Synthesis and Biological Evaluation of Rigid Polycyclic Derivatives of the Diels-Alder Adduct Tricyclo[6.2.1.02,7]undeca-4,9-dien-3,6-dione

Abstract
:Introduction
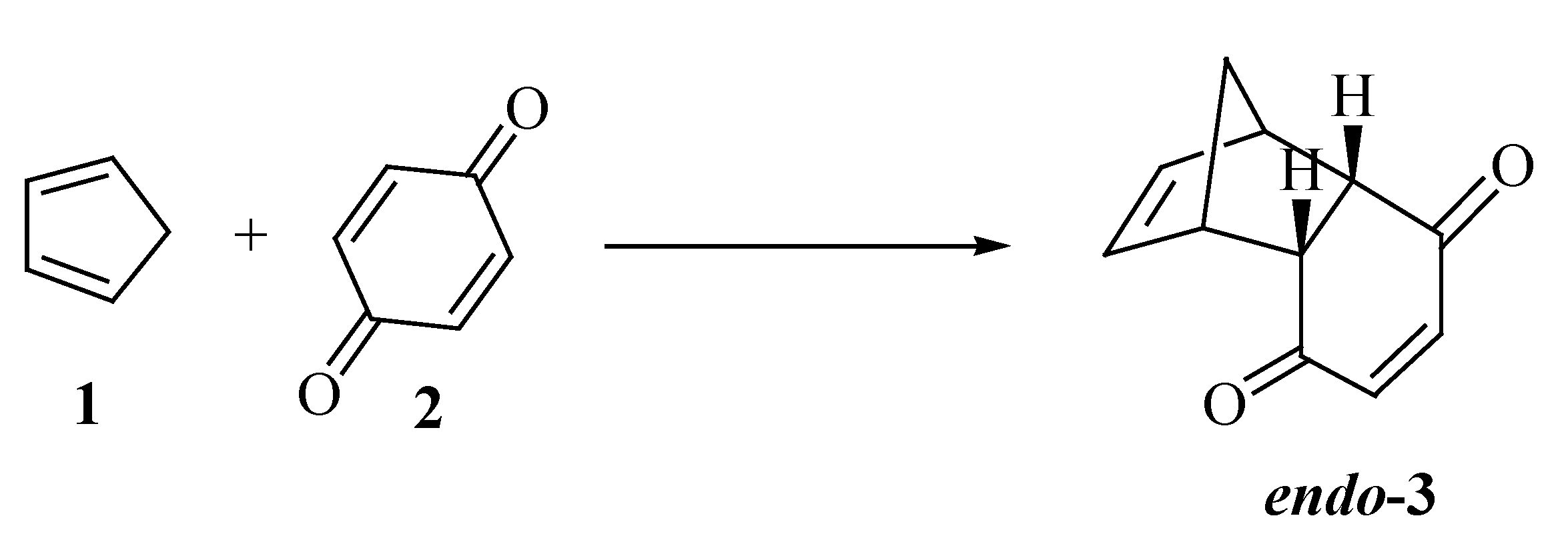

Results and Discussion
Chemistry
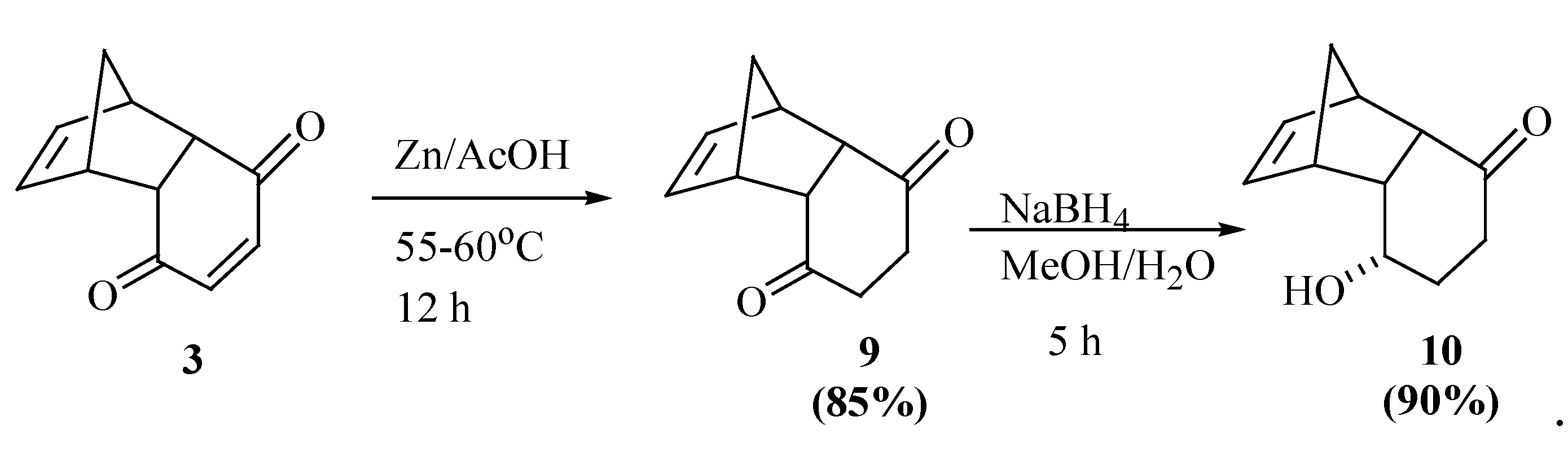
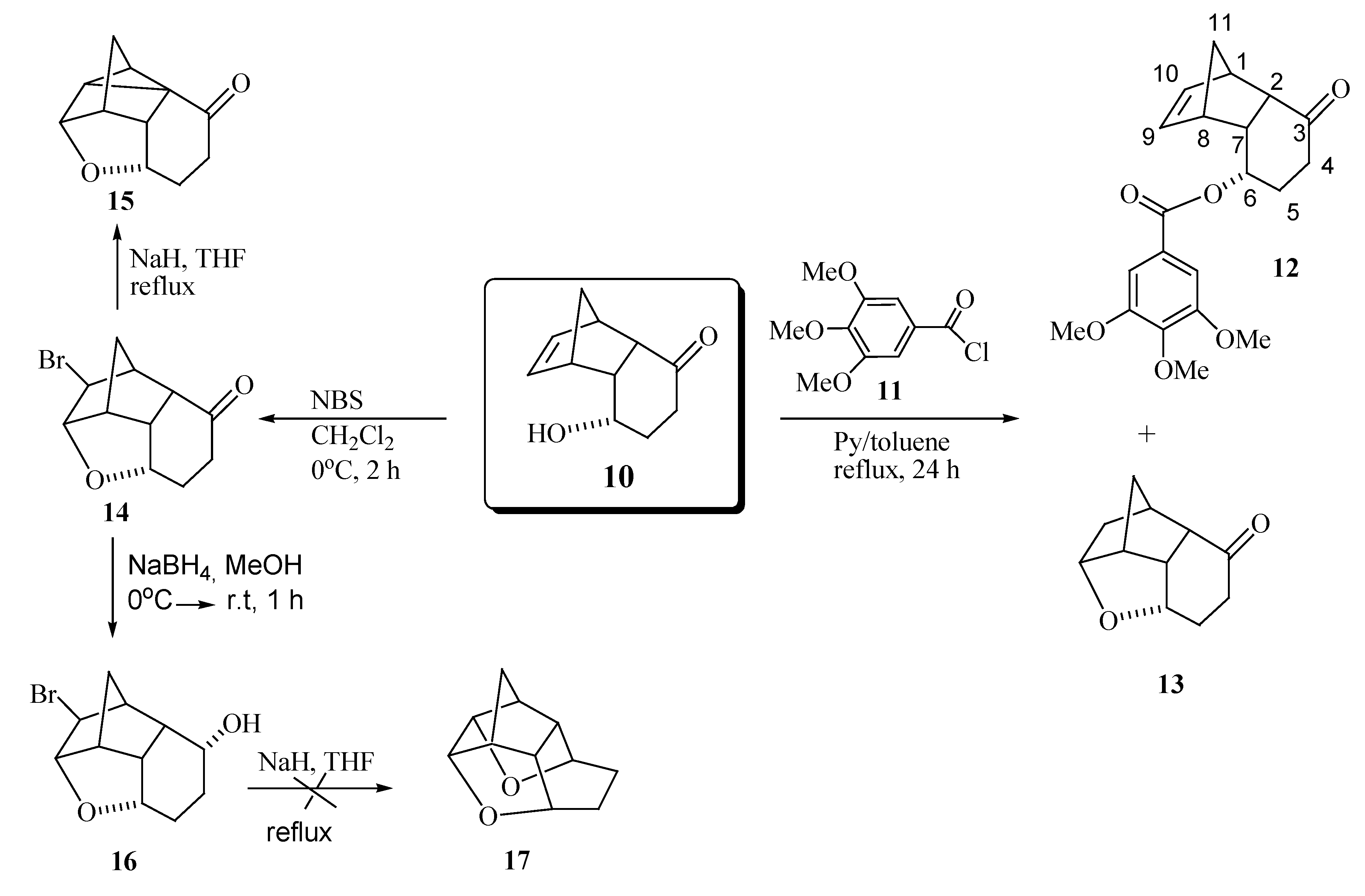
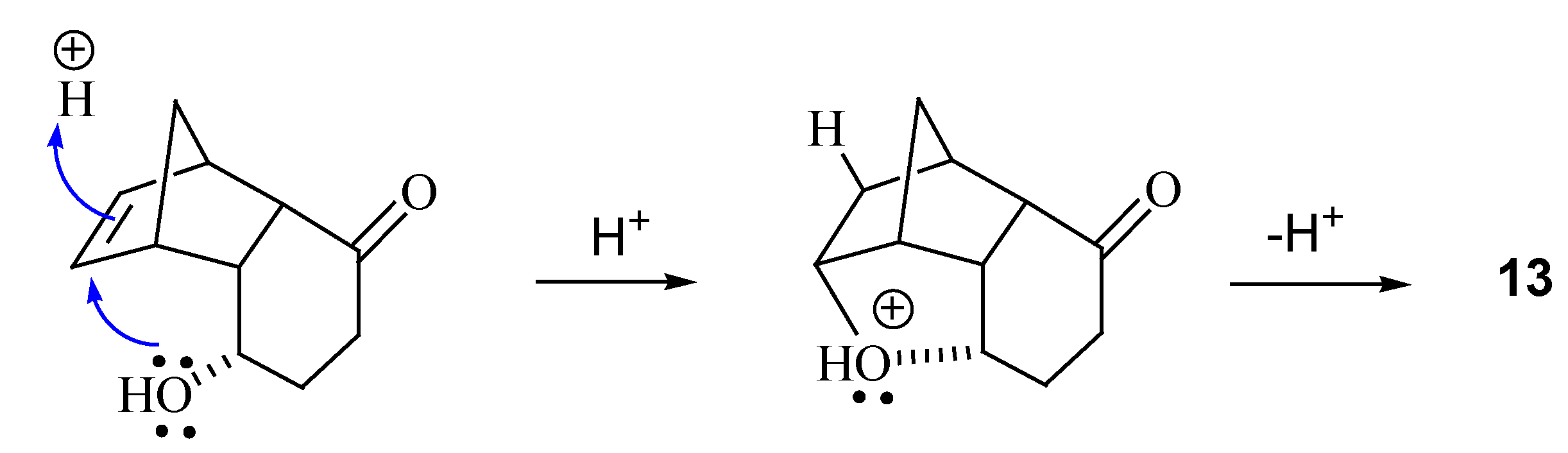

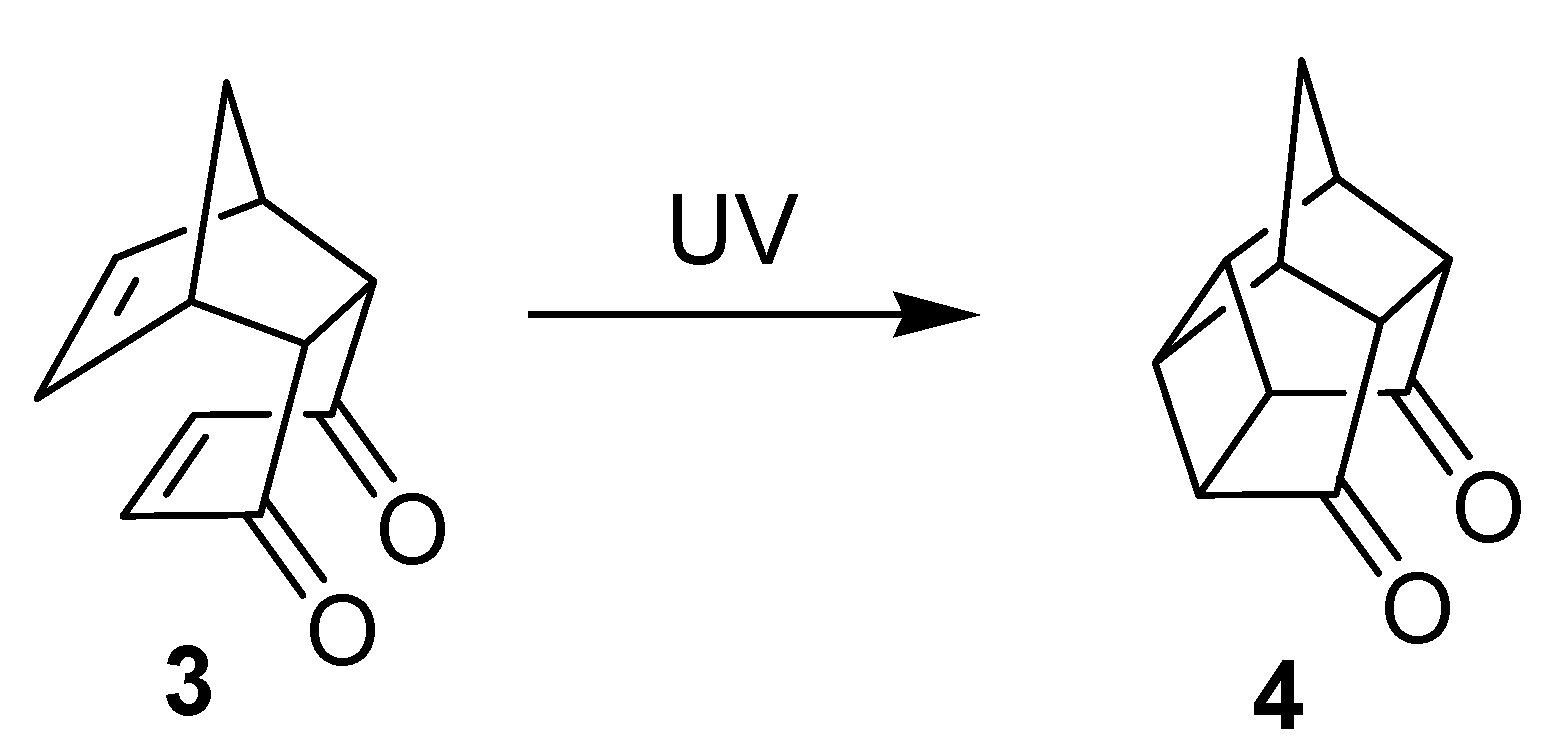
{kind=link}
{kind=link}
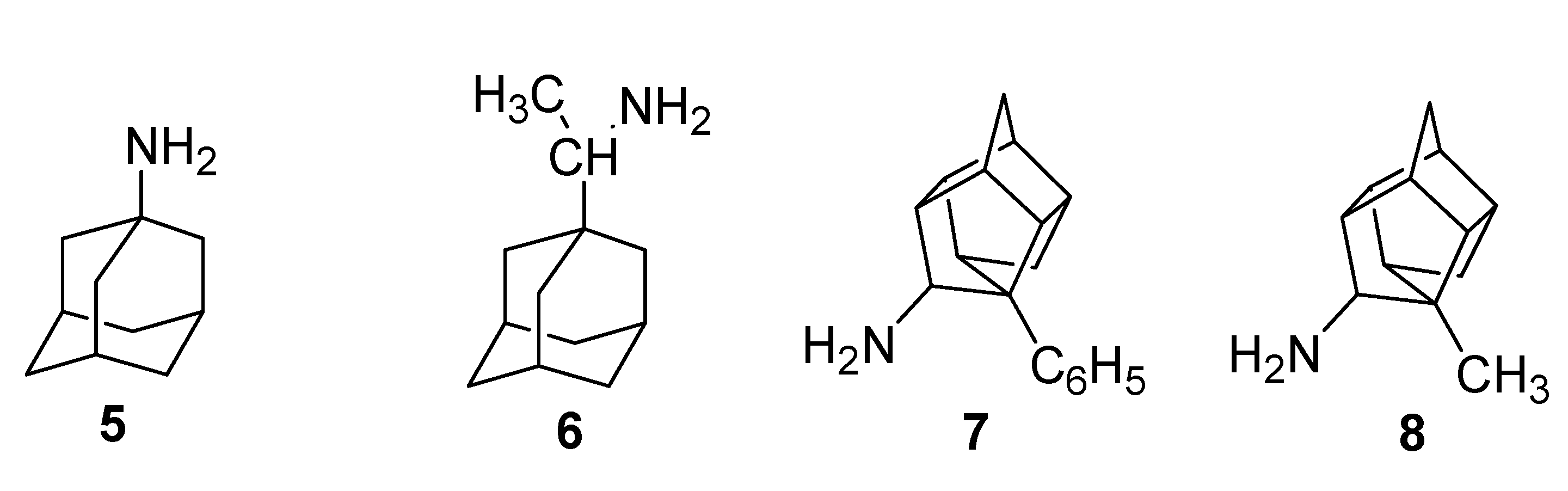
{kind=link}
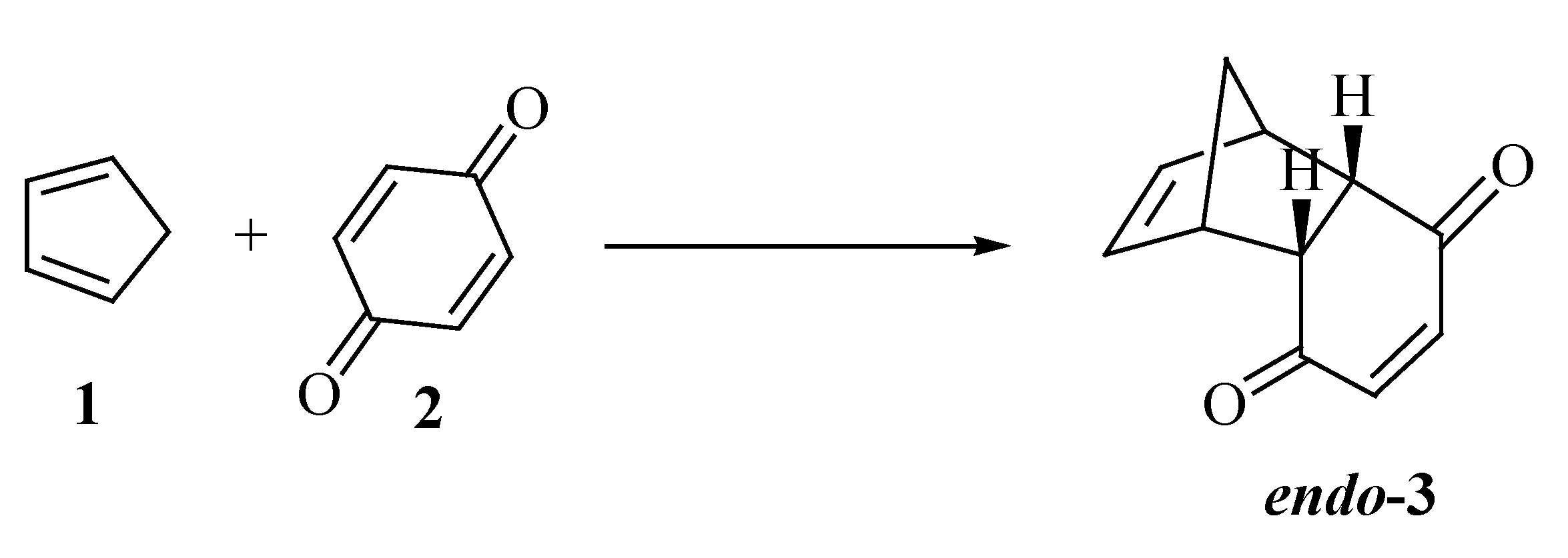{kind=link}
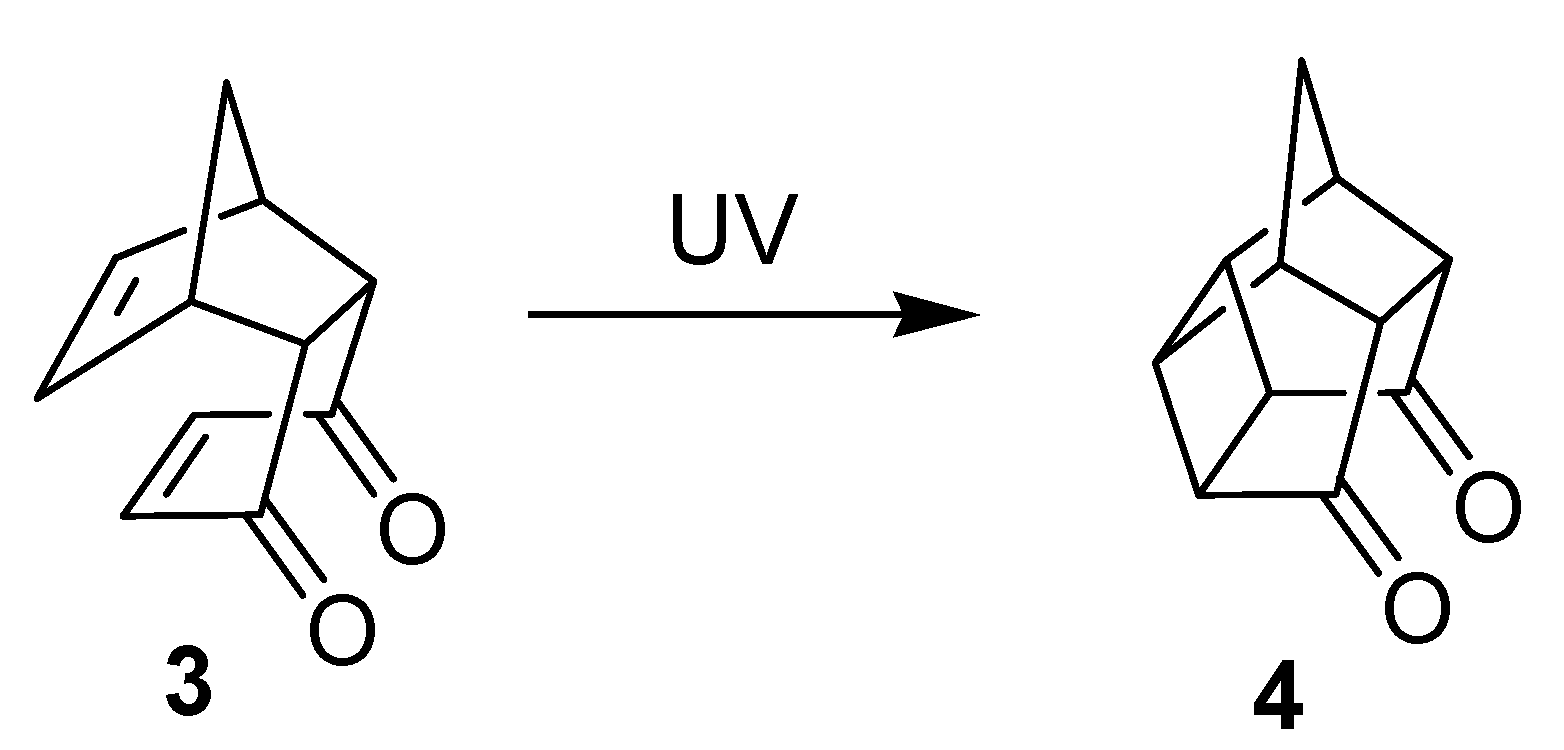{kind=link}
{kind=link}
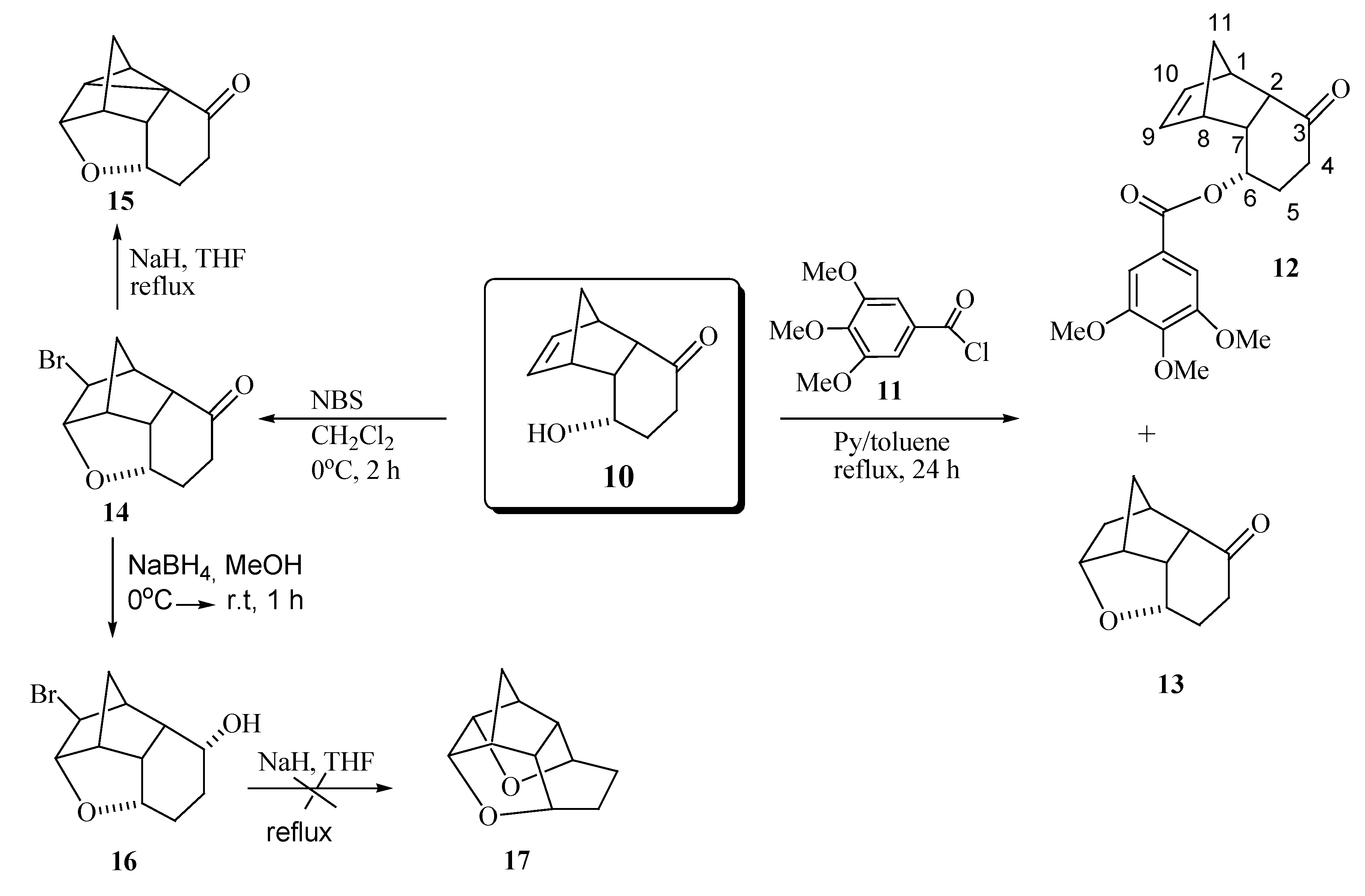{kind=link}
