Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction

Instituto Universitario de Bio-Orgánica “Antonio González”, Departamento de Química Orgánica, Universidad de La Laguna, 38206 La Laguna, Tenerife, Spain
*
Author to whom correspondence should be addressed.
Molecules 2007, 12(2), 194-204; https://doi.org/10.3390/12020194
Submission received: 25 January 2007
/
Revised: 13 February 2007
/
Accepted: 13 February 2007
/
Published: 19 February 2007
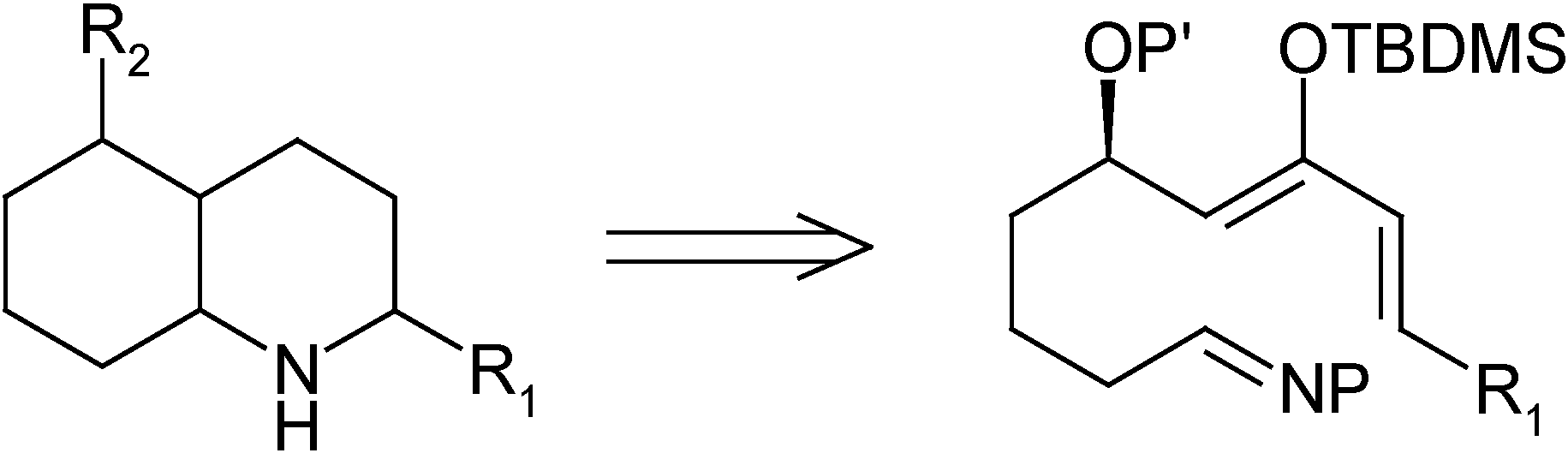{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:A route for the preparation of 2,5-disubstituted octahydroquinolin-4-ones, synthetic precursors of the decahydroquinoline-type toxins, is presented. The key steps are an asymmetric epoxidation and an intramolecular hetero Diels-Alder reaction between an activated diene and an imine. The presence of an allylic stereogenic center induces some selectivity and thus only two cycloadducts are obtained in 70:30 ratio and good yield.
Introduction
Interest in the biological activity of the analogs of naturally occurring toxins, such as those isolated from skin extracts of dendrobatid frogs [1], has prompted the development of new synthetic strategies or has led to the improvement of older ones for these targets [2]. One of the most powerful strategies is based on intramolecular Diels-Alder reactions, which have been used in different approaches, usually with high selectivity [3]. Grieco and Parker [4] have presented a synthesis of (-)-8a-epi-pumiliotoxin based on an hetero Diels-Alder reaction between a diene and an imine carried out under Mannich conditions and they found that the selectivity of the process can be influenced by an allylic substituent.
Within a project aimed at the synthesis of bicyclic alkaloids, we were interested in the development of a synthetic strategy for the preparation of 2,5-disubstituted decahydroquinolines [1], which could allow us to prepare different analogs of the natural toxins. We envisioned that the basic skeleton could be constructed by the intramolecular hetero Diels-Alder reaction of a suitable precursor containing a diene and an imine, in an approach similar to the one used by Grieco and Parker [4].
For our synthesis, and in order to increase the reactivity, we decided to place an activating group such as a tert-butyldimethylsilyloxy moiety on the diene, in such a position as to favor the desired regiochemistry of the process. To achieve the required substitution pattern, a substituent is needed at the allylic position and, in order to have a more flexible synthesis, it must be possible to transform said substituent into the different functional groups present in the natural toxins. Thus, it was decided to place a protected hydroxy group at the allylic position. It was also expected that the 1,3-non-bonded interaction between that substituent and the silyloxy group would increase the selectivity of the process by favoring one of the possible reacting conformations [5]. Therefore, the goal of this work was to establish a synthetic route to an activated diene-imine system, such as the one shown in Scheme 1, and to test its reactivity under hetero Diels-Alder reaction conditions.
Scheme 1.
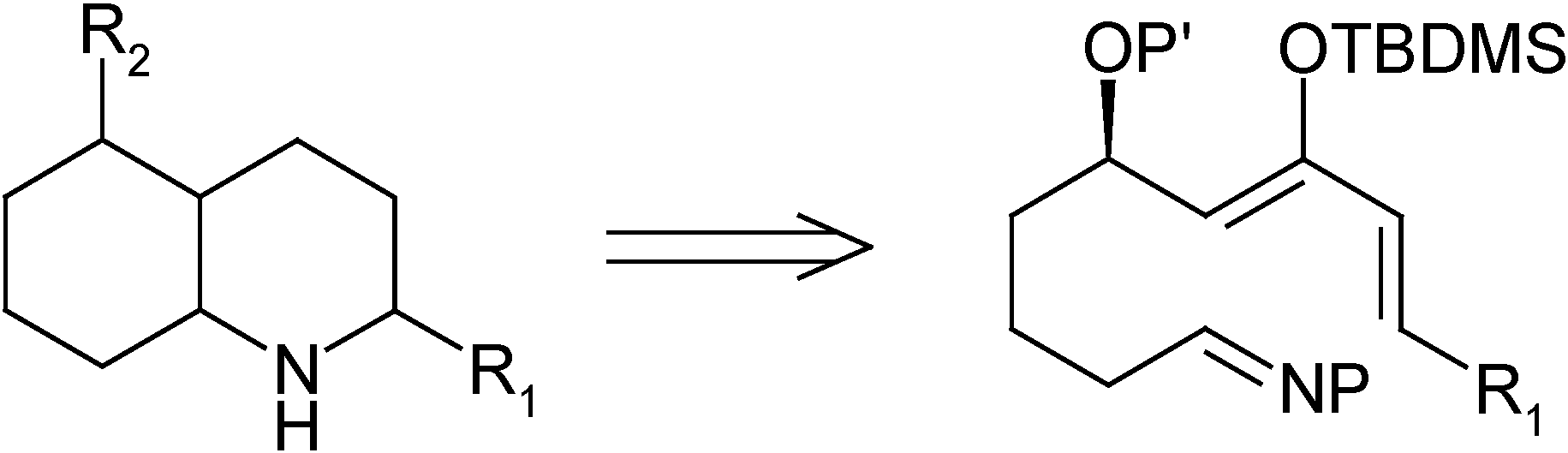
Results and Discussion
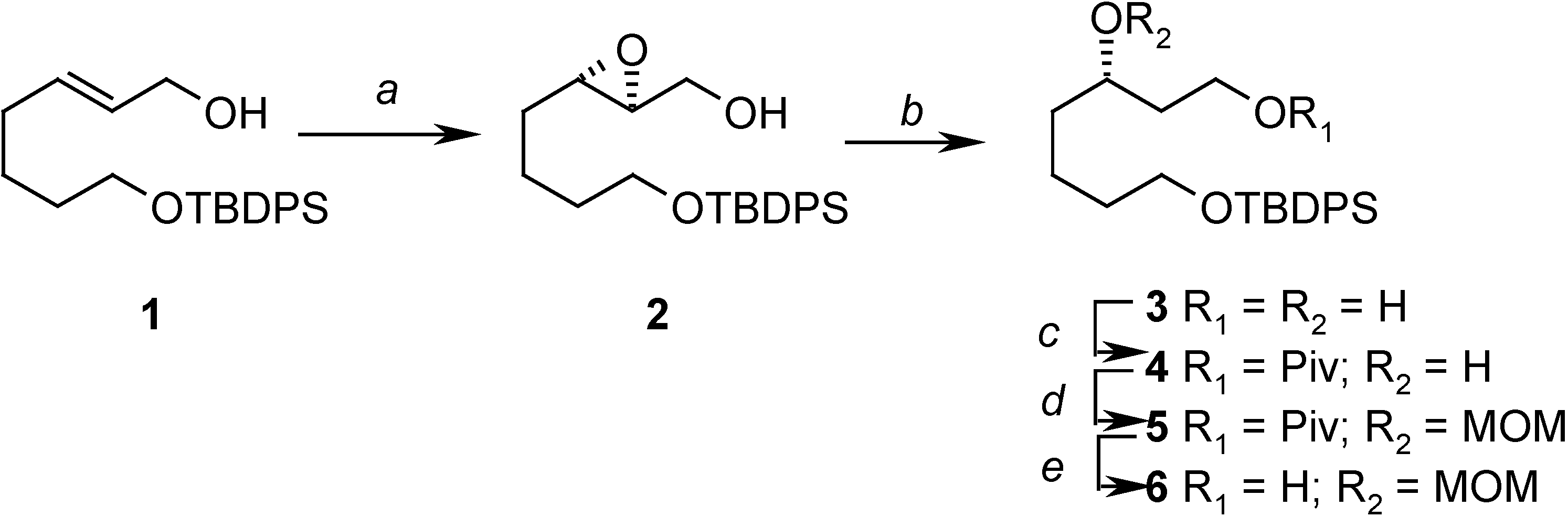
In order to achieve this goal, and to do so in an enantioselective way, we decided to use the Katsuki-Sharpless asymmetric epoxidation reaction [6] as the source of the allylic stereogenic center, and thus the allylic alcohol 1, which can be prepared easily from 1,5-pentanediol [7] was selected as our starting compound. The asymmetric epoxidation gave the expected epoxide 2 in good yield and high enantioselectivity, and in the subsequent steps it was regioselectively opened to give a diol. The secondary ydroxyl group was then protected as the corresponding methoxymethyl ether by first masking the primary one as the bulky pivaloyl ester. Finally, the pivaloyl group was removed, yielding 6 (Scheme 2).
