of 11 revealed a great degree of similarity between them. This suggested that we had obtained a compound like 11, but acetylated at the 2β position. The main spectroscopic data for the confirmation of the presence of an acetate group in the structure of 12, was the appearance of a downfield signal observed at δH= 4.96 ppm for H-2β (1H, ddt, J=11.8, 11.8 and 3.9 Hz) and a singlet signal at 2.02 ppm (3H, s, CH3CO). This was also confirmed from the 13C spectrum, where the signals of the acetate group were observed at δC = 170.6 ppm (CH3CO) and 21.5 ppm (CH3CO).
In a similar way as for compound 11, the structure confirmation for 12 was suggested by the combined NMR spectral analysis: in the 1H-NMR spectrum, three carbinolic hydrogen signals at δ = 3.77 (2H, dd, J=8.4 and 6.4 Hz), 3.38 (1H, d, J=10.8 Hz) and 3.20 (1H, d, J=10.8 Hz) were assigned to the H-12α-β, H-17a and H-17b hydrogens, respectively, which were correlated by a 2D HSQC experiment with 13C signals at δ 65.5 (C-12), and 68.1 (C-17). On the other hand, in the combined 13C and 13C-DEPT-135 spectra, a fourth signal at δ 83.9 ppm was observed, assigned to a quaternary carbinolic carbon (C-8). In this way, the molecular formula C18H30O4 was proposed for compound 12 from the combined 1H-, 13C- and 13C-DEPT-135 data. We did observe a weak molecular ion peak (<1%) in the MS spectrum, but a diagnostic peak at 279 m/z (100%) attributed to the loss of a CH2=OH fragment was observed, suggesting the presence of a primary alcohol in the structure. In addition, our calculation of the unsaturation degree gave a value of four; two are attributed to the A and B rings, one to an acetate group (CH3C=O) and since no signals corresponding to C=C or double bonds were observed in the 13C-NMR spectrum, therefore the formation of a third ring C (pyran ring) fused with B, formed by the union between the carbons C-8-O-C-12, C-11, C-9 was suggested, with C-17 only bonding with C-8. The tricyclic structure of compound 12 was confirmed mainly by the data obtained from heteronuclear 2D HSQC and HMBC correlations; in the latter, correlations between H-12α-β with C-11, C-9 and C-8 were observed, whereas H-17a shows correlations with C-7, C-9 and C-8, while the H-17b only showed correlations with C-7. In addition, the H-12α-β, H-11α, H-11β and H-9 spin sub system was also unequivocally assigned by a gs-sel-1H 1D-TOCSY experiment, when H-12α-β were selectively irradiated. The stereochemistry assigned to the C-8 carbon was deduced from gs-sel-1H 1D-NOESY correlations experiments; long range interactions between H-12α with H-17b and H-17a with Me-20 were observed.
Because the desired result in the opening of the oxirane ring in compound
10 was not obtained when routes
a and
b (
Scheme 2) were used, we decided to make a last attempt. This time we reacted compound
10 in the presence of 6% HClO
4 (a method used for acid-catalyzed solvolytic epoxide opening for the generation of 1,2-diols [
23]). After 24 hours of reaction, the TLC analysis demonstrated a very clear and more polar spot, and a second diffuse and less polar spot. We then decided to stop the reaction, carried out the corresponding workup and purified the crude product by CC. The most polar spot turned out to be compound
11 (52% yield), as was deduced by inspection and comparison of routine
1H- and
13C- NMR spectra of this compound with the spectra of
11, obtained previously through route
b in
Scheme 2. Nevertheless, the routine
1H-NMR spectrum of the less polar spot, revealed the presence of a complex mixture of at least two compounds, which were impossible to separate by CC. Under these circumstances, we thought that acetylation of this mixture would enable us to obtain a greater separation of these compounds. Thus acetylation of the mixture under standard conditions (Ac
2O/CH
2Cl
2/DMAP) followed by CC purification, allowed us to isolate the compound
13 (37% yield) as the major product.
The main spectroscopic data for the confirmation of the structure of 13 were the appearance of four signals in the 1H-NMR spectrum at δ = 5.23 (1H, d, J=8.8 Hz, H-17), 5.00 (1H, ddt, J=11.7, 11.7 and 4.4 Hz), presumably of carbinolic hydrogens and thus assigned to H-2, δ = 4.05 (1H, dddd, J=11.3, 4.4, 4.4 and 2.0 Hz) and 3.53 (1H, ddd, J=11.3, 11.3 and 4.4 Hz); two singlet signals for an acetate group at δ = 2.09 and 2.01 ppm were also observed. The signals at δ = 5.23, 4.05 and 3.53 were correlated by a 2D HSQC experiment with with 13C-NMR signals at δC = 98.0 (CH) and 66.1 (CH2), respectively. On the other hand, the chemical downfield shift observed for doublet signal (δH = 5.23 and δC = 98.0), suggested that presumably this signal could be a hemiacetalic hydrogen.