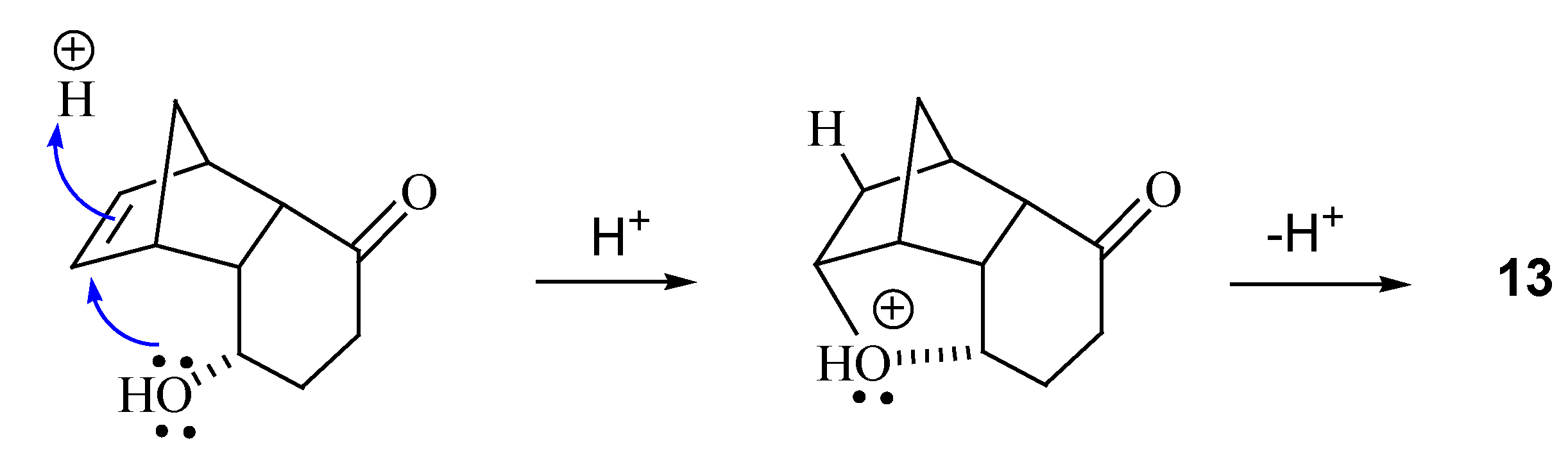{kind=link}
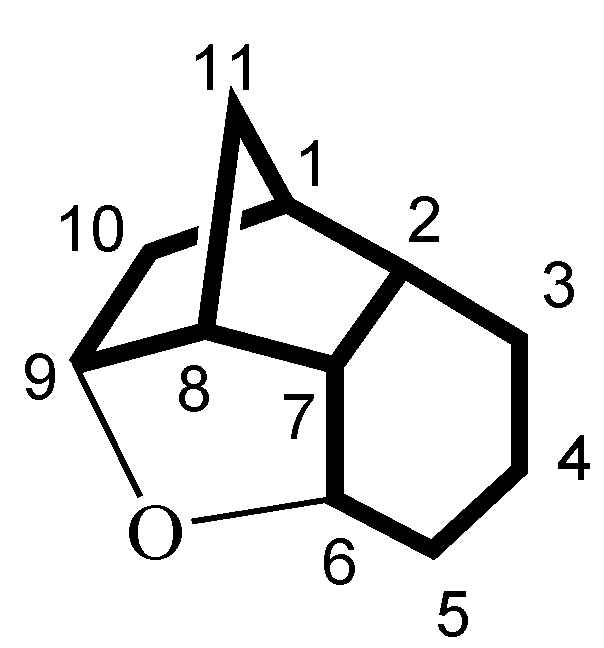
| Positionb | 13 Cδ | 14 Cδ | 16 Cδ | 15 Cδ |
| 1 | 39.3 | 42.2 | 42.9 | 21.8 |
| 2 | 51.2 | 49.8 | 39.5 | 33.1 |
| 3 | 214.0 | 211.8 | 69.7 | 209.4 |
| 4 | 37.4 | 35.0 | 26.7 | 38.3 |
| 5 | 26.5 | 25.9 | 26.1 | 30.4 |
| 6 | 74.4 | 75.3 | 76.1 | 74.1 |
| 7 | 43.5 | 46.9 | 44.2 | 42.0 |
| 8 | 49.1 | 48.6 | 49.1 | 46.3 |
| 9 | 79.7 | 88.4 | 88.7 | 82.2 |
| 10 | 35.1 | 55.1 | 56.5 | 35.3 |
| 11 | 37.2 | 34.9 | 35.4 | 30.3 |
Biological Evaluation
1. Assay for cytotoxic activity
| Compound | Cellsa IC50b [µg/mL, confidence interval] | ||||||