Scheme 2.
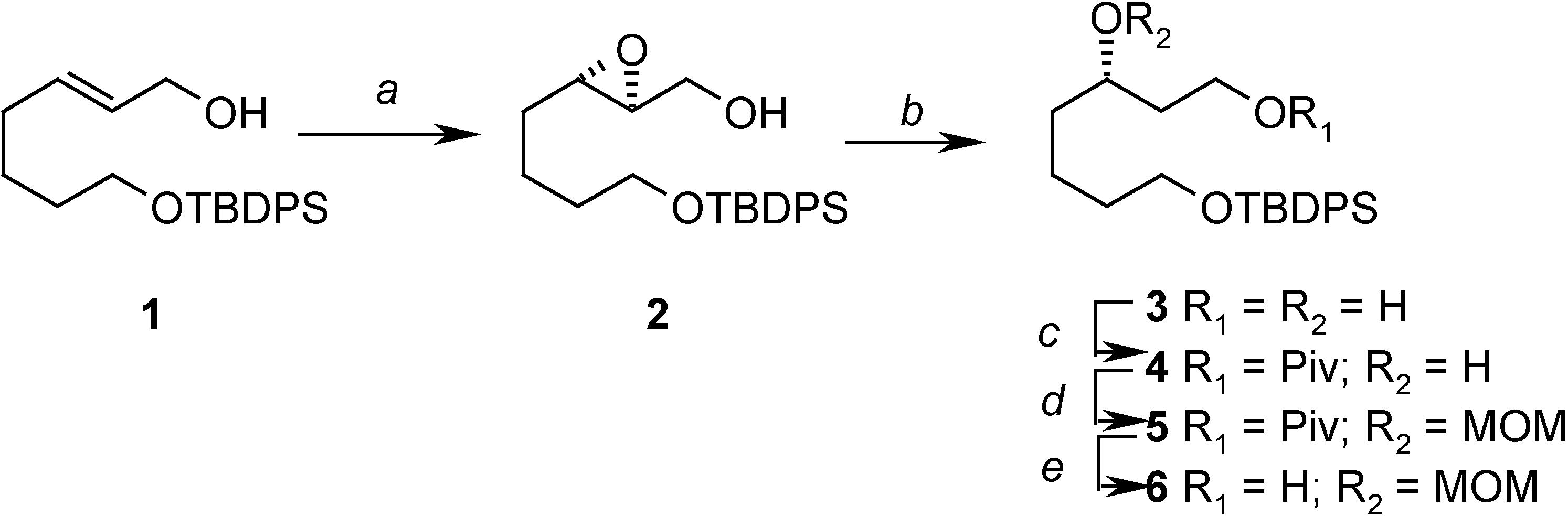
Reagents: (a) (+)-diethyl tartrate, Ti(OiPr)4, t-BuOOH, CH2Cl2, MS 4Å (90%); (b) Red-Al, THF, 0°C to r.t. (95%); (c) PivCl, Py, 0°C to r.t. (88%); (d) MOMCl, i-Pr2NEt, 0°C to r.t. (82%); (e) DIBAL, Et2O, -60°C (100%)
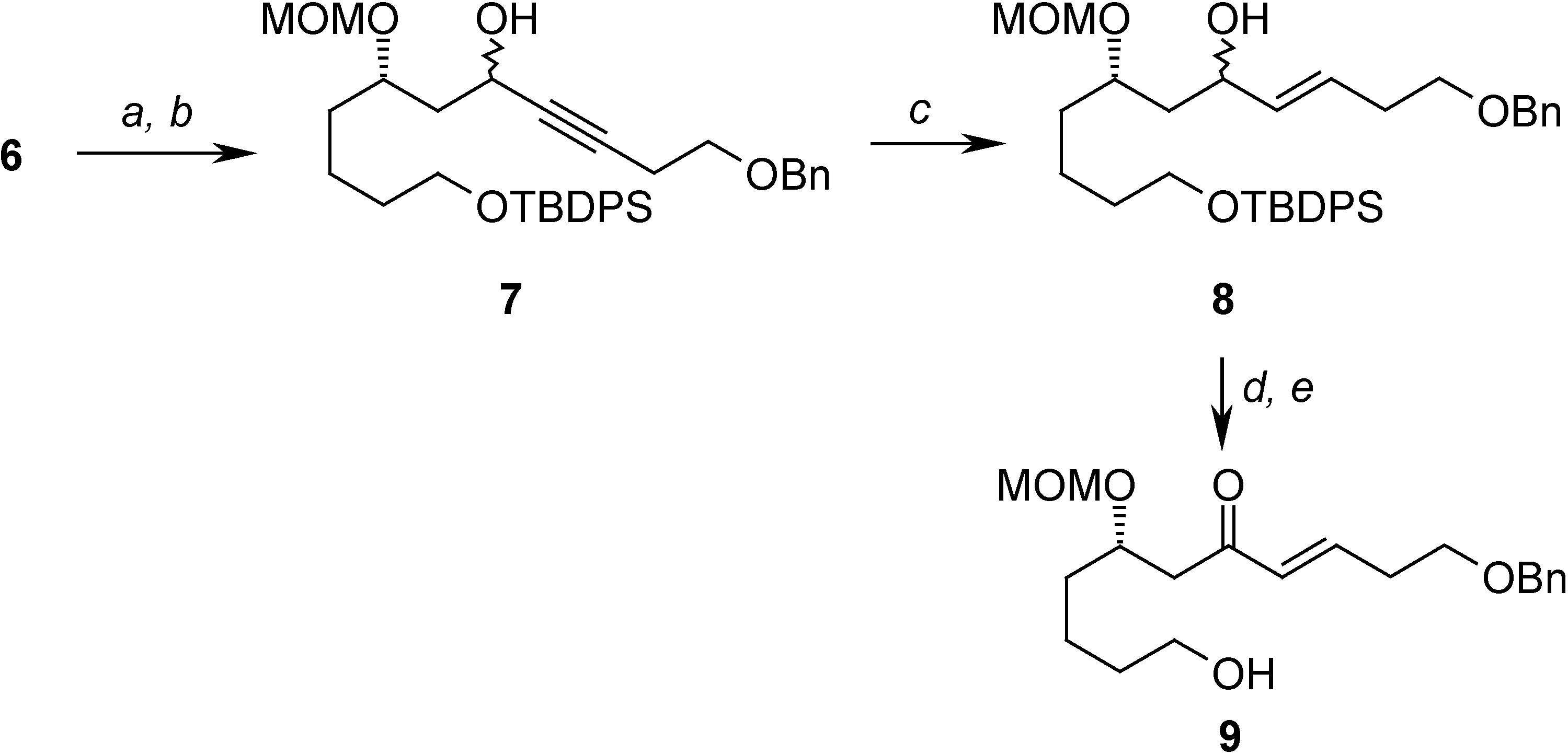
Oxidation of 6 and treatment with the anion of benzyl protected homopropargyl alcohol allowed the construction of 7, which has the carbon skeleton of the target compound. In order to transform 7 into the desired compound, and after several attempts, the following sequence was found to be the most efficient: partial reduction of the triple bond, removal of the silicon protecting group and selective oxidation of the allylic alcohol formed to give the α, β-unsaturated ketone 9, precursor of the activated diene system (Scheme 3).
Scheme 3.