The molecular formula for compound 13 was proposed to be C20H32O5, based on the combined 1H, 13C, 13C-DEPT-135 spectra. It was not possible to observe a molecular ion peak in the MS spectrum. Our calculation of the unsaturation degree gave a value of five: two are attributed to the A and B rings, other two attributed to the carbonyls of acetate groups. On the other hand, since no signals corresponding to double bonds (C=C) were observed in the 13C-NMR spectrum, the formation of a third ring C, presumably fused with B, was suggested.
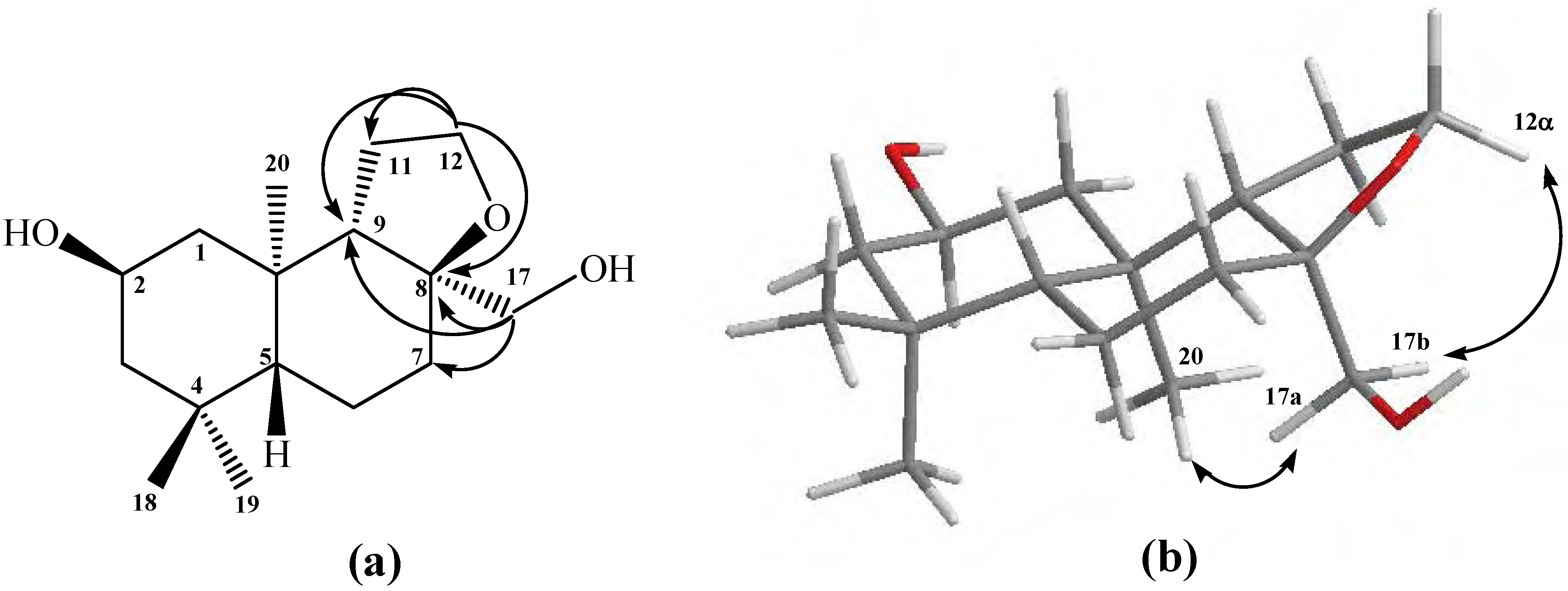
In addition to the antecedents described above, the tricyclic structure proposal for compound
13 was confirmed mainly by the data obtained from heteronuclear 2D HSQC and HMBC correlations; from the latter, the signal at
δΗ = 5.23 was assigned to H-17 and showed
2J correlations with C-8 and
3J with C-7 (
δC = 27.6), C-9 (
δC = 52.0), C-12 (
δC = 66.1) and CH
3C=O (
δC = 169.9) (see
Figure 3a). The stereochemistry assigned to C-17 was deduced by the value of
3JH-H = 8.8Hz corresponding to an axial-axial coupling constant between H-17 and H-8, and from gs-sel-
1H 1D-NOESY correlation experiments. With these last the following long range interactions were observed: H-17 with
δΗ = 3.53 (H-12β), 2.09 (CH
3CO-C-17), 1.54 (H-8), 1.10 (H-9) and 0.97 (H-7β), important data that confirm a β-orientation for H-17. In addition, H-12β (
δΗ = 3.53) showed long range interactions with H-9 (see
Figure 3b).
Figure 3.
Structure of compound 13. (a) HMBC correlations. (b) NOE correlations.
Figure 3.
Structure of compound 13. (a) HMBC correlations. (b) NOE correlations.
