Novel Aflatoxin Derivatives and Protein Conjugates



1
Institute of Hydrochemistry, Chair for Analytical Chemistry, Technische Universität München, Marchioninistrasse 17, 81377 München, Germany;
2
Federal Institute for Materials Research and Testing (BAM), Division I.5 (Bioanalytics), Richard-Willstätter-Strasse 11, 12489 Berlin, Germany
*
Author to whom correspondence should be addressed.
Molecules 2007, 12(3), 641-653; https://doi.org/10.3390/12030641
Submission received: 14 March 2007
/
Revised: 25 March 2007
/
Accepted: 26 March 2007
/
Published: 27 March 2007
Abstract
:Aflatoxins, a group of structurally related mycotoxins, are well known for their toxic and carcinogenic effects in humans and animals. Aflatoxin derivatives and protein conjugates are needed for diverse analytical applications. This work describes a reliable and fast synthesis of novel aflatoxin derivatives, purification by preparative HPLC and characterisation by ESI-MS and one- and two-dimensional NMR. Novel aflatoxin bovine serum albumin conjugates were prepared and characterised by UV absorption and MALDI-MS. These aflatoxin protein conjugates are potentially interesting as immunogens for the generation of aflatoxin selective antibodies with novel specificities.
Introduction
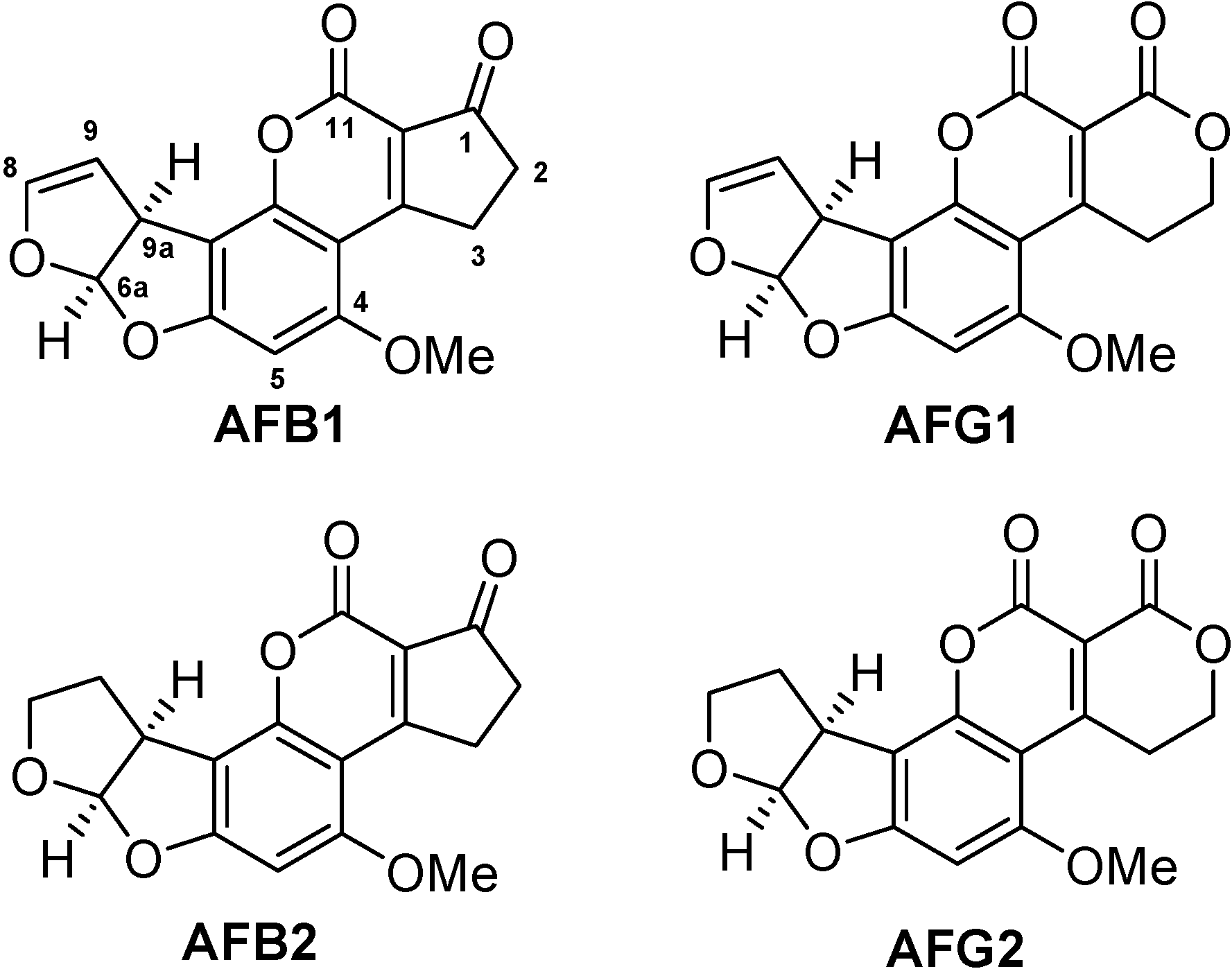
Mycotoxins are secondary metabolic products of low molecular weight generated by diverse moulds. Aflatoxins are a subclass of mycotoxins mainly produced by Aspergillus flavus and Aspergillus parasiticus that can contaminate various kinds of foodstuffs. Due to their high toxicity and carcinogenicity, aflatoxins are of major concern for food producers, the food processing industry, traders and consumers [1]. Legislation in most countries has set strict maximum permissible limits for aflatoxins in the low µg per kg range for many food matrices, e.g. in the United States and the European Union [2,3]. Two types of maximum permissible values exist: one refers to aflatoxin B1, the other to the sum of the four structurally similar aflatoxins B1, B2, G1, and G2 (Figure 1). Aflatoxin M1 and M2, which are contaminants of dairy products, are not considered in this work.
Figure 1.
Structures of the most important aflatoxins B1, B2, G1, G2.

Aflatoxin protein conjugates are required for bioanalytical applications, e.g. as immunogens for the generation of selective polyclonal and monoclonal antibodies against aflatoxins and as protein/enzyme conjugates for enzyme-linked immunosorbent assays (ELISAs). Aflatoxin selective antibodies are needed not only for immunoanalytical screening methods but also for instrumental analysis with immunoaffinity chromatography being a standard cleanup procedure prior to high performance liquid chromatography with fluorescence or mass spectrometric detection [4,5,6]. As aflatoxins, aflatoxin protein conjugates, aflatoxin-selective antibodies, complete ELISA kits for food screening, and immunoaffinity columns for sample cleanup are commercially available by a variety of distributors, the economic importance becomes evident.