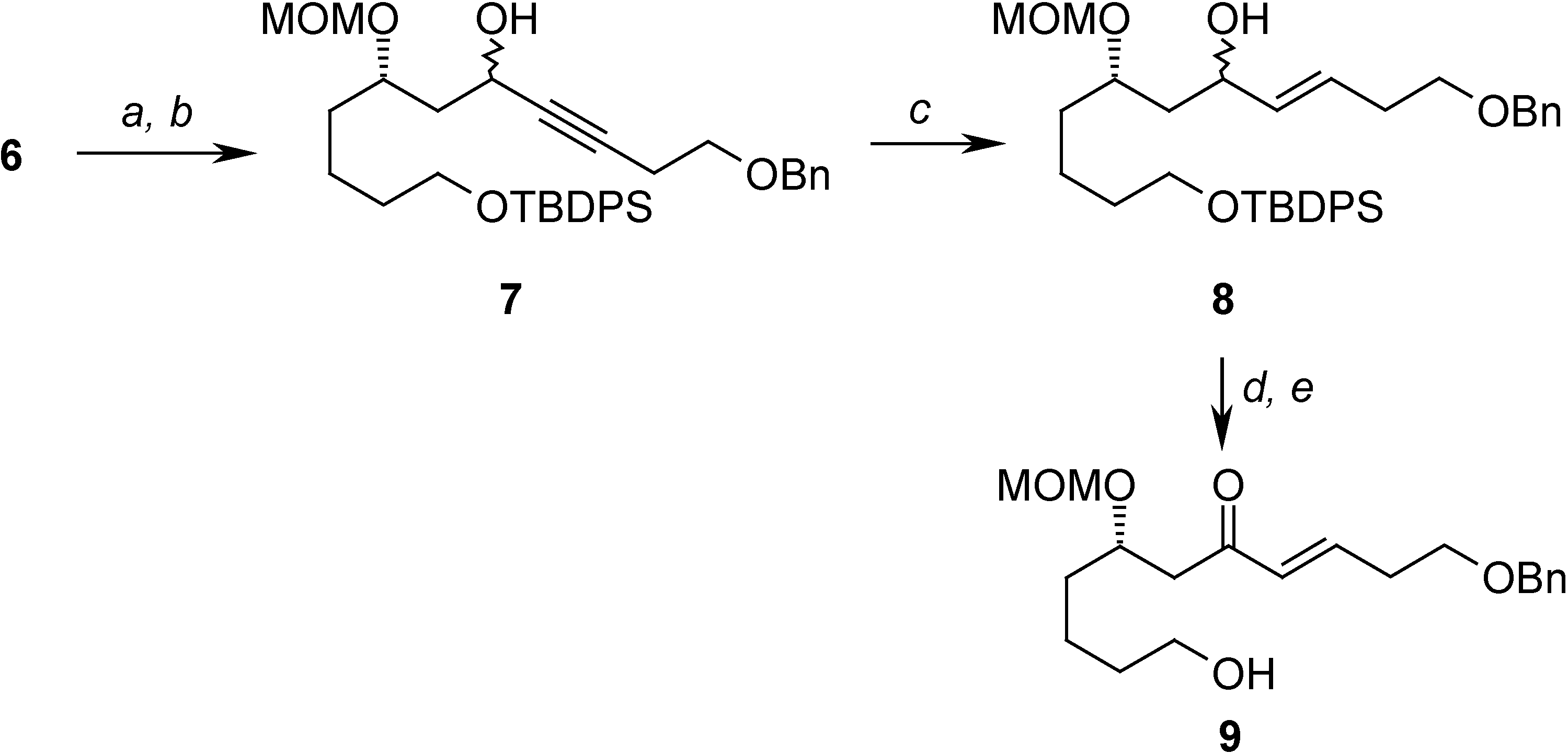
Reagents: (a) Oxalyl chloride, DMSO, -78°C, Et3N; (b) n-BuLi, HC≡CCH2CH2OBn, THF, -78°C to r.t. (88% from 6); (c) LiAlH4 (80%) THF, reflux; (d) TBAF, THF, 0°C to r.t. (87%); (e) MnO2, CH2Cl2, r.t. (81%)
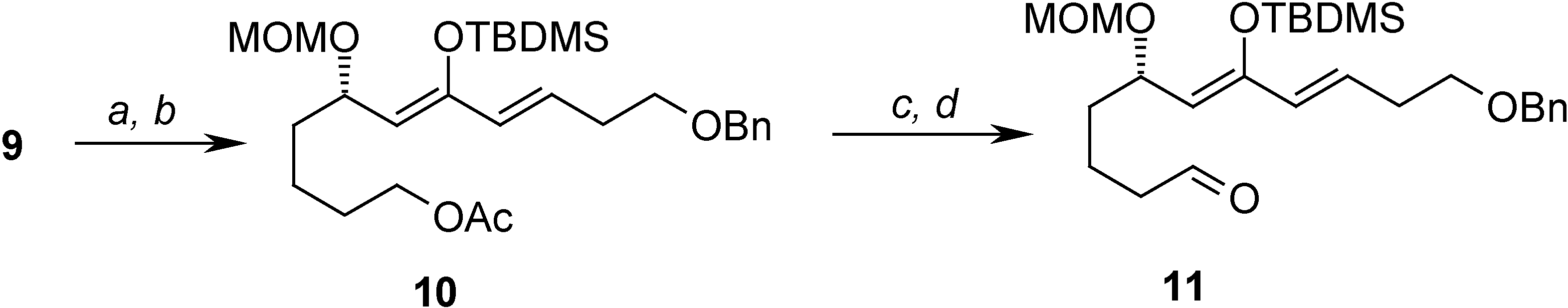
After acetylation of the primary ydroxyl group the compound 10 containing the activated diene system was prepared by LDA enolization of the α,β-unsaturated ketone 9, in a process hindered by side reactions such as elimination of the –OMOM group or the abstraction of the γ proton instead of the α one. The last step on the synthesis was the transformation of the acetylated primary ydroxyl group into the imine, and this was achieved by reductive deprotection of the acetate and SO3·Py oxidation of the free ydroxyl group to give the aldehyde 11, which was pure enough to be used without purification. When 11 was subjected to column chromatography, it was obtained pure, but in only a 40% yield (Scheme 4).
Scheme 4.
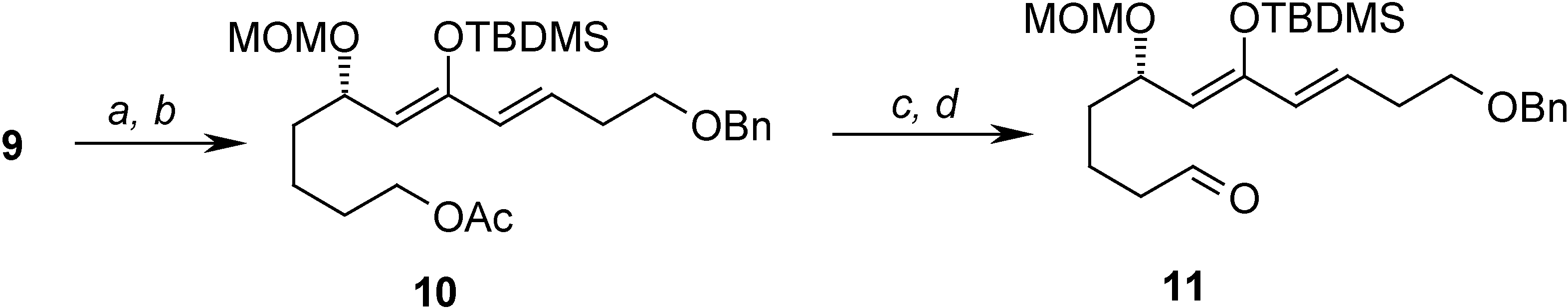
Reagents: (a) Ac2O, Py, 0°C to r.t. (99%); (b) LDA, TBDMSOTf, THF, -78°C (62%); (c) DIBAL, Et2O, -70°C (76%); (d) SO3·Py, DMSO, Et3N, 0°C to r.t. (40%)
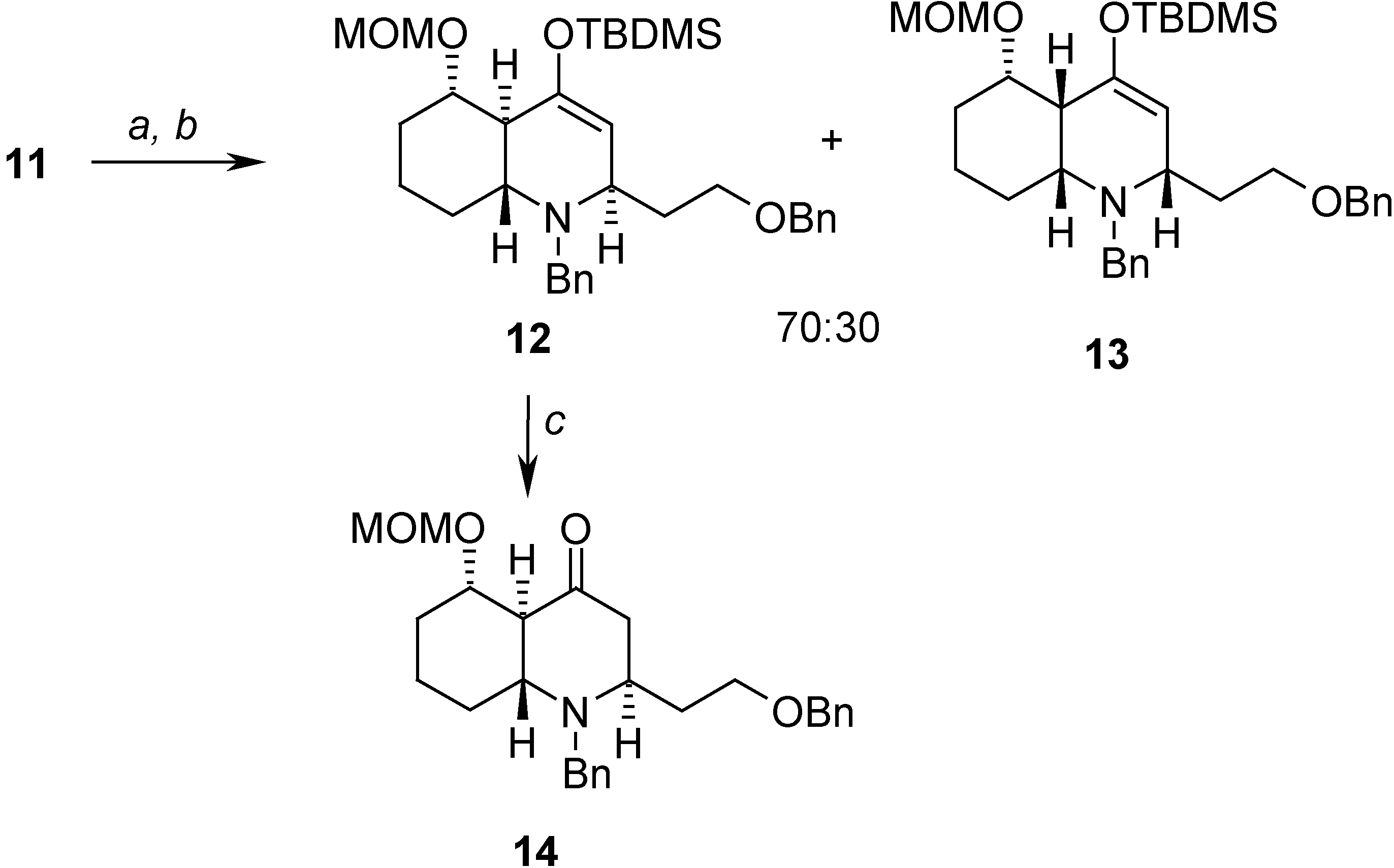
The cycloaddition reaction was carried out by first converting 11 into an imine by treatment with benzylamine in the presence of anhydrous magnesium sulfate and, without purification, the crude reaction mixture was dissolved in CH3CN and indium triflate was added as the Lewis acid. This way, two compounds were isolated in a 70:30 ratio and 79% yield and their structures were established spectroscopically. Both compounds presented the structure expected for the cycloadducts, differing only in the relative stereochemistry at the newly formed stereogenic centers. The major compound 12, results from the exo approach of the imine to the diene portion of the molecule in the most favorable conformation, whereas the minor one 13 arises from the endo approach. The relative stereochemistry was deduced from goesy experiments on 12 and 13 [8] and was confirmed, using also goesy experiments, on the more stable decahydroquinolone 14, prepared from 12 by treatment with tetrabutyl ammonium fluoride (Scheme 5).