Experimental
General
Unless otherwise stated, all chemical reagents purchased (Merck or Aldrich) were of the highest commercially available purity and were used without previous purification. Melting points were measured (in triplicate) on a Stuart-Scientific SMP3 apparatus and are uncorrected. IR spectra were recorded as thin films in a Nicolet Impact 420 spectrometer and frequencies are reported in cm-1. Optical rotations were measured with a sodium lamp (λ=589 nm, D line) on a Perkin Elmer 241 digital polarimeter equipped with 1 dm cells at the temperature indicated in each case. Low resolution mass spectra were recorded on a Shimadzu QP-2000 spectrometer at 70eV ionising voltage and are given as m/z (% rel. int.) 1H-, 13C- (DEPT 135 and DEPT 90), sel. 1D 1H NOESY, sel. 1D 1H TOCSY, 2D HSQC and 2D HMBC spectra were recorded in CDCl3 solutions and are referenced to the residual peaks of CHCl3 at δ 7.26 ppm and δ 77.0 ppm for 1H- and 13C-, respectively, on a Bruker Avance 400 Digital NMR spectrometer, operating at 400.1MHz for 1H and 100.6MHz for 13C. Chemical shifts are reported in δ ppm and coupling constants (J) are given in Hz. Silica gel (Merck 200-300 mesh) was used for C.C. and silica gel plates HF-254 for TLC. TLC spots were detected by heating after spraying with 25% H2SO4 in H2O.
Synthesis of 2β,12-diacetoxy-13,14,15,16,17-pentanor-ent-labdane-8-one (5), 2β,12-diacetoxy-13,14, 15,16,17-pentanor-ent-labdane-8-oxa-9-oxo (6) and 2β−acetoxy-14,15,17-trinor-ent-labdane-8-oxa-9-oxo-12-one (7).
To a solution of 3 (1.31 g, 4.06 mmol) in CH2Cl2 (100 mL), m-CPBA (0.47 g, 2.72 mmol) and NaHCO3 (0.228 g, 2.72 mmol) were added and the mixture was stirred at room temperature for 24 h. Then a saturated solution (2 x 50 mL) of NaHCO3 was added, the organic layer was extracted and washed with H2O (2 x 50 mL). The water layer was discarded and the organic layer dried over Na2SO4 filtered, evaporated and chromatographed on silica-gel with petroleum ether/EtOAc mixtures of increasing polarity (19.8:0.2→7:13) to give three fractions: Fraction I (190.3 mg, 14%) Compound 5: viscous oil, [α]D25 = +40.8° (c 1.46, CHCl3); 1H-NMR: 4.96 (1H, ddt, J=12.0, 12.0 and 3.9 Hz, H-2), 4.09 (1H, m, H-12a), 3.87 (1H, m, H-12b), 2.45 (1H, ddd, J=13.7, 4.9 and 2.0 Hz, H-7α), 2.32 (1H, dd, J=13.7 and 7.1 Hz, H-7β), 2.20 (1H, bd, J=10.0 Hz, H-9), 2.07 (3H, m, H-1α, H-6β and H-11a), 2.02 (3H, s, OAc), 2.01 (3H, s, OAc), 1.81 (1H, ddd, J=12.0, 3.9 and 2.1 Hz, H-3α), 1.63 (1H, dd, J=12.9 and 4.7 Hz, H-6α), 1.54 (1H, dd, J=12.9 and 2.5 Hz, H-5), 1.49 (1H, m, H-11b), 1.31 (1H, dd, J=12.0 and 12.0 Hz, H-3β), 1.20 (1H, dd, J=12.0 and 12.0 Hz, H-1β), 1.02 (3H, s, Me-18), 0.93 (3H, s, Me-19), 0.79 (3H, s, Me-20); 13C-NMR: 44.0 (C-1), 68.3 (C-2), 46.5 (C-3), 34.9 (C-4), 53.5 (C-5), 23.0 (C-6), 41.8 (C-7), 210.4 (C-8), 59.9 (C-9), 43.3 (C-10), 21.5 (C-11), 63.8 (C-12), 33.5 (C-18), 22.4 (C-19), 15.4 (C-20), 171.1 (CH3CO), 21.4 (CH3CO), 170.5 (CH3CO), 21.0 (CH3CO); IR: 2960, 1731, 1714, 1465, 1368, 1240, 1025; MS: 338 ([M+] <1%), 278 (13.4%), 252 (11.9%), 218 (9.0%), 204 (8.7%), 203 (54.7%), 192 (9.5%), 178 (13.4%), 177 (100%), 136 (11.0%), 135 (41.5%), 134 (12.4%), 119 (10.7%), 107 (11.9%), 93 (10.2%), 79 (7.5%) 69 (8.2%). Fraction II (395.7 mg, 27.5%) Compound 6: viscous oil, [α]D23 = -17.7° (c 7.9, CHCl3); 1H-NMR: 5.00 (1H, ddt, J=11.7, 11.7 and 4.1 Hz, H-2), 4.25 (1H, m, H-12a), 4.16 (1H, dd, J=9.6 and 4.1 Hz, H-9), 4.11 (1H, m, H-12b), 2.70 (1H, dd, J=14.0 and 6.8 Hz, H-7α), 2.53 (1H, dd, J=14.0 and 14.0 Hz, H-7β), 2.04 (3H, s, OAc), 2.02 (3H, s, OAc), 1.97 (1H, m, H-6β), 1.87 (2H, m, H-11a and H-11b), 1.79 (2H, m, H-1α and H-3α), 1.56 (1H, m, H-6α), 1.28 (1H, dd, J=12.3 and 12.3 Hz, H-3β), 1.19 (1H, dd, J=12.0 and 12.0 Hz, H-5), 1.04 (1H, dd, J=11.7 and 11.7 Hz, H-1β), 1.00 (6H, s, Me-18 and Me-20), 0.92 (3H, s, Me-19), 13C-NMR: 41.4 (C-1), 67.9 (C-2), 46.5 (C-3), 35.9 (C-4), 58.3 (C-5), 19.5 (C-6), 34.3 (C-7), 174.5 (C-8), 84.3 (C-9), 41.7 (C-10), 28.6 (C-11), 61.2 (C-12), 33.3 (C-18), 22.3 (C-19), 14.1 (C-20), 170.9 (CH3CO), 20.9 (CH3CO), 170.5 (CH3CO), 21.3 (CH3CO); IR: 2966, 1737, 1440, 1368, 1245, 1194, 1173, 1035; MS: 354 ([M+] <1%), 223 (4.6%), 179 (17.2%), 178 (100%), 163 (18.9%), 150 (5.0%), 145 (12.7%), 137 (49.8%), 134 (43.5%), 122 (20.7%), 121 (54.5%), 120 (30.1%), 119 (61.9%), 109 (5.5%), 108 (5.3%), 107 (30.0%), 105 (7.4%), 97 (6.5%), 93 (13.8%), 79 (8.5%), 67 (8.8%), 55 (10.6%). Fraction III (212.1 mg, 57%) Compound 7: viscous oil, [α]D25 = -42.5° (c 8.12, CHCl3); 1H-NMR: 5.00 (1H, ddt, J=11.8, 11.8 and 4.1 Hz, H-2), 4.10 (1H, dd, J=11.5 and 2.5 Hz, H-9), 2.66 (2H, m, H-7α and H-12a), 2.51 (2H, m, H-7β and H-12b), 2.12 (3H, s, H-16), 2.02 (3H, s, OAc), 1.94 (2H, m, H-6β and H-11a), 1.82 (1H, ddd, J=12.0, 3.6 and 2.4 Hz, H-1α), 1.77 (1H, ddd, J=12.4, 4.0 and 2.2 Hz, H-3α), 1.67 (1H, m, H-11b), 1.53 (1H, m, H-6α), 1.28 (1H, dd, J=12.3 and 12.3 Hz, H-3β), 1.17 (1H, dd, J=12.2 and 2.4 Hz, H-5), 1.15 (1H, dd, J=11.8 and 11.8 Hz, H-1β), 0.98 (6H, s, Me-18 and Me-20), 0.91 (3H, s, Me-19); 13C-NMR: 41.2 (C-1), 67.9 (C-2), 46.5 (C-3), 35.9 (C-4), 58.2 (C-5), 19.5 (C-6), 34.3 (C-7), 175.0 (C-8), 86.5 (C-9), 41.9 (C-10), 23.0 (C-11), 39.2 (C-12), 208.9 (C-13), 30.2 (C-16), 33.4 (C-18), 22.3 (C-19), 13.9 (C-20), 170.5 (CH3CO), 21.3 (CH3CO); IR: 2966, 1737, 1721, 1440, 1363, 1245, 1178, 1030, 958; MS: 338 ([M+] <1%), 223 (4.0%), 179 (19.5%), 178 (100%), 163 (17.8%), 150 (5.0%), 145 (12.0%), 137 (48.3%), 135 (15.4%), 134 (44.6%), 123 (6.6%), 122 (20.3%), 121 (53.5%), 120 (28.4%), 119 (59.9%), 109 (5.7%), 108 (5.1%), 107 (29.6%), 105 (7.4%), 97 (7.1%), 95 (7.2%), 94 (5.2%), 93 (13.0%), 91 (8.1%), 85 (4.7%), 82 (5.6%), 81 (7.6%), 79 (8.2%), 69 (8.2%), 67 (9.0%), 55 (11.1%).
Synthesis of 2β,12-dihydroxy-13,14,15,16,17-pentanor-ent-labdane-8-one (8).