A lot of efforts have been made for developing syntheses of useful aflatoxin derivatives and protein conjugates. For the use of protein conjugates as immunogens in general, the hapten design has a strong influence on the characteristics of the resulting antibodies, such as sensitivity (affinity constant) and cross-reactivity pattern concerning structurally similar analytes, here the aflatoxins B1, B2, G1, and G2. In any case, the structure of the toxin should be affected as little as possible by the conjugation procedure. This is necessary for generation of highly affine antibodies. Furthermore, the antibody is likely to recognize the toxin preferentially at the moiety opposite to where it has been attached to the immunogenic protein [7]. Figure 1 shows the relevant aflatoxins B1, B2, G1, and G2 with their pairwise structural differences at opposite moieties of the molecules with aflatoxins 1 and 2 differing in the 8,9-position on one side and aflatoxins B and G differing in the 1-position on the other side.
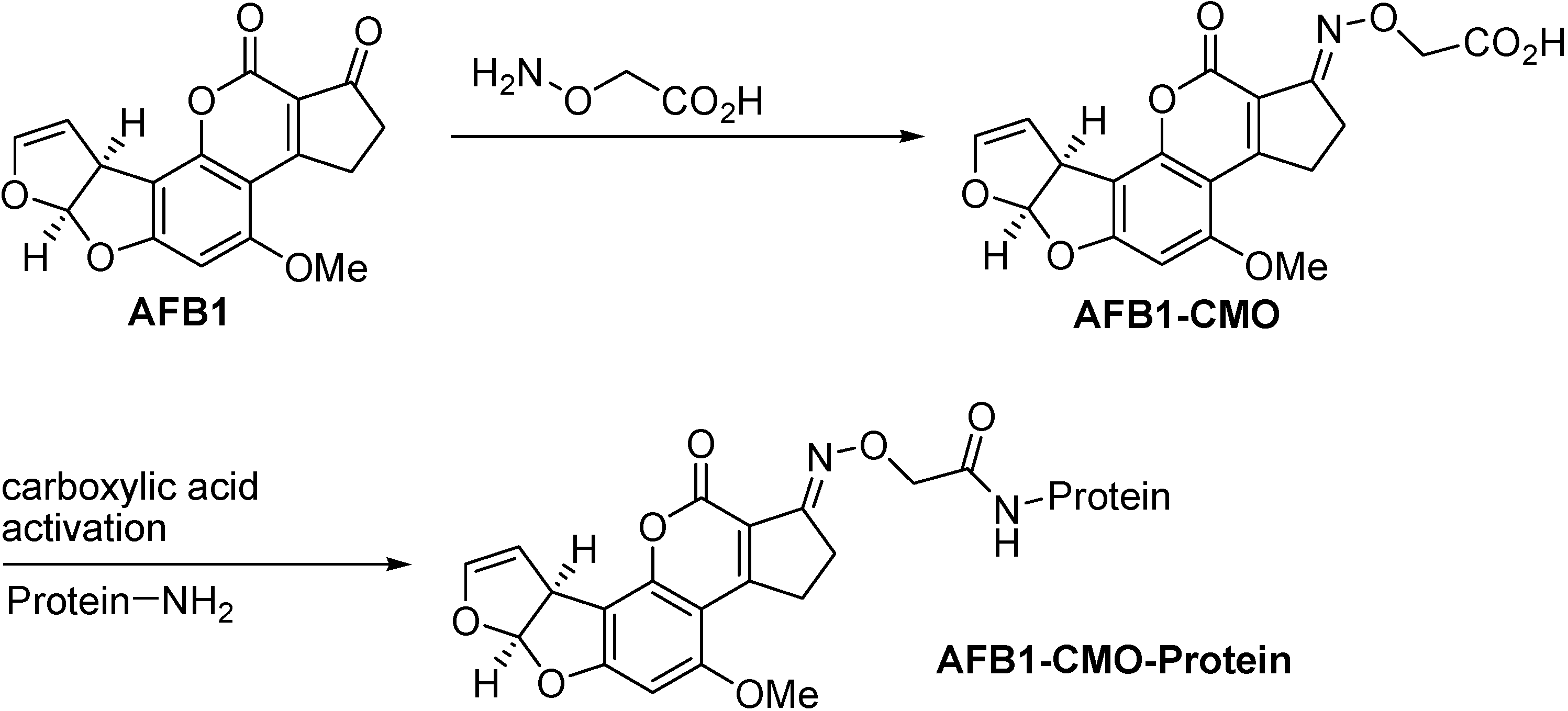
Most established is the conjugation of aflatoxin B1 to a protein in the 1-position by means of a carboxymethyloxime (CMO) spacer (Figure 2) [8,9]. By far most of all aflatoxin selective antibodies produced during the last decade have been generated by immunisations with this (commercially available) conjugate. The resulting monoclonal antibodies all show similar selectivities. The affinity to the aflatoxins usually follows the order AFB1 > AFG1 > AFB2 > AFG2 [10,11,12,13]. For this type of conjugate the cross-reactivity pattern is in accordance with the hapten structure shown in Figure 2. The corresponding hapten synthesis and antibody production has also been accomplished with aflatoxin B2 [14,15].
Figure 2.
Conventional aflatoxin B1 conjugation via its carboxymethyl oxime developed by Chu and Ueno [8,9].

The motivation for developing novel aflatoxin antibodies is to achieve significantly different cross-reactivity patterns. Such antibodies would be potentially useful for detecting multiple toxins on a microarray [16,17] and for developing antibody-based multidimensional analytical methods for the determination of cross-reacting toxins as accomplished elsewhere [18,19,20]. However, this is hardly possible by using only the conventional immunogen AFB1-CMO-protein. Aflatoxin G specific antibodies can hardly be obtained by using the standard coupling method because the corresponding hapten synthesis is not possible with aflatoxin G1/G2. For this purpose, it would also be favorable to have a conjugation procedure, which leaves untouched the moiety where aflatoxins B and G differ. Indeed, efforts to conjugate aflatoxins in the 8,9-position have been reported [21,22,23,24]. Although it was shown that aflatoxin bovine serum albumin (BSA) conjugates prepared by aflatoxin derivatisation in the 8,9-position are useful immunogens that lead to antibodies with interesting specificities [22,23], these methods are hardly used today. This may be due to inconvenient hapten/conjugate synthesis, insufficient hapten/conjugate characterisation, hydrolysis sensitivity of the resulting haptens/conjugates and/or partial destruction of the toxin structure during the conjugation procedure [21,22,23,24].
In this work we present a novel, simple and reliable synthesis for activating aflatoxins in the 8-position, a simple purification of the haptens by preparative HPLC, conjugation to BSA, and characterisation of the protein conjugates by MALDI-MS. The syntheses can easily be carried out with 10 mg or less of aflatoxin B1/G1.