Scheme 5.

Reagents: (a) BnNH2, MgSO4, Et2O, 0°C; (b) In(OTf)3, CH3CN, 0°C to r.t. (79%); (c) TBAF, THF, 0°C to r.t. (90%)
The isolated yield in our case (79%) was higher than that reported by Grieco and Parker [4] (55%), which may be due to the activation provided by the silyloxy group, but the selectivity of the process is similar to that previously described, affording the two cycloadducts in a 70:30 ratio and with the same relative stereochemistry in both of them.
Conclusions
In summary, an enantioselective route to the octahydroquinolinone skeleton with substitution on the 2 and 5 positions has been described. The key steps are a Katsuki-Sharpless asymmetric epoxidation, used for the introduction of the controlling stereogenic center, and an intramolecular hetero Diels-Alder reaction of a molecule containing an activated diene and an imine. The resulting cycloadducts can be used for the synthesis of analogs of naturally occurring toxins of the 2,5-decahydroquinoline family.
Experimental
General
1H-NMR and 13C-NMR spectra were recorded in CDCl3 or C6D6 at 400 MHz (1H) and 100 MHz (13C) on a Bruker AVANCE 400 instrument. The chemical shifts are quoted in ppm using the residual solvent signal as reference. Multiplicities were determined by DEPT experiments. IR spectra were recorded using a Shimadzu 408 spectrophotometer. Optical rotations were determined using a Perkin-Elmer 241 polarimeter. Mass spectra were run on a VG (Fisons) spectrometer. Flash chromatography was performed on columns packed with Merck silica gel 60 (260-400 mesh). Solvents and reagents were generally distilled prior to use, and purified when necessary according to standard procedures.
Preparation of ((2S, 3S)-3-(4-(tert-butyldiphenylsilyloxy)butyl)oxiran-2-yl)methanol (2)
To a mixture of crushed molecular sieves (MS 4Å) and (+)-DET (3.06 mL, 17.8 mmol) in dry CH2Cl2 under argon at -20 °C was added Ti(OPr-i)4 (4.52 mL, 15.32 mmol). After 15 min, compound 1 was added (4.7 g, 12.77 mmol), and 30 min later, a 5.3 M solution of TBHP in benzene (4.3 mL, 23 mmol) was added. The reaction was stirred at -20°C overnight. The reaction was quenched by the addition of tartaric acid solution (15% in water), and further extraction was done with CH2Cl2 (3 × 50 mL). The combined organic extracts were dried (MgSO4), concentrated, and chromatographed on silica gel (70:30 Hex-EtOAc) yielding 4.4 g (90%) of 2 as a colorless oil; ![Molecules 12 00194 i001]() = -14.23 (c 2.08, CHCl3); 1H- NMR (CDCl3) δ 7.70-7.68 (m, 4H), 7.43-7.38 (m, 6H), 3.90 (dd, J = 1.7 Hz, 12.5 Hz, 1H), 3.69 (m, 2H), 3.60 (dd, J = 4.4 Hz, 12.5 Hz, 1H), 3.50 (bs, 1H), 2.95-2.91 (m, 2H), 1.64-1.57 (m, 6H), 1.08 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 63.6 (t), 61.8 (t), 58.6 (d), 56.0 (d), 32.2 (t), 31.3 (t), 26.9 (q), 22.3 (t), 19.2 (s).
= -14.23 (c 2.08, CHCl3); 1H- NMR (CDCl3) δ 7.70-7.68 (m, 4H), 7.43-7.38 (m, 6H), 3.90 (dd, J = 1.7 Hz, 12.5 Hz, 1H), 3.69 (m, 2H), 3.60 (dd, J = 4.4 Hz, 12.5 Hz, 1H), 3.50 (bs, 1H), 2.95-2.91 (m, 2H), 1.64-1.57 (m, 6H), 1.08 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 63.6 (t), 61.8 (t), 58.6 (d), 56.0 (d), 32.2 (t), 31.3 (t), 26.9 (q), 22.3 (t), 19.2 (s).
 = -14.23 (c 2.08, CHCl3); 1H- NMR (CDCl3) δ 7.70-7.68 (m, 4H), 7.43-7.38 (m, 6H), 3.90 (dd, J = 1.7 Hz, 12.5 Hz, 1H), 3.69 (m, 2H), 3.60 (dd, J = 4.4 Hz, 12.5 Hz, 1H), 3.50 (bs, 1H), 2.95-2.91 (m, 2H), 1.64-1.57 (m, 6H), 1.08 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 63.6 (t), 61.8 (t), 58.6 (d), 56.0 (d), 32.2 (t), 31.3 (t), 26.9 (q), 22.3 (t), 19.2 (s).
= -14.23 (c 2.08, CHCl3); 1H- NMR (CDCl3) δ 7.70-7.68 (m, 4H), 7.43-7.38 (m, 6H), 3.90 (dd, J = 1.7 Hz, 12.5 Hz, 1H), 3.69 (m, 2H), 3.60 (dd, J = 4.4 Hz, 12.5 Hz, 1H), 3.50 (bs, 1H), 2.95-2.91 (m, 2H), 1.64-1.57 (m, 6H), 1.08 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 63.6 (t), 61.8 (t), 58.6 (d), 56.0 (d), 32.2 (t), 31.3 (t), 26.9 (q), 22.3 (t), 19.2 (s).Preparation of (S)-7-(tert-butyldiphenylsilyloxy)heptane-1,3-diol (3)
To a solution of 2 (4.2 g, 10.9 mmol) in THF (110 mL) at 0 °C under argon was added Red-Al (9.19 mL, 32.9 mmol, 70%). The reaction was allowed to reach room temperature and stirred for 5 h. The reaction mixture was quenched at 0 °C by slowly addition of MeOH (1.5 mL). The solution was poured into an aqueous saturated solution of potassium sodium tartrate and it was vigorously stirred until the phases became clear. After phase separation, the combined organic extracts were dried (MgSO4), concentrated, and chromatographed on silica gel (60:40 Hex-EtOAc) yielding 4.0 g (95%) of 3 as a colorless oil; 1H-NMR (CDCl3) δ 7.69-7.68 (m, 4H), 7.43-7.37 (m, 6H), 3.68-3.67 (m, 3H), 3.71-3.67 (m, 2H), 3.25 (m, 2H), 1.67-1.58 (m, 2H), 1.51-1.41 (m, 4H), 1.07 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 72.0 (d), 63.8 (t), 61.6 (t), 38.0 (t), 37.4 (t), 32.4 (t), 26.9 (q), 21.8 (t), 19.2 (s); EIMS m/z (%): 329 (M+-tBu, 0.1), 311 (M+-tBu-H2O, 0.57); HRMS (EI): C19H25O3Si (M+-tBu): 329.1573, found 329.1577.
Preparation of (S)-7-(tert-butyldiphenylsilyloxy)-3-hydroxyheptyl pivalate (4)
To a solution of 3 (1.95 g, 5.05 mmol) in pyridine (2.0 mL) at 0°C under argon, was added pivaloyl chloride (0.68 mL, 5.55 mmol). The reaction mixture was stirred at room temperature until TLC showed disappearance of the starting material. The reaction was diluted with Et2O and then ice and finally a saturated aqueous solution of NaCl was added. The mixture was extracted with Et2O (3 × 20 mL). The combined organic layers were dried (MgSO4), concentrated, and chromatographed on silica gel (85:15 Hex-EtOAc) yielding 2.1 g (88%) of 4 as a colorless oil; ![Molecules 12 00194 i001]() = +1.08 (c 3.5, CHCl3); 1H-NMR (CDCl3) δ 7.71-7.76 (m, 4H), 7.45-7.38 (m, 6H), 4.35 (m, 1H), 4.16 (m, 1H), 3.71 (t, J = 6.0 Hz, 2H), 3.66 (m, 1H), 2.45 (s, 1H), 1.84-1.80 (m, 1H), 1.70-1.52 (m, 7H), 1.20 (s, 9H), 1.04 (s, 9H); 13C-NMR (CDCl3) δ 179.0 (s), 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 68.6 (d), 63.8 (t), 61.7 (t), 38.8 (s), 37.1 (t), 36.4 (t), 32.0 (t), 27.2 (q), 26.9 (q), 21.9 (t), 19.2 (s); EIMS m/z (%): 413 (M+-tBu, 5.0), 395 (M+-tBu-H2O, 2.0); HRMS (EI): C24H33O4Si (M+-tBu): 413.2148, found 413.2174.
= +1.08 (c 3.5, CHCl3); 1H-NMR (CDCl3) δ 7.71-7.76 (m, 4H), 7.45-7.38 (m, 6H), 4.35 (m, 1H), 4.16 (m, 1H), 3.71 (t, J = 6.0 Hz, 2H), 3.66 (m, 1H), 2.45 (s, 1H), 1.84-1.80 (m, 1H), 1.70-1.52 (m, 7H), 1.20 (s, 9H), 1.04 (s, 9H); 13C-NMR (CDCl3) δ 179.0 (s), 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 68.6 (d), 63.8 (t), 61.7 (t), 38.8 (s), 37.1 (t), 36.4 (t), 32.0 (t), 27.2 (q), 26.9 (q), 21.9 (t), 19.2 (s); EIMS m/z (%): 413 (M+-tBu, 5.0), 395 (M+-tBu-H2O, 2.0); HRMS (EI): C24H33O4Si (M+-tBu): 413.2148, found 413.2174.