K2CO3 (110.0 mg, 0.796 mmol) was added to a solution of ketone 5 (224.8 mg, 0.664 mmol) in MeOH (50 mL), and the mixture stirred under reflux for 30 min. The solvent was removed until a volume of approximately 5 mL and 30 mL of water were added and the mixture extracted with EtOAc (3 x 30 mL) and the combined organic layers washed successively with water, dried over Na2SO4, filtered, evaporated and chromatographed on silica-gel with mixtures of EtOAc/MeOH of increasing polarity (18:2→15.2:4.8) to give 160.5 mg (95%) of compound 8: white solid, mp = 190-191.6°C (Et2O/MeOH); [α]D25 = +28.8° (c 2.68, MeOH); 1H-NMR: 3.75 (1H, ddt, J=12.0, 12.0, 4.0 Hz, H-2), 3.54 (1H, ddd, J=10.8, 5.5 and 5.5 Hz, H-12a), 3.32 (1H, m, H-12b), 2.38 (1H, ddd, J=13.0, 4.8 and 2.0 Hz, H-7α), 2.30 (1H, dd, J=13.0 and 7.0 Hz, H-7β), 2.25 (1H, d, J=9.6 Hz, H-9), 1.99 (2H, m, H-6β and H-1α), 1.89 (1H, ddd, J=9.4, 9.4 and 5.0 Hz, H-11a), 1.73 (1H, ddd, J= 12.4, 3.8 and 2.1 Hz, H-3α), 1.57 (1H, dd, J=12.9 and 5.0 Hz, H-6α), 1.47 (1H, dd, J=12.9 and 2.1 Hz, H-5), 1.41 (1H, m, H-11b), 1.15 (1H, dd, J=12.0 and 12.0 Hz, H-3β), 1.06 (1H, dd, J=12.0 and 12.0 Hz, H-1β), 0.96 (3H, s, Me-18), 0.83 (3H, s, Me-19), 0.69 (3H, s, Me-20); 13C-NMR: 47.4 (C-1), 64.4 (C-2), 50.2 (C-3), 34.7 (C-4), 53.2 (C-5), 23.0 (C-6), 41.9 (C-7), 213.2 (C-8), 60.1 (C-9), 43.4 (C-10), 24.8 (C-11), 61.4 (C-12), 33.4 (C-18), 22.4 (C-19), 15.6 (C-20); IR: 3339, 2935, 1697, 1470, 1373; MS: 254 ([M]+ <1%), 288 (16.1%), 236 (18.1%), 222 (16.2%), 221 (100%), 203 (47.0%), 137 (5.1%), 135 (5.7%), 121 (5.47%), 91 (6.4%), 79 (5.0%), 55 (5.2%).
Synthesis of methyl 2β,9,12-triacetoxy-13,14,15,16,17-pentanor-8,9-seco-ent-labdane-8-oate (9).
To a solution of 6 (321.6 mg, 0.907 mmol) in MeOH (50 mL), finely divided K2CO3 (150.2 mg, 1.09 mmol) was added and the mixture stirred at room temperature for 3 h. The solvent was removed until a volume of approximately 5 mL and 30 mL of water were added, then 5% HCl (15 mL) was added, the mixture was extracted with EtOAc (3 x 30 mL) and the combined organic layers were washed successively with 10% NaHCO3 and water, dried over Na2SO4, filtered and evaporated. The crude (256.4 mg) was redissolved in CH2Cl2 (50 mL) and pyridine (5 mL), then Ac2O (3 mL) and DMAP (20 mg) were added and the mixture stirred at room temperature for 1.5 h. The solvent was removed until a volume of approximately 20 mL and 30 mL of water and 10% KHSO4 (10 mL) were added, the organic layer was extracted with EtOAc (3 x 20 mL). The combined organic layers were washed to neutrality with a saturated solution of NaHCO3 and water, dried over Na2SO4, filtered, evaporated and chromatographed eluting with mixtures of petroleum ether/EtOAc of increasing polarity (18:2→9:11), then two fractions were obtained: Fraction I (25.3 mg, 6.5% from 6) compound 9: viscous oil; [α]D25 = -6.0° (c 0.65, CHCl3); 1H-NMR: 5.09 (1H, d, J=10.2 Hz, H-9), 4.98 (1H, ddt, J=12.0, 12.0 and 3.7 Hz, H-2), 4.06 (1H, ddd, J=10.4, 6.2 and 4.8 Hz, H-12a), 3.95 (1H, ddd, J=10.4, 10.4 and 4.8 Hz, H-12b), 3.67 (3H, s, OCH3), 2.56 (1H, ddd, J=15.7, 10.0 and 7.0 Hz, H-7a), 2.32 (1H, ddd, J=13.7, 10.0 and 7.0 Hz, H-7b), 2.07 (3H, s, OAc), 2.05 (1H, m, H-11a), 2.04 (3H, s, OAc), 2.02 (3H, s, OAc), 1.85 (1H, ddd, J=12.0, 3,7 and 2.7 Hz, H-3α), 1.77 (2H, m, H-1α and H-11b), 1.70 (2H, m, H-6a and H-6b), 1.29 (1H, dd, J=12.0 and 12.0 Hz, H-3β), 1.18 (1H, dd, J=12.0 and 12.0 Hz, H-1β), 1.04 (3H, s, Me-19), 0.96 (6H, s, Me-19 and Me-20); 13C NMR: 46.7 (C-1), 68.0 (C-2), 38.0 (C-3), 43.2 (C-4), 48.2 (C-5), 21.1 (C-6), 36.0 (C-7), 173.4 (C-8), 75.0 (C-9), 36.7 (C-10), 28.2 (C-11), 61.1 (C-12), 33.7 (C-18), 22.9 (C-19), 18.1 (C-20), 51.6 (CH3O), 171.0 (CH3CO), 21.4 (CH3CO), 170.8 (CH3CO), 20.9 (CH3CO), 170.5 (CH3CO), 20.7 (CH3CO); IR: 2960, 1731, 1434, 1373, 1240, 1178, 1024, 958; MS: 428 ([M]+ <1%), 210 (22.9%), 209 (100%), 195 (14.3%), 177 (27.7%), 159 (54.2%), 154 (13.3%), 136 (14.0%), 135 (20%), 133 (13.6%), 129 (14.7%), 123 (10.7%), 121 (10.3%), 119 (10.0%), 117 (19.7%), 99 (16%), 93 (10.7%), 81 (8.5%). Fraction II (14.2 mg) Compound 6.