Results and Discussion
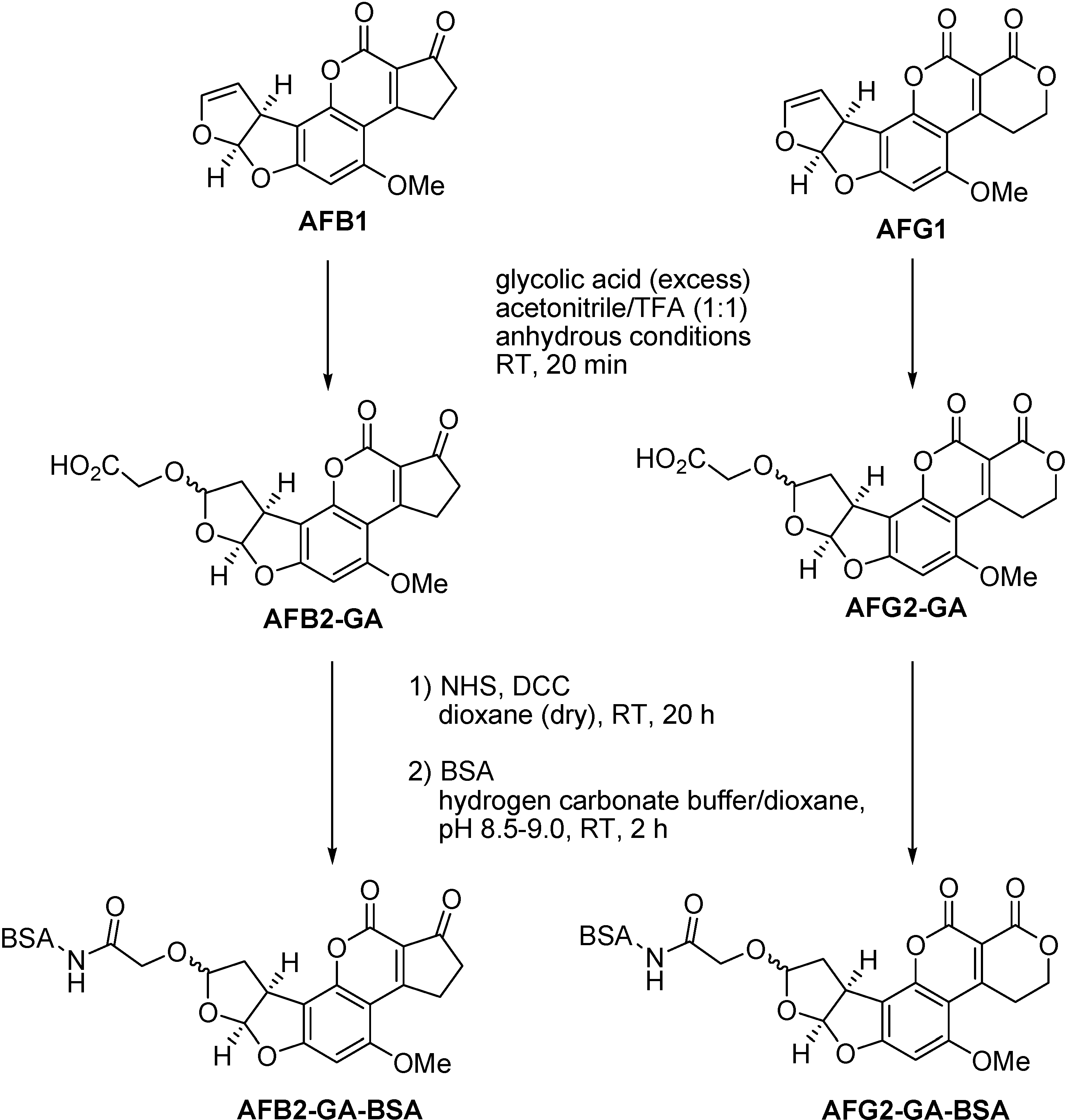
A simple, fast and reliable procedure for coupling aflatoxin B1 and G1 in the 8-position by reaction of the aflatoxin’s enol ether group with glycolic acid is presented. A related reaction has been used for aflatoxin derivatisation in analytical food chemistry. The detection limits of aflatoxins B1 and G1 could be lowered for HPLC with fluorescence detection by conversion to their respective hemiacetals AFB2a and AFG2a during the sample preparation by an enol ether hydrolysis with concentrated trifluoroacetic acid (TFA) [25,26,27]. In this work, we modified the procedure by working preparatively under dry conditions and using glycolic acid as a spacer providing a nucleophilic hydroxyl group for formation of the aflatoxin acetal in the 8-position on one side and a carboxylic acid group for subsequent coupling to proteins on the other side (Figure 3).
Figure 3.
Conversion of AFB1 and AFG1 to AFB2-GA and AFG2-GA and subsequent coupling to BSA via N-hydroxysuccinimide-ester (NHS-ester).
Figure 3.
Conversion of AFB1 and AFG1 to AFB2-GA and AFG2-GA and subsequent coupling to BSA via N-hydroxysuccinimide-ester (NHS-ester).

Figure 4A shows a LC-MS run of an aliquot of the reaction mixture with AFB1 as starting material. The chromatogram (absorption at 355 nm) shows two diastereomeric AFB2-GA products ([M+H]+=389, diastereomeric ratio ~4:1) and only a little AFB2a (~3%, [M+H]+=331) as a reaction by-product of AFB1 with water (although working under anhydrous conditions, traces of water could not be completely excluded). No additional peaks were found with UV detection at lower wavelengths. A similar chromatogram and mass spectrum (not shown) were obtained with AFG2-GA as product with the retention times 16.40 min (major diastereomer) and 17.37 min (minor diastereomer) and [M+H]+=405, [M+H]+=406 (isotopologue) and fragment ion [AFG1+H]+=329 (only a little AFG2a, ~3%, was detected as reaction by-product of AFG1 with water).
Figure 4.
A) HPLC run of the reaction mixture with AFB1 as starting material showing absorption at 355 nm. a: AFB2a, b: AFB2-GA (major diastereomer), c: AFB2-GA (minor diastereomer), u: not identified. B) ESI-TOF mass spectrum of AFB2-GA (major diastereomer) showing [M+H]+=389, [M+H]+=390 (isotopologue) and [AFB1+H]+=313 (fragment ion). The mass spectrum of AFB2-GA (minor diastereomer) gives the same m/z and peak ratios.
Figure 4.
A) HPLC run of the reaction mixture with AFB1 as starting material showing absorption at 355 nm. a: AFB2a, b: AFB2-GA (major diastereomer), c: AFB2-GA (minor diastereomer), u: not identified. B) ESI-TOF mass spectrum of AFB2-GA (major diastereomer) showing [M+H]+=389, [M+H]+=390 (isotopologue) and [AFB1+H]+=313 (fragment ion). The mass spectrum of AFB2-GA (minor diastereomer) gives the same m/z and peak ratios.