= +1.08 (c 3.5, CHCl3); 1H-NMR (CDCl3) δ 7.71-7.76 (m, 4H), 7.45-7.38 (m, 6H), 4.35 (m, 1H), 4.16 (m, 1H), 3.71 (t, J = 6.0 Hz, 2H), 3.66 (m, 1H), 2.45 (s, 1H), 1.84-1.80 (m, 1H), 1.70-1.52 (m, 7H), 1.20 (s, 9H), 1.04 (s, 9H); 13C-NMR (CDCl3) δ 179.0 (s), 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 68.6 (d), 63.8 (t), 61.7 (t), 38.8 (s), 37.1 (t), 36.4 (t), 32.0 (t), 27.2 (q), 26.9 (q), 21.9 (t), 19.2 (s); EIMS m/z (%): 413 (M+-tBu, 5.0), 395 (M+-tBu-H2O, 2.0); HRMS (EI): C24H33O4Si (M+-tBu): 413.2148, found 413.2174.Preparation of (S)-7-(tert-butyldiphenylsilyloxy)-3-(methoxymethoxy)heptyl pivalate (5)
To a solution of 4 (2.1 g, 4.46 mmol) in i-Pr2NEt (1.16 mL) at 0°C under argon, was added MOMCl (0.37 mL, 4.91 mmol). The reaction mixture was stirred at room temperature until TLC showed disappearance of the starting material (2 h). The reaction was diluted with Et2O and then ice and a saturated aqueous solution of NaCl were added. The mixture was extracted with Et2O (3 × 50 mL). The combined organic layers were dried (MgSO4), concentrated, and chromatographed on silica gel (95:5 Hex-EtOAc) yielding 1.89 g (82%) of 5 as a colorless oil; ![Molecules 12 00194 i001]() = +12.13 (c 2.06, CHCl3); 1H-NMR (CDCl3) δ 7.69-7.67 (m, 4H), 7.43-7.37 (m, 6H), 4.64 (d, J = 6.9 Hz, 1H), 4.62 (d, J = 6.9 Hz, 1H), 4.18 (m, 2H), 3.66 (m, 3H), 3.36 (s, 3H), 1.85-1.40 (m, 8H), 1.21 (s, 9H), 1.06 (s, 9H); 13C-NMR (CDCl3) δ 178.5 (s), 135.6 (d), 134.0 (s), 129.5 (d), 127.6 (d), 95.6 (t), 74.4 (d), 63.7 (t), 61.2 (t), 55.6 (q), 38.7 (s), 34.3 (t), 33.4 (t), 32.6 (t), 27.2 (q), 26.9 (q), 21.5 (t), 19.2 (q); EIMS m/z (%): 457 (M+-tBu, 3.1); HRMS (EI): C26H37O5Si (M+-tBu): 457.2410, found 457.2404.
= +12.13 (c 2.06, CHCl3); 1H-NMR (CDCl3) δ 7.69-7.67 (m, 4H), 7.43-7.37 (m, 6H), 4.64 (d, J = 6.9 Hz, 1H), 4.62 (d, J = 6.9 Hz, 1H), 4.18 (m, 2H), 3.66 (m, 3H), 3.36 (s, 3H), 1.85-1.40 (m, 8H), 1.21 (s, 9H), 1.06 (s, 9H); 13C-NMR (CDCl3) δ 178.5 (s), 135.6 (d), 134.0 (s), 129.5 (d), 127.6 (d), 95.6 (t), 74.4 (d), 63.7 (t), 61.2 (t), 55.6 (q), 38.7 (s), 34.3 (t), 33.4 (t), 32.6 (t), 27.2 (q), 26.9 (q), 21.5 (t), 19.2 (q); EIMS m/z (%): 457 (M+-tBu, 3.1); HRMS (EI): C26H37O5Si (M+-tBu): 457.2410, found 457.2404.
= +12.13 (c 2.06, CHCl3); 1H-NMR (CDCl3) δ 7.69-7.67 (m, 4H), 7.43-7.37 (m, 6H), 4.64 (d, J = 6.9 Hz, 1H), 4.62 (d, J = 6.9 Hz, 1H), 4.18 (m, 2H), 3.66 (m, 3H), 3.36 (s, 3H), 1.85-1.40 (m, 8H), 1.21 (s, 9H), 1.06 (s, 9H); 13C-NMR (CDCl3) δ 178.5 (s), 135.6 (d), 134.0 (s), 129.5 (d), 127.6 (d), 95.6 (t), 74.4 (d), 63.7 (t), 61.2 (t), 55.6 (q), 38.7 (s), 34.3 (t), 33.4 (t), 32.6 (t), 27.2 (q), 26.9 (q), 21.5 (t), 19.2 (q); EIMS m/z (%): 457 (M+-tBu, 3.1); HRMS (EI): C26H37O5Si (M+-tBu): 457.2410, found 457.2404.Preparation of (S)-7-(tert-butyldiphenylsilyloxy)-3-(methoxymethoxy)heptan-1-ol (6)
DIBAL (1M in hexanes, 11 mL, 11 mmol) was added to a solution of 5 (1.89 g, 3.67 mmol) in Et2O (129 mL) under argon at -60°C. After stirring for 2 h, MeOH (5 mL) was slowly added and the reaction mixture was allowed to reach room temperature. Then it was poured onto a saturated aqueous solution of sodium potassium tartrate, stirred until the solution becomes clear and extracted with Et2O. The combined organic layers were dried (MgSO4) and concentrated, affording after column chromatography (silica gel, 70:30 Hex-EtOAc), 1.58 g (100%) of alcohol 6, as a colorless oil; ![Molecules 12 00194 i001]() = +22.67 (c 2.67, CHCl3); 1H-NMR (CDCl3) δ 7.68-7.61 (m, 4H), 7.43-7.36 (m, 6H), 4.67 (d, J = 6.8 Hz, 1H), 4.65 (d, J = 6.8 Hz, 1H), 3.81-3.65 (m, 4H), 3.39 (s, 3H), 1.67-1.40 (m, 8H), 1.04 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 95.9 (t), 76.4 (d), 63.7 (t), 59.8 (t), 55.7 (q), 35.6 (t), 34.4 (t), 32.6 (t), 26.9 (q), 21.6 (t), 19.2 (s); EIMS m/z (%): 342 (M+-tBu-OCH3, 1.85).
= +22.67 (c 2.67, CHCl3); 1H-NMR (CDCl3) δ 7.68-7.61 (m, 4H), 7.43-7.36 (m, 6H), 4.67 (d, J = 6.8 Hz, 1H), 4.65 (d, J = 6.8 Hz, 1H), 3.81-3.65 (m, 4H), 3.39 (s, 3H), 1.67-1.40 (m, 8H), 1.04 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 95.9 (t), 76.4 (d), 63.7 (t), 59.8 (t), 55.7 (q), 35.6 (t), 34.4 (t), 32.6 (t), 26.9 (q), 21.6 (t), 19.2 (s); EIMS m/z (%): 342 (M+-tBu-OCH3, 1.85).
= +22.67 (c 2.67, CHCl3); 1H-NMR (CDCl3) δ 7.68-7.61 (m, 4H), 7.43-7.36 (m, 6H), 4.67 (d, J = 6.8 Hz, 1H), 4.65 (d, J = 6.8 Hz, 1H), 3.81-3.65 (m, 4H), 3.39 (s, 3H), 1.67-1.40 (m, 8H), 1.04 (s, 9H); 13C-NMR (CDCl3) δ 135.6 (d), 134.0 (s), 129.6 (d), 127.6 (d), 95.9 (t), 76.4 (d), 63.7 (t), 59.8 (t), 55.7 (q), 35.6 (t), 34.4 (t), 32.6 (t), 26.9 (q), 21.6 (t), 19.2 (s); EIMS m/z (%): 342 (M+-tBu-OCH3, 1.85).(7S)-1-(benzyloxy)-11-(tert-butyldiphenylsilyloxy)-7-(methoxymethoxy)undec-3-yn-5-ol (7)
To a solution of DMSO (0.63 mL, 8.98 mmol) in CH2Cl2 (30 mL) under argon at -78°C was slowly added a solution of oxalyl chloride (2 M in CH2Cl2, 2.24 mL, 4.48 mmol). After 30 min the alcohol 6 (1.61 g, 3.74 mmol) was added in CH2Cl2 (10 mL). After the mixture was stirred for 1 h, TEA (2.6 mL, 18.7 mmol) was added, and the reaction was allowed to reach to room temperature. Water was added, and the reaction was extracted with CH2Cl2. The combined organic extracts were washed with 10 mL each of 1 % aqueous solution of HCl, 5 % Na2CO3, and a saturated solution of NaCl. The solution was dried (MgSO4) and concentrated. The expected aldehyde was obtained and used without further purification. To a solution of ((but-3-ynyloxy)methyl)benzene (658 mg, 4.11 mmol) in THF (15 mL) under argon at -78°C was added n-BuLi (2.57 mL, 4.11 mmol, 1.6 M in hexane), and the mixture was stirred for 30 min. Then, a solution of the previously obtained aldehyde in THF (5 mL) was added. The reaction mixture was allowed to warm to room temperature for 2 h and then it was quenched by the addition of aqueous saturated NH4Cl solution and extracted with Et2O (3 × 25 mL). The combined organic extracts were washed with brine, dried (MgSO4) concentrated, and chromatographed on silica gel (75:25 Hex-EtOAc) yielding 1.93 g (88%) of an inseparable mixture of diastereomers 7.
(7S,E)-1-(benzyloxy)-11-(tert-butyldiphenylsilyloxy)-7-(methoxymethoxy)undec-3-en-5-ol (8)
A suspension of 131 mg (3.46 mmol) of LiAlH4 in 50 ml of THF was cooled to 0 °C, and a solution of 7 (1.7 g, 2.89 mmol) in THF (10 mL) was added dropwise. The reaction mixture was heated at reflux for 2 h, cooled to 0 °C and quenched by successive addition of H2O (0.13 mL), aqueous solution of NaOH 10% (0.13 mL) and H2O (0.4 mL). The mixture was filtered through Celite®, the residue was washed with EtOAc, the combined filtrates were concentrated, and chromatographed on silica gel (75:25 Hex-EtOAc) yielding 1.36 g (80%) of an inseparable mixture of diastereomers 8.