Synthesis of 2β,12-diacetoxy-8β,17-epoxy-13,14,15,16-tetranor-ent-labdane (10).
A solution of 4 (2.4 g, 7.49 mmol) in 100mL of CH2Cl2 was prepared, then NaHCO3 (0.90 g, 10.7 mmol) and m-CPBA (1.85 g, 8.03 mmol) in four portions of approximately 0.463 g was added. The reaction mixture was stirred at room temperature for 8 h. The mixture was filtered and the organic layer extracted with a saturated solution of NaHCO3 (3 x 30 mL) and the organic layers washed successively with water, dried over Na2SO4, filtered, evaporated and chromatographed eluting with mixtures of petroleum ether/EtOAc of increasing polarity (19.8:0.2→7:13) to give 1.50 g (57%) of 10. compound 10: viscous oil; [α]D25 = +10.0° (c 11.8, CHCl3); 1H-NMR: 4.99 (1H, ddt, J=12.0, 12.0 and 3.9 Hz, H-2), 4.02 (1H, m, H-12b), 3.93 (1H, m, H-12a), 2.73 (1H, d, J=3.9 Hz, H-17a), 2.49 (1H, d, J=3.9 Hz, H-17b), 2.00 (6H, s, 2xOAc), 1.98 (1H, m, H-1α), 1.82 (3H, m, H-3α, H-6β and H-7α), 1.55 (1H, dd, J=7.3 and 3.4 Hz, H-9), 1.41 (3H, m, H-7, H-11b and H-6α), 1.23 (1H, dd, J=12.0 and 12.0 Hz, H-3β), 1.07 (3H, m, H-1β, H-5 and H-11a), 0.92 (3H, s, Me-18), 0.89 (3H, s, Me-19), 0.87 Me-20); 13C NMR: 43.9 (C-1), 68.3 (C-2), 46.5 (C-3), 34.7 (C-4), 54.3 (C-5), 21.2 (C-6), 35.9 (C-7), 58.2 (C-8), 50.4 (C-9), 41.2 (C-10), 21.5 (C-11), 65.1 (C-12), 50.4 (C-17), 33.4 (C-18), 22.3 (C-19), 15.3 (C-20), 170.9 (CH3CO), 21.4 (CH3CO), 170.5 (CH3CO), 21.0 (CH3CO); IR: 2950, 1737, 1465, 1368, 1240, 1024, 958, 892; MS: 352 ([M]+ <1%), 292, (43.5%), 277 (14.5), 232 (22.4%), 218 (17.9%), 217 (100%), 203, (13.8%), 201 (11.8%), 199 (22.3%), 191 (24.8%), 189 (15.8%), 187 (14.7%), 176 (12.9%), 175 (20.3), 173 (11.3%), 163 (11.7%), 161 (16.8%), 159 (13.9%), 157 (15.1%), 156 (23.0%), 150 (13.3%), 149 (32.5%), 148 (14.2%), 147 (15.8%), 145 (18.2%), 137 (13.1%), 136 (25.6%), 135 (92.1%), 134 (18.3%), 133 (29.7%), 131 (17.5%), 123 (18.7%), 122 (22.6%), 121 (50.9%), 120 (15.5%), 119 (40.0%), 114 (26.6%), 110 (15.8%), 109 (37.8%), 107 (47.0%), 105 (31.4%), 97 (18.2%), 95 (27.0%), 93 (45.7%), 91 (29.2%), 81 (24.9%), 79 (28.8%), 77 (12.6%), 69 (21.1%), 67 (22.8%), 55 (23.0%).
Synthesis of 8β,12-epoxy-13,14,15,16-tetranor-ent-labdane-2β,17-diol (11).