| HL-60 | HCT-8 | SF-295 | MDA-MB 435 | ||||
| 3 | 11.34 (8.68 – 14,80) | 13.62 (5.52 – 33.63) | > 25 | 13.52 (11.57 – 15.80) | |||
| 9 | > 25 | > 25 | > 25 | 10.47 (4.80 – 22.05) | |||
| 10 | > 25 | > 25 | > 25 | > 25 | |||
| 12 | > 25 | 6.68 (4.22 – 10.56) | 7.58 (6.12 – 9.39) | 13.51 (11.16 – 16.37) | |||
| 14 | > 25 | > 25 | > 25 | > 25 | |||
| 15 | > 25 | > 25 | > 25 | > 25 | |||
| 16 | > 25 | > 25 | > 25 | > 25 | |||
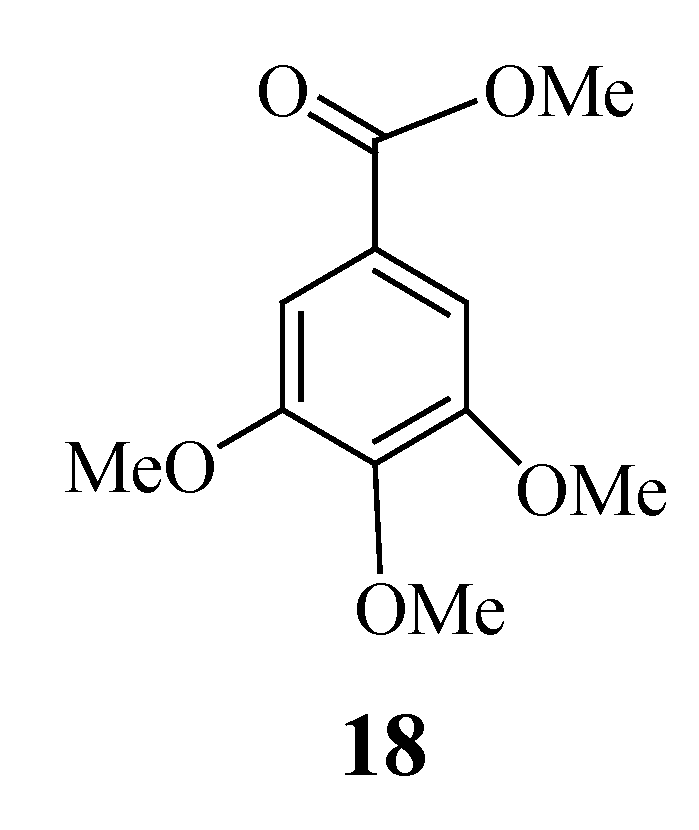
| 18 | > 25 | > 25 | > 25 | > 25 | |||
2. Assay of hemolytic activity in Swiss mouse (Mus musculus) erythrocytes
Conclusions