(S,E)-1-(benzyloxy)-11-hydroxy-7-(methoxymethoxy)undec-3-en-5-one (9)
To a solution of 8 (1.4 g, 2.37 mmol) in THF (40 mL) at 0 °C under argon was added TBAF (1.1 equiv). The reaction mixture was allowed to warm to room temperature and stirred for 12 h. The reaction was quenched by the addition of ice and brine. After extraction with EtOAc (3 × 25 mL), the combined extracts were dried (MgSO4), concentrated, and chromatographed on silica gel (20:80 Hex-EtOAc) yielding 726 mg (87%) of the corresponding diol as an inseparable mixture of diastereomers. To a suspension of MnO2 (6.9 g) in CH2Cl2, under an O2 atmosphere, was added the mixture of alcohols previously obtained (698 mg, 1.98 mmol) in CH2Cl2 (10 mL). After 12 h the reaction mixture was filtered through celite and the solution was concentrated and chromatographed on silica gel (30:70 Hex-EtOAc) yielding 561 mg (81%) of 9 as a colorless oil; ![Molecules 12 00194 i001]() = +1.44 (c 3.60, CHCl3); 1H-NMR (CDCl3) δ 7.37-7.26 (m, 5H), 6.86 (dt, J = 6.9 Hz, 15.9 Hz, 1H), 6.17 (d, J = 15.9 Hz, 1H), 4.61 (d, J = 6.9 Hz, 1H), 4.63 (d, J = 6.9 Hz, 1H), 4.52 (s, 2H), 4.09 (m, 1H), 3.61 (m, 4H), 3.32 (s, 3H), 2.90 (dd, J = 6.9 Hz, 10.0 Hz, 1H), 2.62 (dd, J = 5.9, 10.0 Hz, 1H), 2.55-2.50 (m, 2H), 1.68-1.42 (m 6H); 13C-NMR (CDCl3) δ 198.3 (s), 144.2 (d), 137.7 (s), 131.9 (d), 128.2 (d), 127.5 (d), 127.4 (d), 95.8 (t), 74.1 (d), 72.8 (t), 68.3 (t), 62.2 (t), 55.3 (d), 45.0 (t), 34.5 (t), 32.6 (t), 32.3 (t), 21.1 (t).
= +1.44 (c 3.60, CHCl3); 1H-NMR (CDCl3) δ 7.37-7.26 (m, 5H), 6.86 (dt, J = 6.9 Hz, 15.9 Hz, 1H), 6.17 (d, J = 15.9 Hz, 1H), 4.61 (d, J = 6.9 Hz, 1H), 4.63 (d, J = 6.9 Hz, 1H), 4.52 (s, 2H), 4.09 (m, 1H), 3.61 (m, 4H), 3.32 (s, 3H), 2.90 (dd, J = 6.9 Hz, 10.0 Hz, 1H), 2.62 (dd, J = 5.9, 10.0 Hz, 1H), 2.55-2.50 (m, 2H), 1.68-1.42 (m 6H); 13C-NMR (CDCl3) δ 198.3 (s), 144.2 (d), 137.7 (s), 131.9 (d), 128.2 (d), 127.5 (d), 127.4 (d), 95.8 (t), 74.1 (d), 72.8 (t), 68.3 (t), 62.2 (t), 55.3 (d), 45.0 (t), 34.5 (t), 32.6 (t), 32.3 (t), 21.1 (t).
= +1.44 (c 3.60, CHCl3); 1H-NMR (CDCl3) δ 7.37-7.26 (m, 5H), 6.86 (dt, J = 6.9 Hz, 15.9 Hz, 1H), 6.17 (d, J = 15.9 Hz, 1H), 4.61 (d, J = 6.9 Hz, 1H), 4.63 (d, J = 6.9 Hz, 1H), 4.52 (s, 2H), 4.09 (m, 1H), 3.61 (m, 4H), 3.32 (s, 3H), 2.90 (dd, J = 6.9 Hz, 10.0 Hz, 1H), 2.62 (dd, J = 5.9, 10.0 Hz, 1H), 2.55-2.50 (m, 2H), 1.68-1.42 (m 6H); 13C-NMR (CDCl3) δ 198.3 (s), 144.2 (d), 137.7 (s), 131.9 (d), 128.2 (d), 127.5 (d), 127.4 (d), 95.8 (t), 74.1 (d), 72.8 (t), 68.3 (t), 62.2 (t), 55.3 (d), 45.0 (t), 34.5 (t), 32.6 (t), 32.3 (t), 21.1 (t).Preparation of (S,6Z,8E)-11-(benzyloxy)-7-(tert-butyldimethylsilyloxy)-5-(methoxymethoxy)undeca-6,8-dienyl acetate (10)
To a solution of 9 (560 mg, 1.6 mmol) in pyridine (0.2 mL) at 0 °C under argon was added Ac2O (0.15 mL, 1.6 mmol). The reaction mixture was stirred overnight at room temperature and then ice- water was added. The mixture was extracted with CH2Cl2(3 × 15 mL), and the combined organic phases were dried (MgSO4), concentrated and chromatographed on silica gel (60:40 Hex-EtOAc) yielding 624 mg (99%) of (S,E)-11-(benzyloxy)-5-(methoxymethoxy)-7-oxoundec-8-enyl acetate as a colorless oil; ![Molecules 12 00194 i001]() = -0.85 (c 2.60, CHCl3); 1H-NMR (CDCl3) δ 7.36 (m, 5H), 6.85 (dt, J = 6.8 Hz, 15.9 Hz, 1H), 6.17 (d, J = 15.9 Hz, 1H), 4.64 (d, J = 6.8 Hz, 1H), 4.58 (d, J = 6.8 Hz, 1H), 4.51 (s, 2H), 4.05 (m, 3H), 3.58 (t, J = 6.3 Hz, 2H), 3.31 (s, 3H), 2.88 (dd, J = 6.8 Hz, 15.9 Hz, 1H), 2.61-2.48 (m, 3H), 2.02 (s, 3H), 1.75-1.38 (m, 6H); 13C-NMR (CDCl3) δ 198.3 (s), 171.1 (s), 144.3 (d), 138.0 (s), 132.1 (d), 128.4 (d), 127.7 (d), 96.1 (t), 74.2 (d), 73.0 (t), 68.2 (t), 64.3 (t), 55.5 (q), 45.3 (t), 34.7 (t), 32.9 (t), 28.6 (t), 21.7 (t), 20.9 (q). tert-Butyldimethylsilyl triflate (0.51 mL, 2.23 mmol) was added to a stirred solution of the previously prepared compound (624 mg, 1.59 mmol) in THF (16 mL) at -78 °C under argon. After 15 min a solution of LDA (0.24 M in THF, 7.9 mL, 1.9 mmol) was added. The reaction mixture was stirred at -78 °C for 30 min, and then a saturated aqueous solution of NH4Cl (15 mL) was added. The reaction was allowed to reach room temperature and extracted with Et2O. The combined organic phases were dried (MgSO4), concentrated, and chromatographed on silica gel (80:20 Hex-EtOAc) yielding 500 mg (62%) of 10 as a colorless oil;
= -0.85 (c 2.60, CHCl3); 1H-NMR (CDCl3) δ 7.36 (m, 5H), 6.85 (dt, J = 6.8 Hz, 15.9 Hz, 1H), 6.17 (d, J = 15.9 Hz, 1H), 4.64 (d, J = 6.8 Hz, 1H), 4.58 (d, J = 6.8 Hz, 1H), 4.51 (s, 2H), 4.05 (m, 3H), 3.58 (t, J = 6.3 Hz, 2H), 3.31 (s, 3H), 2.88 (dd, J = 6.8 Hz, 15.9 Hz, 1H), 2.61-2.48 (m, 3H), 2.02 (s, 3H), 1.75-1.38 (m, 6H); 13C-NMR (CDCl3) δ 198.3 (s), 171.1 (s), 144.3 (d), 138.0 (s), 132.1 (d), 128.4 (d), 127.7 (d), 96.1 (t), 74.2 (d), 73.0 (t), 68.2 (t), 64.3 (t), 55.5 (q), 45.3 (t), 34.7 (t), 32.9 (t), 28.6 (t), 21.7 (t), 20.9 (q). tert-Butyldimethylsilyl triflate (0.51 mL, 2.23 mmol) was added to a stirred solution of the previously prepared compound (624 mg, 1.59 mmol) in THF (16 mL) at -78 °C under argon. After 15 min a solution of LDA (0.24 M in THF, 7.9 mL, 1.9 mmol) was added. The reaction mixture was stirred at -78 °C for 30 min, and then a saturated aqueous solution of NH4Cl (15 mL) was added. The reaction was allowed to reach room temperature and extracted with Et2O. The combined organic phases were dried (MgSO4), concentrated, and chromatographed on silica gel (80:20 Hex-EtOAc) yielding 500 mg (62%) of 10 as a colorless oil; ![Molecules 12 00194 i001]() = -49.39 (c 4.90, CHCl3); 1H-NMR (C6D6) δ 7.37-7.10 (m, 5H), 6.02 (dt, J = 6.7 Hz, 15.9 Hz, 1H), 5.88 (d, J = 15.9 Hz, 1H), 4.86 (d, J = 6.6, 1H), 4.75 (m, 2H), 4.46 (d, J = 6.6 Hz, 1H), 4.28 (s, 2H), 4.04-3.96 (m, 2H), 3.30 (t, J = 6.4 Hz, 2H), 3.24 (s, 3H), 2.30-2.26 (m, 2H), 1.67 (s, 3H), 1.66-1.50 (m, 6H), 0.99 (s, 9H), 0.20 (s, 3H), 0.14 (s, 3H); 13C-NMR (CDCl3) δ 169.5 (s), 150.1 (s), 138.5 (s), 129.7 (d), 128.2 (d), 127.9 (d), 127.4 (d), 113.4 (d), 93.2 (t), 72.4 (t), 69.5 (d), 69.2 (t), 63.7 (t), 54.5 (q), 35.3 (t), 32.6 (t), 28.5 (t), 25.6 (q), 21.9 (t), 19.9 (q), 18.0 (s), -4.1 (q).