To a solution of 10 (185.9 mg, 0.527 mmol) in MeOH (30 mL), finely divided K2CO3 (94.8 mg, 0.686 mmol) was added and the mixture stirred at 0°C for 2 hours. Then the mixture was filtered, evaporated and chromatographed with petroleum ether/EtOAc mixtures of increasing polarity (19:1→20:0) yielding 134.4 mg (95 %). Compound 11: viscous oil; [α]D25 = +5.19° (c 0.366, CHCl3); 1H-NMR: 3.85 (1H, ddt, J=11.4, 11.4 and 3.8 Hz, H-2), 3.77 (2H, dd, J=8.4 and 6.5 Hz, H-12α and H-12β) 3.40 (1H, d, J=10.8 Hz, H-17a), 3.20 (1H, d, J=10.8 Hz, H-17b), 2.06 (1H, ddd, J=14.7, 5.9 and 3.9 Hz, H-7α), 2.03 (1H, m, H-11β), 1.99 (1H, m, H-11α), 1.92 (1H, ddd, J=13.2, 7.1 and 2.0 Hz, H-11α), 1.80 (1H, ddd, J=11.4, 3.8 and 2.5 Hz, H-3α), 1.70 (1H, ddd, J=14.7, 10.1 and 6.8 Hz, H-7β), 1.53 (1H, d, J=7.1 Hz, H-9), 1.44 (2H, m, H-6α and H-6β), 1.12 (1H, dd, J=11.4 and 11.4 Hz, H-3β), 0.97 (3H, s, Me-20), 0.95 (3H, s, Me-18), 0.93 (3H, s, Me-19), 0.86 (1H, dd, J=11.4 and 11.4 Hz, H-1β), 0.83 (1H, dd, J=11.7 and 3.9 Hz, H-5); 13C-NMR: 50.7 (C-1), 64.5 (C-2), 51.1 (C-3), 34.9 (C-4), 50.7 (C-5), 17.7 (C-6), 29.7 (C-7), 84.0 (C-8), 53.2 (C-9), 37.7 (C-10), 27.1 (C-11), 65.5 (C-12), 68.0 (C-17), 33.5 (C-18), 22.9 (C-19), 16.5 (C-20); IR: 3375, 2925, 1460, 1368, 1255, 1209, 1153, 1035; MS: 268 ([M]+ <1%), 238 (16.9%), 237, (100%), 219 (14.9%), 135 (23.6%), 107 (7.1%), 97 (32.5%), 95 (7.0%), 93 (7.3%), 85 (9.0%), 69 (8.6%), 55 (8.8%).
Synthesis of 2β−acetoxy-8β,12-epoxy-13,14,15,16-tetranor-ent-labdan-17-ol (12).
A solution of 10 (400 mg, 1.13 mmol) in THF (40 mL) was prepared, then aqueous HIO4 [27 mL, prepared by dissolution of 8.3 g (43.3 mmol) in 27 mL of H2O] in two portions (13.5 mL each) was added and the mixture stirred at room temperature for 2.5 hours. The solvent was removed until a volume of approximately 20 mL and the solution was neutralized with NaHCO3. The organic layer was extracted with EtOAc (3 x 30 mL) and the combined organic layers washed with H2O (3 x 20 mL) dried over Na2SO4, filtered, evaporated and chromatographed eluting with mixtures of petroleum ether/EtOAc of increasing polarity (19.8:0.2→6.8:13.2) to give 7.5 mg (2.1%) Compound 12: viscous oil; [α]D25 = +12.7° (c 0.355, CHCl3); 1H-NMR: 4.96 (1H, ddt, J=11.8, 11.8 and 3.9 Hz, H-2), 3.77 (2H, dd, J=8.4 and 6.4 Hz, H-12α and H-12β) 3.38 (1H, d, J=10.8 Hz, H-17a), 3.20 (1H, d, J=10.8 Hz, H-17b), 2.07 (1H, dd, J=5.8 and 4.5 Hz, H-7α), 2.02 (3H, s, OAc), 2.00 (1H, m, H-11β), 1.99 (1H, m, H-11α), 1.89 (1H, ddd, J=6.7, 6.7 and 1.5 Hz, H-11α), 1.78 (1H, ddd, J=11.8, 3.8 and 2.5 Hz, H-3α), 1.69 (1H, ddd, J=14.7, 10.1 and 6.9 Hz, H-7β), 1.53 (1H, dd, J=7.9 and 1.9 Hz, H-9), 1.51 (1H, m, H-6β), 1.46 (1H, m, H-6α), 1.22 (1H, dd, J=11.8 and 11.8 Hz, H-3β), 1.02 (3H, s, Me-20), 0.97 (3H, s, Me-19), 0.95 (3H, s, Me-18), 0.94 (1H, m, H-1β), 0.86 (1H, dd, J=11.6 and 3.9 Hz, H-5); 13C-NMR: 46.6 (C-1), 68.3 (C-2), 46.8 (C-3), 34.8 (C-4), 50.8 (C-5), 17.6 (C-6), 29.6 (C-7), 83.9 (C-8), 53.1 (C-9), 37.7 (C-10), 27.1 (C-11), 65.5 (C-12), 68.1 (C-17), 33.4 (C-18), 22.8 (C-19), 16.2 (C-20), 170.6 (CH3CO), 21.5 (CH3CO); IR: 3426, 2940, 1737, 1460, 1368, 1239, 1020; MS: 310 ([M]+ <1%), 280 (20.1%), 279 (100%), 220 (8%), 219, (47.5%), 159 (5.2%), 137 (7.8%), 135 (36.2%), 107 (7.8%), 97 (20.8%), 93 (7.4%), 55 (7.8%).