Experimental Section
General
Chemistry Synthesis of rel-(1S,2S,7R,8R)-tricyclo[6.2.1.02,7]undeca-4,9-dien-3,6-dione (3)
Synthesis of rel-(2S,3S,4S,5S,6S,10S,1R,9R)-2-bromine-3,6-epoxytricyclo[6.2.1.05,10]undecan-9-ol (16)
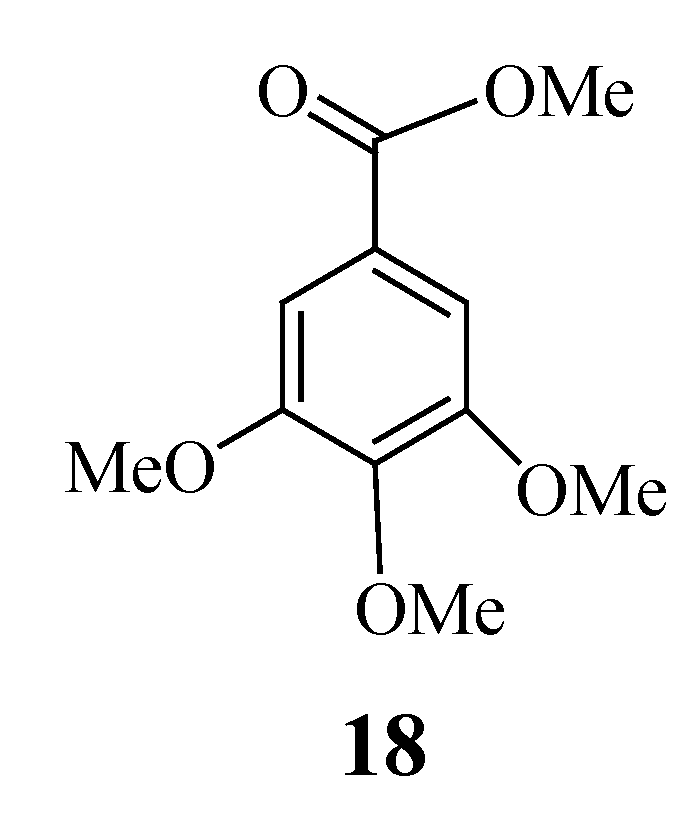
Synthesis of rel-(1S,3S,7S,2R,8R)-6-oxo-3-(3,4,5-trimethoxyphenylcarbonyloxy) tricyclo[6.2.1.02,7]-undec-9-ene (12) and rel- (1S,2S,6S,8S,7R,9R)-6,9-epoxytricyclo[6.2.1.02,7]undecan-3-one (13)
Synthesis of 6,9-epoxytetracyclo[6.2.1.02,7.02,10]undecan-3-one (15)
Biological Evaluation
1. Assay for cytotoxic activity
2. Swiss mouse (Mus musculus) erythrocyte hemolysis assay