= -49.39 (c 4.90, CHCl3); 1H-NMR (C6D6) δ 7.37-7.10 (m, 5H), 6.02 (dt, J = 6.7 Hz, 15.9 Hz, 1H), 5.88 (d, J = 15.9 Hz, 1H), 4.86 (d, J = 6.6, 1H), 4.75 (m, 2H), 4.46 (d, J = 6.6 Hz, 1H), 4.28 (s, 2H), 4.04-3.96 (m, 2H), 3.30 (t, J = 6.4 Hz, 2H), 3.24 (s, 3H), 2.30-2.26 (m, 2H), 1.67 (s, 3H), 1.66-1.50 (m, 6H), 0.99 (s, 9H), 0.20 (s, 3H), 0.14 (s, 3H); 13C-NMR (CDCl3) δ 169.5 (s), 150.1 (s), 138.5 (s), 129.7 (d), 128.2 (d), 127.9 (d), 127.4 (d), 113.4 (d), 93.2 (t), 72.4 (t), 69.5 (d), 69.2 (t), 63.7 (t), 54.5 (q), 35.3 (t), 32.6 (t), 28.5 (t), 25.6 (q), 21.9 (t), 19.9 (q), 18.0 (s), -4.1 (q).
= -0.85 (c 2.60, CHCl3); 1H-NMR (CDCl3) δ 7.36 (m, 5H), 6.85 (dt, J = 6.8 Hz, 15.9 Hz, 1H), 6.17 (d, J = 15.9 Hz, 1H), 4.64 (d, J = 6.8 Hz, 1H), 4.58 (d, J = 6.8 Hz, 1H), 4.51 (s, 2H), 4.05 (m, 3H), 3.58 (t, J = 6.3 Hz, 2H), 3.31 (s, 3H), 2.88 (dd, J = 6.8 Hz, 15.9 Hz, 1H), 2.61-2.48 (m, 3H), 2.02 (s, 3H), 1.75-1.38 (m, 6H); 13C-NMR (CDCl3) δ 198.3 (s), 171.1 (s), 144.3 (d), 138.0 (s), 132.1 (d), 128.4 (d), 127.7 (d), 96.1 (t), 74.2 (d), 73.0 (t), 68.2 (t), 64.3 (t), 55.5 (q), 45.3 (t), 34.7 (t), 32.9 (t), 28.6 (t), 21.7 (t), 20.9 (q). tert-Butyldimethylsilyl triflate (0.51 mL, 2.23 mmol) was added to a stirred solution of the previously prepared compound (624 mg, 1.59 mmol) in THF (16 mL) at -78 °C under argon. After 15 min a solution of LDA (0.24 M in THF, 7.9 mL, 1.9 mmol) was added. The reaction mixture was stirred at -78 °C for 30 min, and then a saturated aqueous solution of NH4Cl (15 mL) was added. The reaction was allowed to reach room temperature and extracted with Et2O. The combined organic phases were dried (MgSO4), concentrated, and chromatographed on silica gel (80:20 Hex-EtOAc) yielding 500 mg (62%) of 10 as a colorless oil; = -49.39 (c 4.90, CHCl3); 1H-NMR (C6D6) δ 7.37-7.10 (m, 5H), 6.02 (dt, J = 6.7 Hz, 15.9 Hz, 1H), 5.88 (d, J = 15.9 Hz, 1H), 4.86 (d, J = 6.6, 1H), 4.75 (m, 2H), 4.46 (d, J = 6.6 Hz, 1H), 4.28 (s, 2H), 4.04-3.96 (m, 2H), 3.30 (t, J = 6.4 Hz, 2H), 3.24 (s, 3H), 2.30-2.26 (m, 2H), 1.67 (s, 3H), 1.66-1.50 (m, 6H), 0.99 (s, 9H), 0.20 (s, 3H), 0.14 (s, 3H); 13C-NMR (CDCl3) δ 169.5 (s), 150.1 (s), 138.5 (s), 129.7 (d), 128.2 (d), 127.9 (d), 127.4 (d), 113.4 (d), 93.2 (t), 72.4 (t), 69.5 (d), 69.2 (t), 63.7 (t), 54.5 (q), 35.3 (t), 32.6 (t), 28.5 (t), 25.6 (q), 21.9 (t), 19.9 (q), 18.0 (s), -4.1 (q).Preparation of (S,6Z,8E)-11-(benzyloxy)-7-(tert-butyldimethylsilyloxy)-5-(methoxymethoxy)undeca-6,8-dienal (11)
To a solution of 10 (500 mg, 0.98 mmol) in Et2O (10 mL) at -70°C under argon was added DIBAL (1 M in hexanes, 4.94 mL, 4.94 mmol). The reaction was stirred until TLC showed disappearance of the starting material (15 min). The reaction mixture was quenched by slowly addition of MeOH (1.0 mL) and the solution was allowed to reach room temperature. The solution was poured into an aqueous saturated solution of potassium sodium tartrate and it was vigorously stirred until the phases became clear. After phase separation, the combined organic extracts were dried (MgSO4), concentrated, and chromatographed on silica gel (70:30 Hex-EtOAc) yielding 345 mg (76%) of (S,6Z,8E)-11-(benzyloxy)-7-(tert-butyldimethylsilyloxy)-5-(methoxymethoxy)undeca-6,8-dien-1-ol as a colorless oil; 1H-NMR (C6D6) δ 7.33-7.06 (m, 5H), 6.05-5.98 (m, 1H), 5.85 (d, J = 15.6 Hz, 1H), 4.85 (d, AB system, J = 6.6 Hz, 1H), 4.75 (m, 2H), 4.47 (d, AB system, J = 6.6 Hz, 1H), 4.27 (s, 2H), 3.45 (t, J = 6.0 Hz, 2H), 3.27 (t, J = 6.5 Hz, 2H), 3.24 (s, 3H), 2.29-2.23 (m, 2H), 1.78-1.30 (m, 7H), 1.05 (s, 9H), 0.19 (s, 3H), 0.14 (s, 3H); 13C-NMR (C6D6) δ 150.6 (s), 139.1 (s), 130.3 (d), 128.5 (d), 127.8 (d), 127.7 (d), 114.2 (d), 93.8 (t), 72.9 (t), 70.4 (t), 69.8 (t), 62.6 (d), 55.1 (q), 36.1 (t), 33.2 (t), 26.2 (q), 22.4 (t), 18.6 (s), -3.4 (q). To a solution of the previously prepared alcohol (340 mg, 0.73 mmol) in CH2Cl2 (7 mL) under argon at 0 °C was added TEA (0.5 mL, 3.65 mmol) and DMSO (0.77 mL, 11.0 mmol). After 15 min SO3·Py (348 mg, 3.65 mmol) was added. The reaction mixture was allowed to reach to room temperature and stirred for 3 h. Water was added, and the reaction was extracted with Et2O (4 × 15 mL). The combined organic extracts were washed with brine. The solution was dried (MgSO4) and concentrated. The expected aldehyde 11 was obtained pure enough, according to 1H-NMR, and was usually used without further purification. This aldehyde could be purified by column chromatography on silica gel (80:20 Hex:EtOAc), although the yield was very low due to decomposition.
Hetero Diels-Alder Reaction
To a solution of the previously purified aldehyde 11 (135 mg, 0.29 mmol) in Et2O (0.6 mL) under argon at 0 °C were added benzylamine (0.03 mL, 0.29 mmol) and a small amount of anhydrous MgSO4. After stirring for 30 min the reaction was diluted with pentane, filtered and concentrated. The crude reaction mixture was dissolved in CH3CN (5.0 mL) and cooled to 0 °C under argon atmosphere. Then In(OTf)3 (81.5 mg, 0.145 mmol) in CH3CN (6 mL) was added and the reaction was allowed to warm to room temperature. After stirring for 12 h, a cold aqueous solution of NaHCO3 was added. After extraction with Et2O, the combined organic layers were dried (MgSO4), concentrated and chromatographed on silica gel (90:10 Hex-EtOAc) yielding 127 mg (79%) of a 70:30 mixture of 12 and 13 which were separated by HPLC.
(2R,4aS,5S,8aR)-1-benzyl-2-(2-benzyloxy)ethyl)-4-(tert-butyldimethylsilyloxy)-5-(methoxymethoxy)-1,2,4a,5,6,7,8,8a-octahydroquinoline (12) (major isomer); colorless oil; ![Molecules 12 00194 i001]() = -23.08 (c 1.30, CHCl3); 1H-NMR (CDCl3) δ 7.32-7.13 (m, 10H), 4.81 (m, 2H), 4.61 (d, J = 6.9 Hz, 1H), 4.13 (AB system, J = 11.6, 8.8 Hz, 2H), 3.84 (d, J = 13.3 Hz, 1H), 3.43 (m, 1H), 3.38 (s, 3H), 3.26 (d, J = 13.3 Hz, 1H), 3.22 (m, 1H), 3.08 (m, 1H), 2.67 (m, 1H), 2.15 (m, 2H), 1.80 (m, 1H), 1.75-1.69 (m, 3H), 1.60-1.55 (m, 2H), 1.44 (m, 2H), 0.98 (s, 9H), 0.22 (s, 3H), 0.20 (s, 3H); 13C-NMR (CDCl3) δ 151.5 (s), 140.8 (s), 138.6 (s), 129.1 (d), 128.2 (d), 128.0 (d), 127.8 (d), 127.3 (d), 126.7 (d), 108.0 (d), 97.5 (t), 80.5 (d), 72.7 (t), 68.0 (t), 56.2 (d), 55.3 (q), 53.7 (d), 51.1 (t), 44.2 (d), 35.4 (t), 34.4 (t), 30.4 (t), 26.2 (q), 23.0 (t), 18.7 (s), -4.9 (q).
