Synthesis of (R)-Dihydropyridones as Key Intermediates for an Efficient Access to Piperidine Alkaloids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction

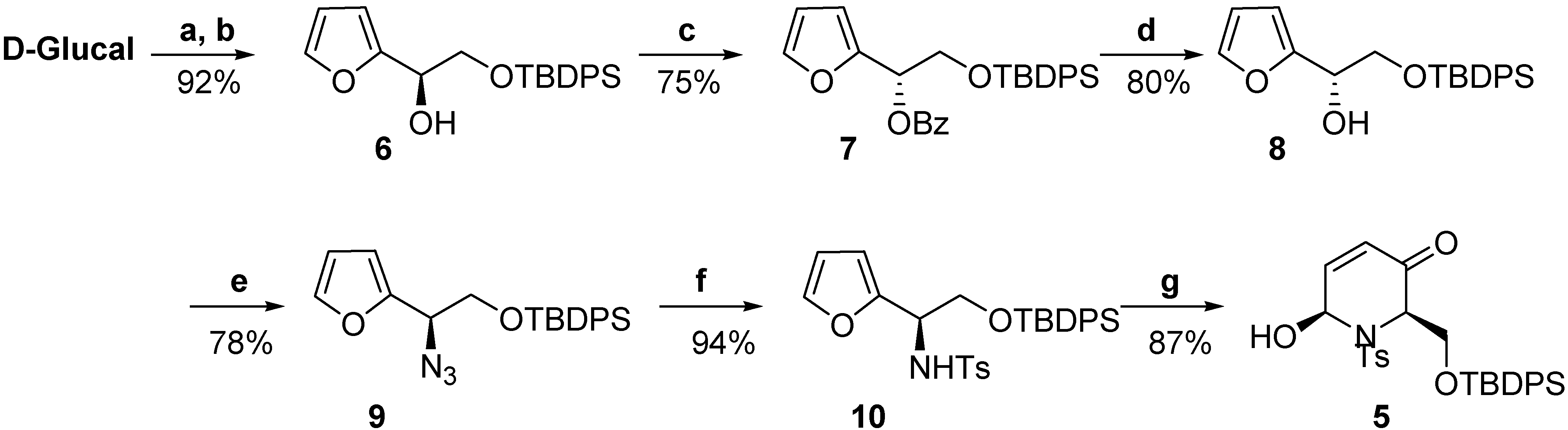
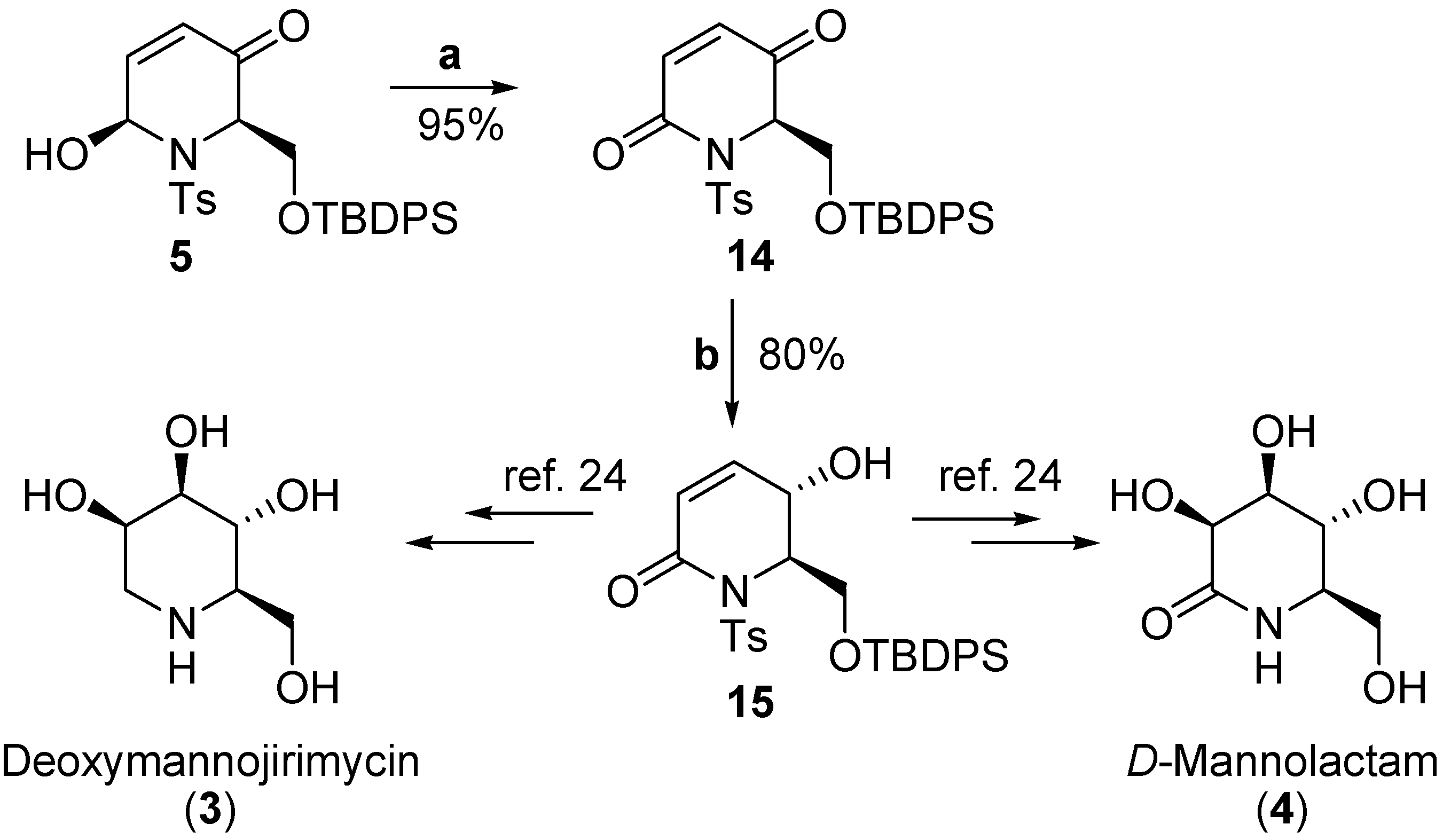
Results and Discussion