Statistical Analysis
Acknowledgments
References and Notes
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen reihe. Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Oliveira, K.T. Estudos sintéticos e teóricos sobre anulenos e baquenolidas. Ph.D. Thesis, University of São Paulo, 2006. [Google Scholar] (for online access see: http://www.teses.usp.br/teses/disponiveis/59/59138/tde-18082006-181618/)
- Marchand, P.; Xing, D.; Wang, Y.; Bott, S.G. Improved synthesis of racemic and optically active 4-hydroxycyclohex-2-en-1-one. Tetrahedron Asymmetr. 1995, 6, 2709–2711. [Google Scholar] [CrossRef]
- Cookson, R.C.; Grundwell, E.; Hudec, J. Synthesis of cage-like molecules by irradiation of Diels-Alder adducts. Chem. Ind. (London) 1958, 1003–1004. [Google Scholar]
- Govender, T.; Hariprakasha, H.K.; Kruger, H.G.; Raasch, T. Synthesis of Trishomocubane Amino Acid Derivatives. S. Afr. J. Chem. 2005, 58, 37–40. [Google Scholar]
- Geldenhuys, W.J.; Malan, S. F.; Bloomquist, J.R.; Marchand, A.P.; Van der Schyf, C.J. Pharmacology and structure – activity relationships of bioactive polycyclic cage compounds: a focus on pentacycloundecane derivatives. Med. Res. Rev. 2005, 25, 22–48. [Google Scholar]
- Kitagawa, K.; Mizobuchi, N.; Hama, T.; Hibi, T.; Konishi, R.; Futaki, S. Synthesis and antinociceptive activity of [D-Ala2]Leu-enkephalin derivatives conjugated with the adamantane moiety. Chem. Pharm. Bull. 1997, 45, 1782–1787. [Google Scholar] [CrossRef]
- Marchand, A.P.; Allen, R.W. Improved synthesis of pentacyclo[5.4.0.02,6.03,10.05,9]undecane. J. Org. Chem. 1974, 39, 1596–1596. [Google Scholar] [CrossRef]
- Wassermann, A. The mechanism of additions to double bonds. Part I. Thermochemistry and kinetics of a diene synthesis. J. Chem. Soc. 1935, 828–838. [Google Scholar] [CrossRef]
- Oda, M.; Kawase, T.; Okada, T.; Enomoto, T. Org. Synth. Coll. Vol. IX 1998, 186.