= -23.08 (c 1.30, CHCl3); 1H-NMR (CDCl3) δ 7.32-7.13 (m, 10H), 4.81 (m, 2H), 4.61 (d, J = 6.9 Hz, 1H), 4.13 (AB system, J = 11.6, 8.8 Hz, 2H), 3.84 (d, J = 13.3 Hz, 1H), 3.43 (m, 1H), 3.38 (s, 3H), 3.26 (d, J = 13.3 Hz, 1H), 3.22 (m, 1H), 3.08 (m, 1H), 2.67 (m, 1H), 2.15 (m, 2H), 1.80 (m, 1H), 1.75-1.69 (m, 3H), 1.60-1.55 (m, 2H), 1.44 (m, 2H), 0.98 (s, 9H), 0.22 (s, 3H), 0.20 (s, 3H); 13C-NMR (CDCl3) δ 151.5 (s), 140.8 (s), 138.6 (s), 129.1 (d), 128.2 (d), 128.0 (d), 127.8 (d), 127.3 (d), 126.7 (d), 108.0 (d), 97.5 (t), 80.5 (d), 72.7 (t), 68.0 (t), 56.2 (d), 55.3 (q), 53.7 (d), 51.1 (t), 44.2 (d), 35.4 (t), 34.4 (t), 30.4 (t), 26.2 (q), 23.0 (t), 18.7 (s), -4.9 (q).
= -23.08 (c 1.30, CHCl3); 1H-NMR (CDCl3) δ 7.32-7.13 (m, 10H), 4.81 (m, 2H), 4.61 (d, J = 6.9 Hz, 1H), 4.13 (AB system, J = 11.6, 8.8 Hz, 2H), 3.84 (d, J = 13.3 Hz, 1H), 3.43 (m, 1H), 3.38 (s, 3H), 3.26 (d, J = 13.3 Hz, 1H), 3.22 (m, 1H), 3.08 (m, 1H), 2.67 (m, 1H), 2.15 (m, 2H), 1.80 (m, 1H), 1.75-1.69 (m, 3H), 1.60-1.55 (m, 2H), 1.44 (m, 2H), 0.98 (s, 9H), 0.22 (s, 3H), 0.20 (s, 3H); 13C-NMR (CDCl3) δ 151.5 (s), 140.8 (s), 138.6 (s), 129.1 (d), 128.2 (d), 128.0 (d), 127.8 (d), 127.3 (d), 126.7 (d), 108.0 (d), 97.5 (t), 80.5 (d), 72.7 (t), 68.0 (t), 56.2 (d), 55.3 (q), 53.7 (d), 51.1 (t), 44.2 (d), 35.4 (t), 34.4 (t), 30.4 (t), 26.2 (q), 23.0 (t), 18.7 (s), -4.9 (q).(2S,4aR,5S,8aR)-1-benzyl-2-(2-benzyloxy)ethyl)-4-(tert-butyldimethylsilyloxy)-5-(methoxymethoxy)-1,2,4a,5,6,7,8,8a-octahydroquinoline (13) (minor isomer); colorless oil; 1H-NMR (CDCl3) δ 7.35-7.21 (m, 10H), 4.76-4.74 (m, 1H), 4.64-4.60 (m, 2H), 4.35-4.32 (m, 3H), 3.83 (d, J = 14.0 Hz, 1H), 3.68 (d, J = 14.0 Hz, 1H), 3.50-3.43 (m, 2H), 3.31 (s, 3H), 3.29 (m, 1H), 3.07-3.04 (m, 1H), 2.73 (m, 1H), 1.88 (m, 1H), 1.74-1.37 (m, 7H), 0.94 (s, 9H), 0.18 (s, 3H), 0.17 (s, 3H); 13C-NMR (CDCl3) δ 147.3 (s), 140.8 (s), 138.6 (s), 128.6 (d), 128.3 (d), 128.1 (d), 127.7 (d), 127.4 (d), 126.6 (d), 105.4 (d), 95.2 (t), 73.7 (d), 72.9 (t), 68.4 (t), 59.8 (t), 55.4 (d), 55.2 (q), 39.3 (d), 37.1 (t), 29.5 (t), 27.8 (t), 25.7 (q), 20.1 (t), 18.1 (s), -4.9 (q).
Preparation of (2R,4aS,5S,8aR)-1-benzyl-2-(2-benzyloxy)ethyl)-5-(methoxymethoxy)octahydro-quinolin-4(1H)-one (14)
To a solution of 12 (79 mg, 0.14 mmol) in THF (5 mL) at 0 °C under argon was added TBAF (1.1 equiv). The reaction mixture was allowed to warm to room temperature and stirred for 45 min. The reaction was quenched with ice and brine. After extraction with Et2O, the combined extracts were dried (MgSO4), concentrated, and chromatographed on silica gel (75:25 Hex-EtOAc) yielding 56 mg (90%) of 14 as a colorless oil; 1H-NMR (CDCl3) δ 7.37-7.16 (m, 10H), 4.97 (d, J = 6.7 Hz; 1H), 4.66 (d, J =6.7 Hz, 1H), 4.33 (c, J = 11.7 Hz, 2H), 4.10 (d, J = 14.2 Hz, 1H), 3.85-3.80 (m, 1H), 3.73 (d, J = 14.2 Hz, 1H), 3.43-3.40 (m, 3H), 3.40 (s, 3H), 2.98-2.86 (m, 2H), 2.59 (t, J = 10.2 Hz, 1H), 2.17 (m, 1H), 2.08 (d, J = 12.2 Hz, 1H), 1.80-1.77 (m, 3H), 1.62-1.49 (m, 2H), 1.30-1.20 (m, 2H); 13C-NMR (CDCl3) δ 208.6 (s), 140.4 (s), 138.3 (s), 128.2 (d), 127.5 (d), 127.4 (d), 126.9 (d), 97.0 (t), 73.2 (d), 72.7 (t), 67.2 (t), 60.1 (d), 57.8 (d), 57.5 (d), 55.5 (q), 51.6 (t), 43.2 (t), 32.2 (t), 30.7 (t), 30.6 (t), 21.8 (t); EIMS m/z (%): 437 (M+, 0.7); HRMS (EI): C27H35NO4 (M+): 437.2566, found 437.2554.
Acknowledgments
This work was supported in part by Grant BQU2001-3184 (MEC, Spain) and a University of La Laguna Grant for Consolidated Research Groups.
References and Notes
- Daly, J.W.; Spande, T.F.; Garraffo, H.M. Alkaloids from amphibian skin. A tabulation of over eight-hundred compounds. J. Nat. Prod. 2005, 68, 1556–1575. [Google Scholar]
- Toyooka, N.; Nemoto, H. Synthetic studies on dart-poison frog alkaloids. Rec. Res. Dev. Org. Chem. 2002, 6, 611–624. [Google Scholar]
- Banner, E.J.; Stevens, E.D.; Trudell, M.L. Stereoselective synthesis of the cis-275B decahydroquinoline ring system. Tetrahedron Lett. 2004, 45, 4411–4414. [Google Scholar] Abe, H.; Aoyagi, S.; Kibayashi, C. First total synthesis of the marine alkaloids (±)-Fasicularine and (±)-Lepadiformine based on stereocontroled intramolecular acylnitroso-Diels-Alder. J. Am. Chem. Soc. 2000, 122, 4583–4592. [Google Scholar] Comins, D.L.; Al-Awar, R.S. An intramolecular Diels-Alder / retro-Mannich approach to the cis-perhydroquinoline ring system. Model studies toward the synthesis of Lycopodium alkaloids. J. Org. Chem. 1992, 57, 4098–4103. [Google Scholar] Oppolzer, W.; Flaskamp, E.; Bieber, L.W. Efficient asymmetric synthesis of pumiliotoxin C via intramolecular [4+2] cycloaddition 141-145. Helv. Chim. Acta 2001, 84, 141–145. [Google Scholar]
- Grieco, P.A.; Parker, D.T. Octahydroquinoline synthesis via immonium ion based Diels-Alder chemistry. Synthesis of (-)-8a-epipumiliotoxin C. J. Org. Chem. 1988, 53, 3658–3662. [Google Scholar]
- Adam, W.; Gläser, J.; Peters, K.; Prein, M. Highly like-selective [4+2] cycloadditions of chiral dienols: The importance of 1,3-allylic strain in the hydroxy-directed stereocontrol. J. Am. Chem. Soc. 1995, 117, 9190–9193. [Google Scholar] Roush, W.R.; Kageyama, M.; Riva, R.; Brown, B.B.; Warmus, J.S.; Moriarty, K.J. Enantioselective synthesis of the bottom half of chlorothricolide. 3. Studies of the steric directing group strategy for stereocontrol in intramolecular Diels-Alder reactions. J. Org. Chem. 1991, 56, 1192–1210. [Google Scholar]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Murphy, J.A.; Rasheed, F.; Roome, S.J.; Scott, K.A.; Lewis, N. Intramolecular termination of radical polar crossover reactions. J. Chem. Soc. Perkin Trans. 1. 1998, 15, 2331–2340. [Google Scholar] [CrossRef]
- Stonehouse, J.; Adell, P.; Keeler, J.; Shaka, A.J. Ultrahigh-quality NOE spectra. J. Am. Chem. Soc. 1994, 116, 6037–6038. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds described in this paper are available from authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Ruiz, J.M.; Afonso, M.M.; Palenzuela, J.A. Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction. Molecules 2007, 12, 194-204. https://doi.org/10.3390/12020194
AMA Style
Ruiz JM, Afonso MM, Palenzuela JA. Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction. Molecules. 2007; 12(2):194-204. https://doi.org/10.3390/12020194
Chicago/Turabian StyleRuiz, Juan M., Maria M. Afonso, and J. Antonio Palenzuela. 2007. "Synthesis of 2,5-Disubstituted Octahydroquinolin-4-ones via anIntramolecular Hetero Diels-Alder Reaction" Molecules 12, no. 2: 194-204. https://doi.org/10.3390/12020194