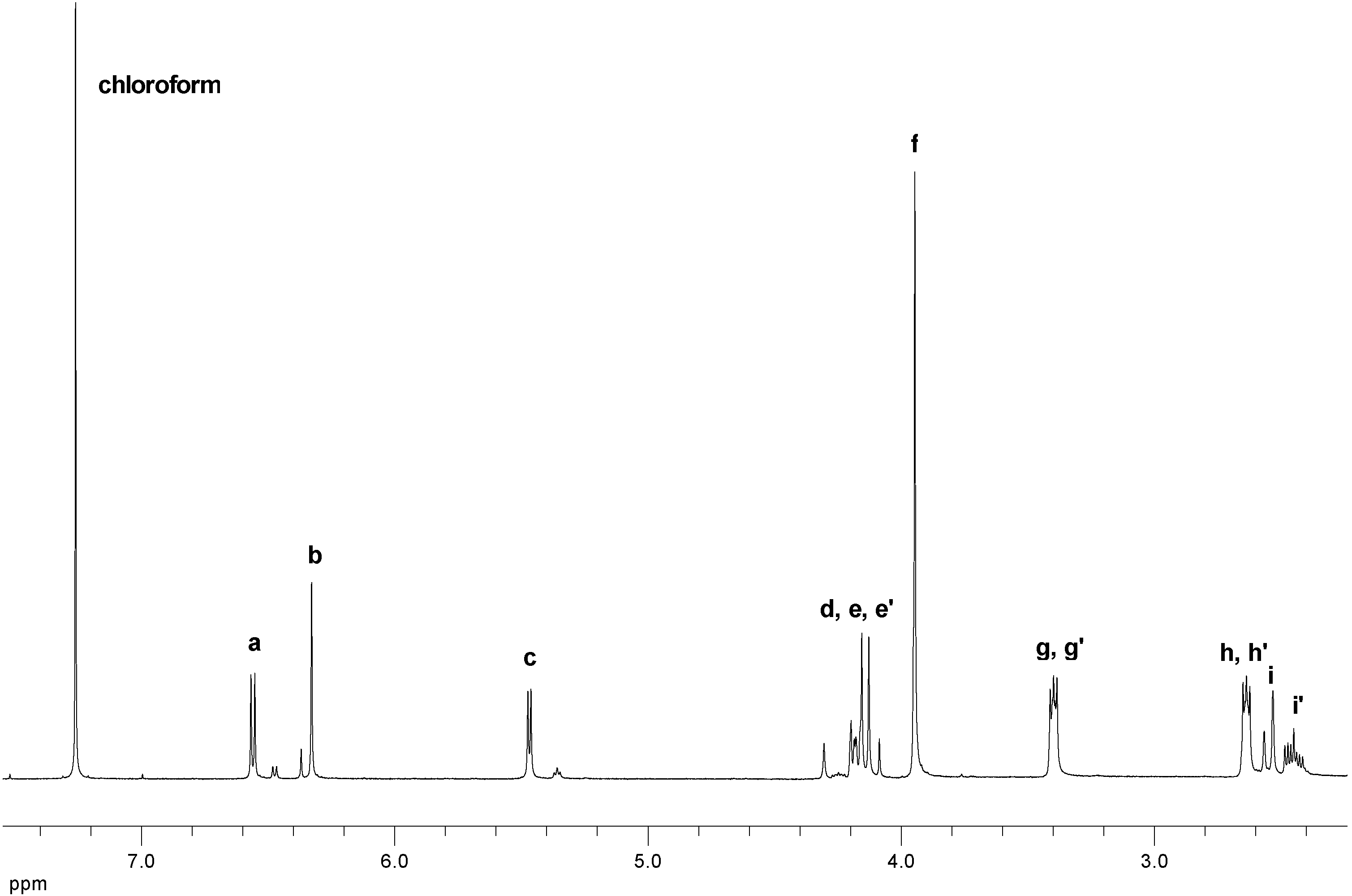
Figure 5 shows the 1H-NMR spectrum of AFB2-GA (two diastereomers, ratio ~4:1) initially purified by preparative HPLC. Only the spectrum of the major diastereomer of AFB2-GA is discussed in detail including two-dimensional NMR spectra (see Supporting Information). Table 1 contains the most important 1H- and 13C-NMR data of AFB2-GA and AFG2-GA. The 1H- spectrum of AFB2-GA (Figure 5 and Table 1) shows some particularities: proton c couples with i’ but not with i, thus resulting in a doublet; i couples with i’ (geminal) but not with c and d (vicinal), whereas i’ couples with i, c and d. This lack of vicinal couplings is caused by torsion angles ~90° and were likewise observed for aflatoxin B2a (the hemiacetal of AFB1) [28]. Protons e and e’ have slightly different shifts and their signals overlap with proton d (clearly visible in the HSQC spectrum, see Supporting Information) thus resulting in a multiplet.
Figure 5.
1H-NMR spectrum of AFB2-GA in CDCl3.


| 1H-NMR signals* | 1H-NMR signals* |
| a: ∂ = 6.56 (d, 1H, J = 6.1 Hz) | a: ∂ = 6.59 (d, 1H, J = 6.1 Hz) |
| b: ∂ = 6.33 (s, 1H) | b: ∂ = 6.32 (s, 1H) |
| c: ∂ = 5.47 (d, 1H, J = 4.8 Hz) | c: ∂ = 5.47 (d, 1H, J = 4.8 Hz) |
| d, e, e’: ∂ = 4.05 - 4.22 (m, 3H) | d, e, e’: ∂ = 4.06 - 4.22 (m, 3H) |
| f: ∂ = 3.95 (s, 3H) | f: ∂ = 3.92 (s, 3H) |
| g, g’: ∂ = 3.40 (m, 2H) | g, g’: ∂ = 3.45 (m, 2H) |
| h, h’: ∂ = 2.64 (m, 2H) | h, h’: ∂ = 4.41 (m, 2H) |
| i: ∂ = 2.55 (d, 1H, J = 13.8 Hz) | i: ∂ = 2.54 (d, 1H, J = 13.6 Hz) |
| i’: ∂ = 2.45 (ddd, 1H, J = 4.9 Hz, J = 9.4 Hz, J = 14.0 Hz) | i’: ∂ = 2.44 (ddd, 1H, J = 4.8 Hz, J = 9.3 Hz, J = 13.9 Hz) |
| 13C NMR signals*, ** | 13C NMR signals*, ** |
| 1: ∂ = 202, 2: ∂ = 177, 3: ∂ = 171, 4: ∂ = 166, 5: ∂ = 162, 6: ∂ = 155, 7: ∂ = 153, 8: ∂ = 117, 9: ∂ = 114, 10: ∂ = 109, 11: ∂ = 106, 12: ∂ = 104, 13: ∂ = 91, 14: ∂ = 64, 15: ∂ = 56, 16: ∂ = 43, 17: ∂ = 38, 18: ∂ = 35, 19: ∂ = 29. | 1: ∂ = 161, 2: ∂ = 162, 3: ∂ = 173, 4: ∂ = 165, 5: ∂ = 162, 6: ∂ = 156, 7: ∂ = 152, 8: ∂ = 107, 9: ∂ = 114, 10: ∂ = 109, 11: ∂ = 106, 12: ∂ = 103, 13: ∂ = 91, 14: ∂ = 64, 15: ∂ = 56, 16: ∂ = 43, 17: ∂ = 38, 18: ∂ = 65, 19: ∂ = 29. |


{kind=link}
{kind=link}
{kind=link}
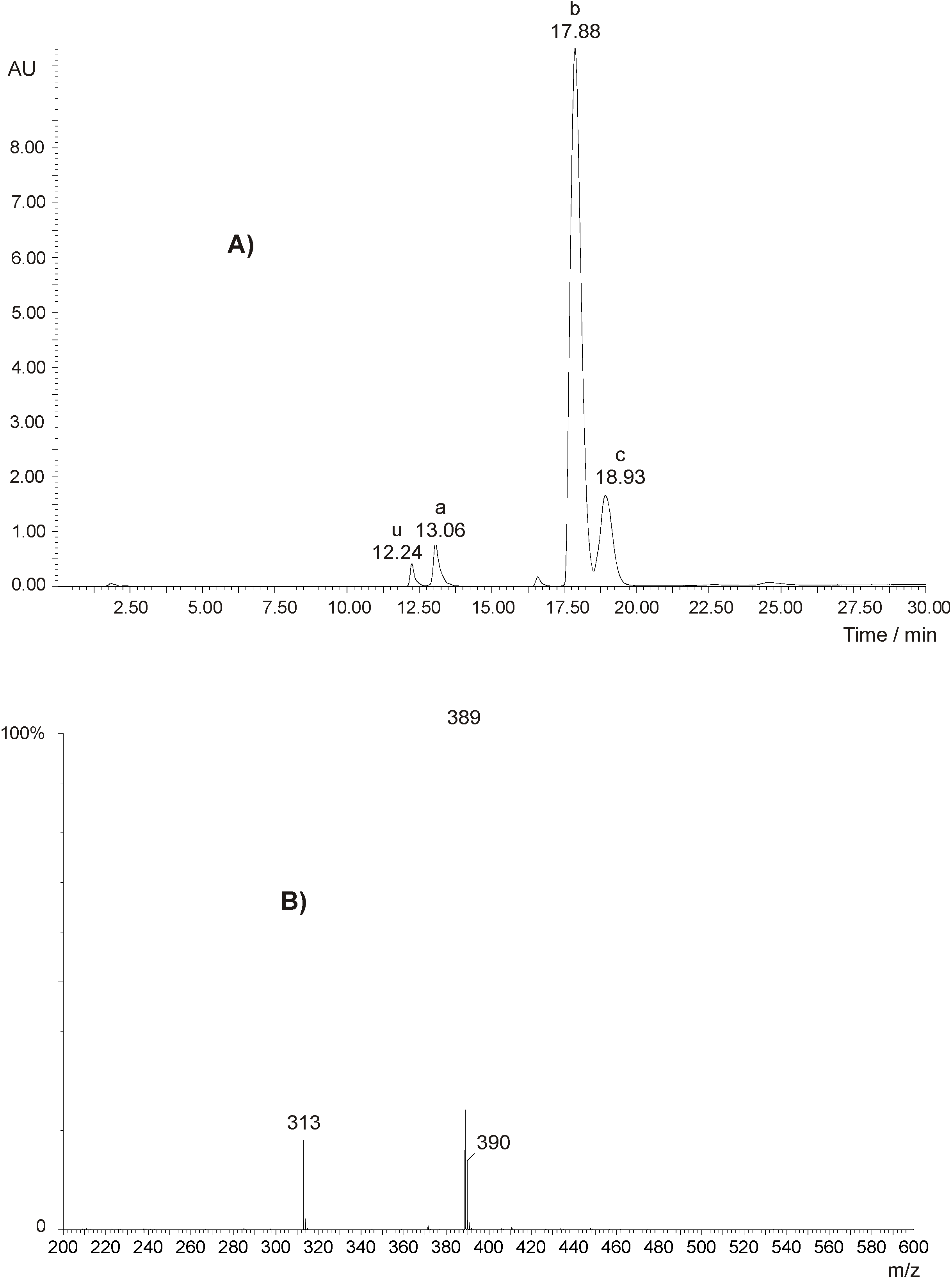{kind=link}
{kind=link}
{kind=link}
* NMR shifts are given in ppm relative to CHCl3 (1H: 7.26 ppm, 13C: 77 ppm).** Carbons 1, 2, 3, 8, 9, 11, 13, 14, 15, 16, 17, 18, and 19 could clearly be identified by means of the chemical shifts of the 13C- spectrum and the DEPT, HSQC and HMBC spectra. Carbons 4, 5, 6, 7, 10, and 12 could be assigned by means of the HMBC spectrum only.
A comparison of the chemical shifts and couplings constants of the proton signals of the two diastereomers of AFB2a (including Karplus analysis) [28,29] and AFB2-GA strongly suggests that the main isomer has an 8S configuration due to attack of glycolic acid on the aflatoxin’s protonated enol ether group from the sterically less hindered side, as observed elsewhere [29,30] (note: the α/β nomenclature in [28] and [29] is not consistent). The NMR spectra of AFG2-GA are similar (not shown, see Supporting Information). Comparison of the 1H- and 13C-NMR spectra of AFB2-GA and AFG2-GA show significantly differing shifts where expected, particularly concerning protons h, h’ and carbons 1, 2, 8 and 18 (Table 1).