- Stroermer, M.J.; Butler, D.N.; Warrener, R.N.; Weerasuria, K.D.V.; Fairlie, D.P. Cycloadditions of isobenzofuran to a constrained template bearing neighboring dienophiles. Chem. Eur. J. 2003, 9, 2068–2071. [Google Scholar]
- Marchand, A.P.; Reddy, G.M. Mild and highly selective ultrasound-promoted zinc/acetic acid reduction of C:C bonds in α,β-unsaturated γ-dicarbonyl compounds. Synthesis 1991, 198–200. [Google Scholar] [CrossRef]
- Constantino, M. G.; Beatriz, A.; Silva, G.V.J.da.; Zukerman-Schpector, J. Synthetic studies on the diels–alder adduct from 3,4-dimethoxyfuran and benzoquinone. Synth. Commun. 2001, 31, 3329–3336. [Google Scholar] [CrossRef]
- Takano, S.; Inomata, K.; Ogasawara, K. A new route to (+)-2,3-(isopropylidenedioxy)-4-cyclopentenone via the optically active dicyclopentadiene intermediate. Chem. Lett. 1989, 359–362. [Google Scholar] [CrossRef]
- Konno, H.; Ogasawara, K. Enantioselective construction and utilization of 2-(cyclohex-2-enyl)phenols. Synlett 1998, 1004–1006. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.A.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bodesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Mosman, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Berridge, M.V.; Tan, A.S.; McCoy, K.D.; Wang, R. The biochemical and cellular basis of cellproliferation assays that use tetrazolium salts. Biochemica 1996, 4, 15–20. [Google Scholar]
- Aki, H.; Yamamoto, M. Drug binding to human erythrocytes in the process of ionic drug-induced hemolysis : flow microcalorimetric approaches. Biochem. Pharmacol. 1991, 41, 133–138. [Google Scholar] [CrossRef]
- Malheiros, S.V.P.; Meirelles, N.C.; de Paula, E. Pathways involved in trifluoperazine-, dibucaine- and praziquantel-induced hemolysis. Biophys. Chem. 2000, 83, 89–100. [Google Scholar] [CrossRef]
- Furniss, B.S.; Hannaford, A.J.; Smith, P.W.G.; Tachell, A.R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman Scientific & Technical: New York, 1989; pp. 1025–1026. [Google Scholar]
- Costa-Lotufo, L.V.; Cunha, G.M.A.; Fontenele, J.B.; Junior, H.V.N.F.; Sousa, C.M.; Silveira, E.R.; Nogueira, N.A.P.; Moraes, M.O.; Viana, G.S.B. Cytotoxic activity of chalcones isolated from Lonchocarpus sericeus. Phytother. Res. 2003, 17, 155–159. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available from authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ito, F.M.; Petroni, J.M.; De Lima, D.P.; Beatriz, A.; Marques, M.R.; De Moraes, M.O.; Costa-Lotufo, L.V.; Montenegro, R.C.; Ferreira Magalhãe, H.I.; Do Ó Pessoa, C. Synthesis and Biological Evaluation of Rigid Polycyclic Derivatives of the Diels-Alder Adduct Tricyclo[6.2.1.02,7]undeca-4,9-dien-3,6-dione. Molecules 2007, 12, 271-282. https://doi.org/10.3390/12020271
Ito FM, Petroni JM, De Lima DP, Beatriz A, Marques MR, De Moraes MO, Costa-Lotufo LV, Montenegro RC, Ferreira Magalhãe HI, Do Ó Pessoa C. Synthesis and Biological Evaluation of Rigid Polycyclic Derivatives of the Diels-Alder Adduct Tricyclo[6.2.1.02,7]undeca-4,9-dien-3,6-dione. Molecules. 2007; 12(2):271-282. https://doi.org/10.3390/12020271
Chicago/Turabian StyleIto, Felicia Megumi, Jacqueline Marques Petroni, Dênis Pires De Lima, Adilson Beatriz, Maria Rita Marques, Manoel Odorico De Moraes, Letícia Veras Costa-Lotufo, Raquel Carvalho Montenegro, Hemerson Iury Ferreira Magalhãe, and Cláudia Do Ó Pessoa. 2007. "Synthesis and Biological Evaluation of Rigid Polycyclic Derivatives of the Diels-Alder Adduct Tricyclo[6.2.1.02,7]undeca-4,9-dien-3,6-dione" Molecules 12, no. 2: 271-282. https://doi.org/10.3390/12020271