Synthesis of 8β,12-epoxy-13,14,15,16-tetranor-ent-labdane-2β,17-diol (11) and (8S)-2β,17-diacetoxy-12,17S-epoxy-13,14,15,16-tetranor-ent-labdane (13).
A solution of 10 (318.8 mg, 0.904 mmol) in acetone (30 mL) was prepared, then HClO4 (40 mL 6% v/v) was added and the mixture stirred at room temperature for 24 hours. The solvent was removed until a volume of approximately 20 mL and the solution was neutralized with NaHCO3. The organic layer was extracted with EtOAc (3 x 30 mL) and the combined organic layers washed with H2O (3 x 20 mL) dried over Na2SO4, filtered, evaporated and chromatographed eluting with mixtures of petroleum ether/EtOAc of increasing polarity (19.0:1.0→16.0:4.0) to give 126.2 mg (more polar fraction, 52%) of compound 11. The less polar fraction, was dissolved in CH2Cl2 (20 mL) and pyridine (0.5 mL), then Ac2O (2 mL) and DMAP (10 mg) were added and the mixture stirred at room temperature for 30 min. The solvent was removed until a volume of approximately 10 mL and 5 mL of water and 10% KHSO4 (5 mL) were added, the organic layer was extracted with EtOAc (3 x 10 mL). The combined organic layers were washed to neutrality with a saturated solution of NaHCO3 and water, dried over Na2SO4, filtered, evaporated and chromatographed eluting with mixtures of petroleum ether/EtOAc of increasing polarity (19.8:0.2→12.0:8.0) to give 117.9 mg (37%) of compound 13: viscous oil; [α]D25 = +23.4° (c 1.60, CHCl3); 1H-NMR: 5.23 (1H, d, J=8.8 Hz, H-17), 5.01 (1H, ddt, J=11.7, 11.7 and 4.4 Hz, H-2), 4.05 (1H, dddd, J=11.3, 4.4, 4.4 and 2.0 Hz, H12α), 3.53 (1H, ddd, J=11.3, 11.3 and 4.4 Hz, H-12β), 2.09 (3H, s, OAc), 2.01 (3H, s, OAc), 1.95 (1H, ddd, J=11.7, 2.5 and 2.0 Hz, H-1α), 1.79 (2H, m, H-7α and H-3α), 1.64 (1H, m, H-6α), 1.54 (1H, ddd, J=12.2, 8.8 and 3.9 Hz, H-8), 1.42 (2H, m, H-11α and H-11β), 1.27 (1H, dd, J=12.2 and 3.9 Hz, H-6β), 1.22 (1H, dd, J=11.7 and 11.7 Hz, H-3β), 1.10 (1H, ddd, J=11.7, 11.7 and 4.9 Hz, H-9), 0.97 (1H, ddd, J=12.2, 12.2 and 4.4 Hz, H-7β), 0.96 (1H, dd, J=11.7 and 11.7 Hz, H-1β), 0.94 (3H, s, Me-20), 0.92 (3H, s, Me-18), 0.91 (3H, s, Me-19), 0.90 (1H, dd, J=12.2 and 2.0 Hz, H-5); 13C-NMR: 43.8 (C-1), 68.7 (C-2), 46.7 (C-3), 34.8 (C-4), 54.2 (C-5), 20.1 (C-6), 27.6 (C-7), 39.0 (C-8), 52.0 (C-9), 37.7 (C-10), 24.2 (C-11), 66.1 (C-12), 98.0 (C-17), 33.3 (C-18), 22.4 (C-19), 14.8 (C-20), 170.5 (CH3CO), 21.4 (CH3CO), 169.9 (CH3CO), 21.0 (CH3CO); IR: 2940, 2848, 1757, 1731, 1465, 1358, 1230, 1127, 1066, 1030, 948; MS: 352 ([M]+ <1%), 293 (7.4%), 292 (38.3%), 232 (12.0%), 217 (16.8%), 163 (11.1%), 150, (13.3%), 149 (87.1%), 135 (29.0%), 122 (7.4%), 121 (9.0%), 110 (24.3%), 109 (7.5%), 107 (10.8%), 105 (7.7%), 98 (7.9%), 97 (100%), 96 (20.6%), 95 (13.9%), 93 (9.2%), 91 (10.5%), 81 (7.6%), 79 (10.7%), 67 (12.8%).

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}