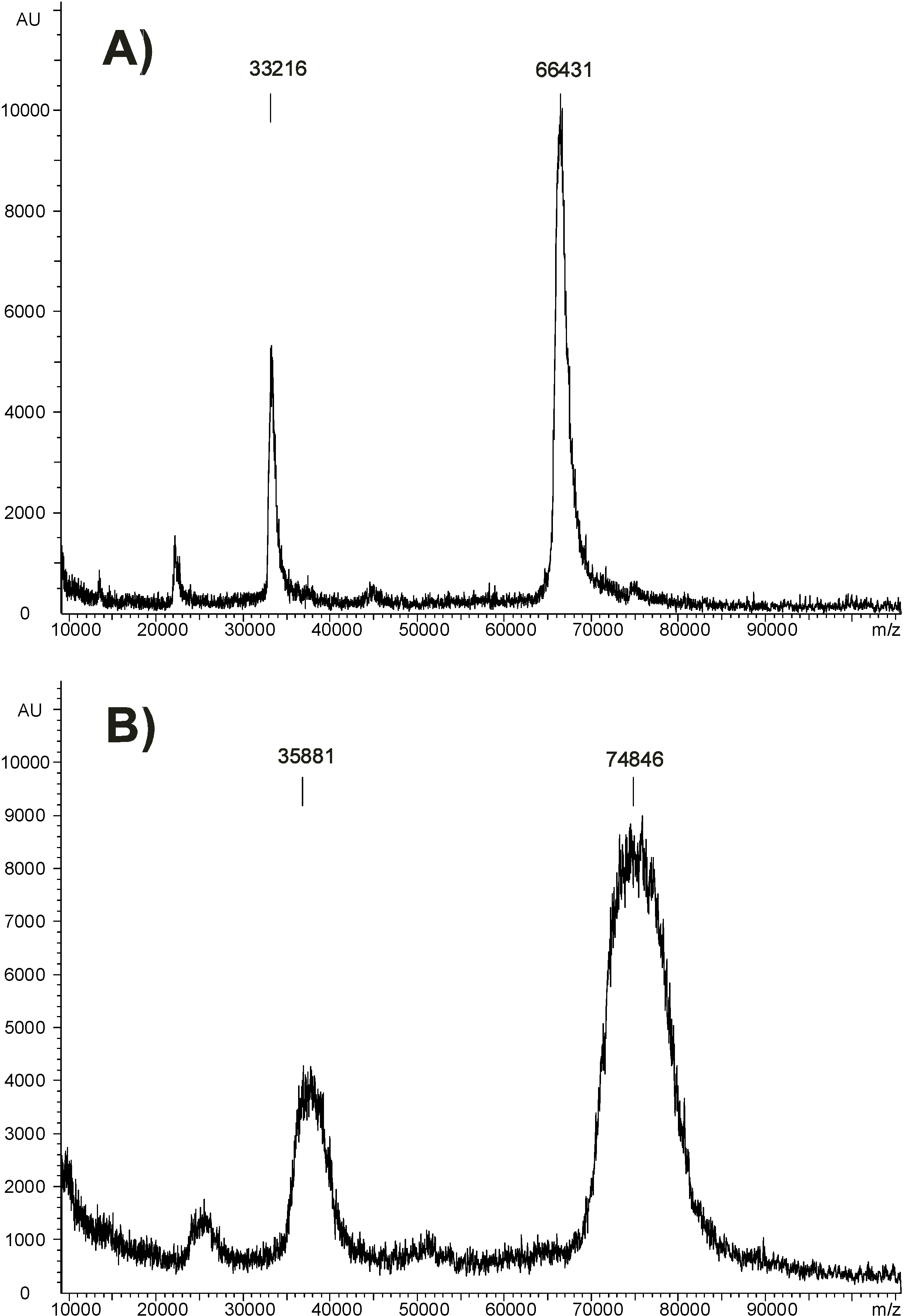
The novel haptens AFB2-GA and AFG2-GA were coupled to BSA by the NHS-ester method (Figure 3). For the formation of the NHS-ester we used a slight excess of hapten over N,N’-dicyclohexylcarbodiimide (DCC) to prevent direct reaction of BSA with DCC. For determination of the mean coupling densities of the BSA conjugates measurement of the UV absorption was compared with MALDI-MS. The UV measurement was carried out as decribed (Experimental Section) by calculating the hapten concentration in the sample using Beer-Lambert’s law. Validated extinction coefficients for AFB2 and AFG2 in methanol were taken as molar absorptivities [31]. The mean coupling densities were then calculated as the ratio of hapten concentration to protein concentration, assuming that no losses of protein occurred during synthesis and purification of the conjugates. For AFB2-GA-BSA a mean coupling density of 13-14 was determined by the UV method, whereas the MALDI method gave 22-23. For AFG2-GA-BSA 11.5-12.5 was determined by UV, whereas MALDI gave 17.5-18.5. Thus, it must be concluded that UV measurements using the molar absorptivities of AFB2 and AFG2 underestimates the real mean coupling densities, as the molar absorptivities of AFB2-GA and AFG2-GA may be significantly higher than those of AFB2 and AFG2. It is therefore suggested to determine the mean coupling densities of these conjugates by MALDI-MS. Figure 6 shows the MALDI spectra obtained from a BSA reference sample and AFB2-GA-BSA. A significant shift of the peak maximum of about ∆m/z=8400 and relative peak broadening due to a distribution of coupling density is visible, indicating that conjugates with hapten densities ranging from about 15 to 35 are present. The MALDI spectrum of AFG2-GA-BSA looks similar with a shift of the peak maximum of approximately ∆m/z=7000 and significant peak broadening due to coupling densities ranging from about 12 to 35 (see Supporting Information).
Figure 6.
MALDI spectra of A) BSA and B) AFB2-GA-BSA showing [M+H]+ and [M+2H]2+ peaks.

We would like to recommend this simple and clean method of aflatoxin conjugation with regard to potential applications for generation of aflatoxin specific antibodies and development of bioanalytical methods for determination of aflatoxins. Generation of monoclonal antibodies with the immunogens decribed here is presently under way in our laboratories.
Experimental Section
Safety note
Aflatoxins, especially aflatoxins B1 and G1, are supposed to be extremely carcinogenic. The procedure described here minimises health risks. Aflatoxin contaminated material should be decontaminated with an aqueous solution of sodium hypochlorite (5%).
General
Aflatoxins B1 and G1 were purchased from Sigma (St. Louis, MO, USA) in amounts of 10 mg per vial. BSA (Fraction V, ~99%, art. no. A3059) was purchased from Sigma. For the hapten syntheses dry acetonitrile (max. 10 ppm H2O) from Merck (Darmstadt, Germany) was used. Glycolic acid (H2O ~1%) was purchased from Fluka (Buchs, Switzerland) and dried in a desiccator at ~10−2 mbar over phosphorous pentoxide. Trifluoroacetic acid (TFA) (H2O ≤0.05%), N-hydroxysuccinimide (NHS), N,N’-dicyclohexylcarbodiimide (DCC), and dioxane (H2O ≤0.01%), were purchased from Fluka. For preparative HPLC, gradient grade acetonitrile, deionized water and high purity acetic acid from Merck was used. LC-MS grade water and methanol was purchased from Riedel-de Haën (Seelze, Germany). Sephadex G-25 columns were purchased from GE Healthcare Bio-Sciences AB (Uppsala, Sweden). The NHS-ester formation was carried out in 1 mL vials (Max Recovery) from Waters with screw caps and non-preslit PTFE/silicone septa. A steel cannula can be pierced through the septum for connection to the vacuum/argon line for working under water and oxygen free conditions.
For solid phase extraction, a piston pump (A41503) from Knauer (Berlin, Germany) and a C18 cartridge (10×10 mm) from Phenomenex (Torrance, CA, USA) was used. The preparative HPLC system consisted of a gradient pump (L-6200A) from Merck (Darmstadt, Germany), a six way manual injection valve (7725) from Rheodyne (Rohnert Park, CA, USA), a C18 column (Gemini, 5 µm, 150×10 mm) from Phenomenex, a UV detector (L-4250) from Merck/Hitachi and a fraction collector (280.6008.76) from Polymer Laboratories (Shropshire, UK).
Synthesis of AFB2-GA and AFG2-GA and purification by preparative HPLC
In a heated and argon flushed 25 mL Schlenk flask with septum and stirring bar, dry glycolic acid (1.0 g) was dissolved in dry TFA (4 mL). Aflatoxin B1 or G1 (10 mg) was dissolved in dry acetonitrile (4 mL) directly in the septum covered vial obtained from the distributor. This solution was added to the glycolic acid/TFA mixture with a syringe at room temperature while stirring. The reaction mixture turned yellow-green within seconds. After 20 min of stirring, the solution was added to water (75 mL) and pumped over a semi-preparative C18 cartridge with subsequent flushing with 1% acetic acid in water (10 mL). The C18 cartridge was screwed into the six way injection valve of the HPLC system described above. Gradient elution was applied using acetonitrile and water, both containing 0.1% of acetic acid, at a flow rate of 4.7 mL/min and the product fraction collected. The linear gradient started at 20% of acetonitrile for 1 min, then going to 50% of acetonitrile within 25 min. UV absorption was monitored at 360 nm. The product was collected as a mixture of diastereomers from approximately 10 to 13 min. The solvent of the product fraction was removed in vacuo and the pale yellow, cristalline product dried overnight under high vacuum. About 10-11 mg of purified product were obtained (an accurate relative yield of isolated product is not given due to the small amounts of material and the fact that distributors usually “overfill” their toxin vials). Characterisation by LC-MS and NMR has been discussed before.
LC-MS analysis of the reaction mixture
The reaction mixture of AFB2-GA or AFG2-GA (10 µL) was added to water (100 µL). For LC-MS analysis this mixture (30 µL) was injected into the system consisting of a separations module (2695) from Waters (Milford, MA, USA), a C18 column (Gemini, 5 µm, 100×4.6 mm) from Phenomenex (Torrance, CA, USA), a post-column split, a UV photodiode array detector (996) from Waters and an ESI-TOF mass spectrometer (LCT, Waters/Micromass) equipped with a Z-spray source. Methanol and water, both containing 0.1% of acetic acid, were used as mobile phase. The linear gradient started with 10% of methanol for 1 min, going to 90% of methanol within 20 min at a total flow rate of 0.8 mL/min. By means of the post-column split 160 µL/min was pumped into the ESI source, the rest into the UV detector. The cone voltage of the ESI source was 3000 V, the source temperature 90 °C, the desolvation gas temperature 130 °C at a nitrogen flow rate of 800 L/h.
Preparation of AFB2-GA-BSA and AFG2-GA-BSA via NHS-ester
In a heated and argon flushed 1 mL vial with a PTFE coated septum screw cap a 0.29 m solution of NHS in dry dioxane (20 µL, 5.87·10−6 mol, 1.14 eq) was added to a 12.9 mm solution of AFB2-GA in dry dioxane (400 µL, 5.15·10−6 mol, 1.00 eq) or to a 12.9 mm solution of AFG2-GA in dry dioxane (416 µL, 5.15·10−6 mol, 1.00 eq). A 0.19 m solution of DCC in dry dioxane (25 µL, 4.75·10−6 mol, 0.92 eq) was added and the reaction mixture was shaken for 20 h at room temperature (the dicyclohexylurea by-product starts crystallising out after approximately 12 h). This solution (100 µL) was then added in one portion to an ice cold 0.13 mm solution of BSA in buffer (0.71 m solution of NaHCO3 in water brought to pH 7.5 by addition of semi-concentrated hydrochloric acid, 227 µL, 2.95·10−8 mol, 5.73·10−3 eq; addition of dioxane raises the pH to 8.5-9.0). The mixture was shaken for 2 h while warming up to room temperature. The conjugation can be followed qualitatively by bare eye, as the conjugates show a strong yellow-green fluorescence. The conjugates were purified on a Sephadex G-25 column and PBS (phosphate buffered saline, pH 7.4) as mobile phase. Characterisation by UV absorption and MALDI-MS has been discussed before.
Measurement of the mean coupling density by UV absorption
An aliquot of a solution of 2.0 mg of purified AFB2-GA-BSA or AFG2-GA-BSA in 1.5 mL PBS (100 µL) was made up to 1 mL with PBS in a UV transparent cuvette. Absorption spectra were recorded from 200 to 600 nm with a Beckmann DU 650 UV spectrometer (Fullerton, CA, USA) with PBS as blank. The absorption at the maximum around 360 nm was taken for further calculation of the conjugates’ mean coupling densities by Beer-Lambert law.
Measurement of the mean coupling density by MALDI-MS
For MALDI measurements, a solution of 2.0 mg of AFB2-GA-BSA or AFG2-GA-BSA in 1.5 mL PBS (100 µL) was desalted with a Sephadex G-25 column and deionized water as solvent. This solution (100 µL) was mixed with saturated sinapic acid solution in acetonitrile/0.1% TFA (100 µL, 2:1, v/v). This mixture (0.5 µL) was applied onto the stainless steel target. Sample drying was carried out at room temperature. Spectra were recorded after evaporation of the solvent. Molecular mass determination was performed on a Bruker REFLEX III time-of-flight spectrometer (Bruker-Daltonics, Bremen, Germany). The mass spectrometer was equipped with a pulsed UV-laser (N2-laser, 337 nm) and a 2-GHz digitiser. All measurements were carried out in positive ionisation mode. Mass spectra were acquired as sums of ion signals generated by the sample irradiation with 60 laser pulses. The singly and doubly charged ion signals from BSA were used for external mass calibration of all mass spectra.
NMR spectroscopy
NMR spectra were recorded on a Varian Inova 400 and on a Varian NMR system 400 (Palo Alto, CA, USA) at 400 MHz (1H) and 100 MHz (13C). CDCl3 was used as solvent.
Supplementary Material
Supplementary material file: http://www.mdpi.org/molecules/papers/12030641sm.pdf.
Supplementary File 1Acknowledgments
We thank Dr. Markus Wunderlin and Markus Fischer from the Universität Ulm for recording MALDI spectra and Claudia Dubler and Dr. David Stephenson from the Ludwig-Maximilians-Universität München for providing NMR spectra.
References
- Eaton, D.L.; Groopman, J.D. The Toxicology of Aflatoxins: Human Health, Veterinary, and Agricultural Significance; Academic Press: San Diego, CA, 1994. [Google Scholar]
- Food and Agriculture Organisation. Worldwide Regulations for Mycotoxins in Food and Feed 2003. FAO Food and Nutr. Paper 2004, 81. [Google Scholar]
- van Egmond, H.P. Natural Toxins: Risks, Regulations and the Analytical Situation in Europe. Anal. Bioanal. Chem. 2004, 378, 1152–1160. [Google Scholar]
- Krska, R.; Welzig, E.; Berthiller, A.; Molinelli, A.; Mizaikoff, B. Advances in the Analysis of Mycotoxins and its Quality Assurance. Food Add. Contam. 2005, 22, 345–353. [Google Scholar] [CrossRef]
- Castegnaro, M.; Tozlovanu, M.; Wild, C.; Molinié, A.; Sylla, A.; Pfohl-Leszkowicz, A. Advantages and Drawbacks of Immunoaffinitiy Columns in Analysis of Mycotoxins in Food. Mol. Nutr. Food Res. 2006, 50, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.M.; Trucksess, M.W. Application of Immunoaffinity Columns to Mycotoxin Analysis. J. Assoc. Off. Anal. Chem. Int. 1997, 80, 941–949. [Google Scholar]
- Landsteiner, K. Serologische Reaktionen mit künstlichen Komplexantigenen und einfachen chemischen Substanzen; Springer: Berlin, 1933. [Google Scholar]
- Chu, F.S.; Ueno, I. Production of Antibody against Aflatoxin B1. Appl. Environ. Microbiol. 1977, 33, 1125–1128. [Google Scholar] [PubMed]
- Chu, F.S.; Hsia, M.T.S.; Sun, P.S. Preparation and Characterization of Aflatoxin B1-1-(O-carboxymethyl)-oxime. J. Assoc. Off. Anal. Chem. Int. 1977, 60, 791–794. [Google Scholar]
- Devi, K.T.; Mayo, M.A.; Reddy, K.L.N.; Delfosse, P.; Reddy, G.; Reddy, S.V.; Reddy, D.V.R. Production and Characterization of Monoclonal Antibodies for Aflatoxin B1. Lett. Appl. Microbiol. 1999, 29, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Kolosova, A.Y.; Shim, W.-B.; Yang, Z.-Y.; Eremin, S.A.; Chung, D.-H. Direct Competitive ELISA Based on Monoclonal Antibody for Detection of Aflatoxin B1. Stabilization of ELISA Kit Components and Application to Grain Samples. Anal. Bioanal. Chem. 2006, 384, 286–294. [Google Scholar]
- Lee, N.A.; Wang, S.; Allan, R.D.; Kennedy, I.R. A Rapid Aflatoxin B1 ELISA: Development and Validation with Reduced Matrix Effects for Peanuts, Corn, Pistaccio, and Soybeans. J. Agric. Food Chem. 2004, 52, 2746–2755. [Google Scholar]
- Lipigorngoson, S.; Limtrakul, P.; Suttajit, M.; Yoshizawa, T. In-house Direct cELISA for Determining Aflatoxin B1 in Thai Corn and Peanuts. Food Add. Contam. 2003, 20, 838–845. [Google Scholar] [CrossRef]
- Hastings, K.L.; Hagler, W.M.; Harris, T.M.; Voyksner, R.D.; Dean, J.H. Production and Characterization of Aflatoxin B2 Oximinoacetate. J. Agric. Food Chem. 1989, 37, 393–400. [Google Scholar] [CrossRef]
- Hastings, K.L.; Tulis, J.J.; Dean, J.H. Production and Characterization of a Monoclonal Antibody to Aflatoxin B2. J. Agric. Food Chem. 1988, 36, 404–408. [Google Scholar]
- Fall, B.I.; Eberlein-König, B.; Behrendt, H.; Niessner, R.; Ring, J.; Weller, M.G. Microarrays for the Screening of Allergen-specific IgE in Human Serum. Anal. Chem. 2003, 75, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Knecht, B.G.; Strasser, A.; Dietrich, R.; Märtlbauer, E.; Niessner, R.; Weller, M.G. Automated Microarray System for the Simultaneous Detection of Antibiotics in Milk. Anal. Chem. 2004, 76, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Winklmair, M.; Schuetz, A.J.; Weller, M.G.; Niessner, R. Immunochemical Array for the Identification of Cross-reacting Analytes. Fresenius J. Anal. Chem. 1999, 363, 731–737. [Google Scholar] [CrossRef]
- Wortberg, M.; Jones, G.; Kreissig, S.B.; Rocke, D.M.; Gee, S.J.; Hammock, B.D. An Approach to the Construction of an Immunoarray for Differentiating and Quantitating Cross Reacting Analytes. Anal. Chim. Acta 1996, 319, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Wortberg, M.; Hammock, B.D.; Rocke, D.M. A Procedure for the Immunoanalysis of Samples Containing One or More Members of a Group of Cross-reacting Analytes. Anal. Chim. Acta 1996, 336, 175–183. [Google Scholar] [CrossRef]
- Swenson, D.H.; Miller, J.A.; Miller, E.C. The Reactivity and Carcinogenicity of Aflatoxin B1-2,3-Dichloride, a Model for the Putative 2,3-Oxide Metabolite of Aflatoxin B1. Cancer Res. 1975, 35, 3811–3823. [Google Scholar] [PubMed]
- Lau, H.P.; Gaur, P.K.; Chu, F.S. Preparation and Characterization of Aflatoxin B2a-Hemiglutarate and its Use for the Production of Antibody against Aflatoxin B1. J. Food Safety 1981, 3, 1–13. [Google Scholar] [CrossRef]
- Chu, F.S.; Steinert, B.W.; Gaur, P.K. Production and Characterization of Antibody against Aflatoxin G1. J. Food Safety 1985, 7, 161–170. [Google Scholar] [CrossRef]
- Kononenko, G.P.; Burkin, A.A.; Soboleva, N.A. Comparative Characterization of Immune Reagents Based on Hemiacetals of Aflatoxin B1 and Sterigmatocystine. Appl. Biochem. Microbiol. 2002, 38, 487–492. [Google Scholar] [CrossRef]
- Kok, W.T. Derivatization Reactions for the Determination of Aflatoxins by Liquid Chromatography with Fluorescence Detection. J. Chromatogr. B 1994, 659, 127–137. [Google Scholar] [CrossRef]
- Takahashi, D.M. Reversed-phase High-performance Liquid Chromatography Analytical System for Aflatoxins in Wines with Fluorescence Detection. J. Chromatogr. 1977, 131, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Official Methods of Analysis of the Association of Official Analytical Chemists, 15th Edition ed; AOAC: Arlington, 1990; Ch. 26.
- Trost, B.M.; Toste, F.D. Palladium Catalyzed Kinetic and Dynamic Kinetic Asymmetric Transformations of γ-Acyloxybutenolides. Enantioselective Total Synthesis of (+)-Aflatoxin B1 and B2a. J. Am. Chem. Soc. 2003, 125, 3090–3100. [Google Scholar]
- Ashley, D.L.; Orti, D.L.; Hill, R.H. Proton Nuclear Magnetic Resonance Evidence for Two Configurations of the Hemiacetals of Aflatoxin B1 and Sterigmatocystin. J. Agric. Food Chem. 1987, 35, 782–785. [Google Scholar] [CrossRef]
- Baertschi, S.W.; Raney, K.D.; Stone, M.P.; Harris, T.M. Preparation of the 8,9-Epoxide of the Mycotoxin Aflatoxin B1: The Ultimate Carcinogenic Species. J. Am. Chem. Soc. 1988, 110, 7929–7931. [Google Scholar] [CrossRef]
- Nesheim, S.; Trucksess, M.W.; Page, S.W. Molar Absorptivities of Aflatoxins B1, B2, G1 and G2 in Acetonitrile, Methanol, and Toluene-Acetonitrile (9+1) (Modification of AOAC Official Method 971.22): Collaborative Study. J. Assoc. Off. Anal. Chem. Int. 1999, 82, 251–258. [Google Scholar]
- Sample Availability: Contact the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Cervino, C.; Knopp, D.; Weller, M.G.; Niessner, R. Novel Aflatoxin Derivatives and Protein Conjugates. Molecules 2007, 12, 641-653. https://doi.org/10.3390/12030641
AMA Style
Cervino C, Knopp D, Weller MG, Niessner R. Novel Aflatoxin Derivatives and Protein Conjugates. Molecules. 2007; 12(3):641-653. https://doi.org/10.3390/12030641
Chicago/Turabian StyleCervino, Christian, Dietmar Knopp, Michael G. Weller, and Reinhard Niessner. 2007. "Novel Aflatoxin Derivatives and Protein Conjugates" Molecules 12, no. 3: 641-653. https://doi.org/10.3390/12030641