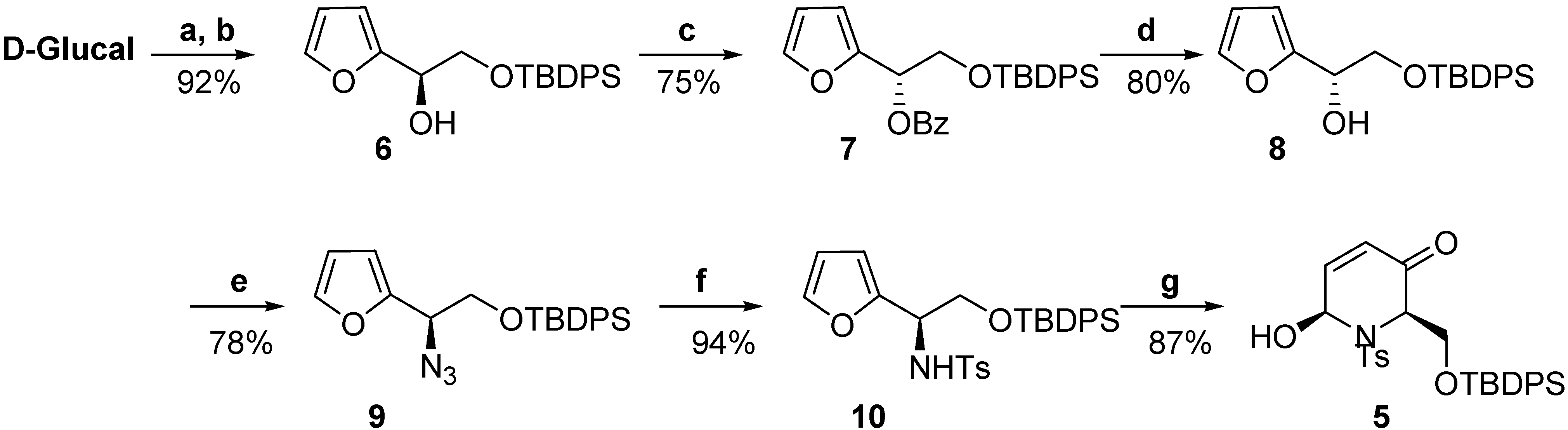
 –25.5) constitutes additional proof of the assigned configuration, since its enantiomer displays the opposite sign ( +27.9) [20b].
–25.5) constitutes additional proof of the assigned configuration, since its enantiomer displays the opposite sign ( +27.9) [20b].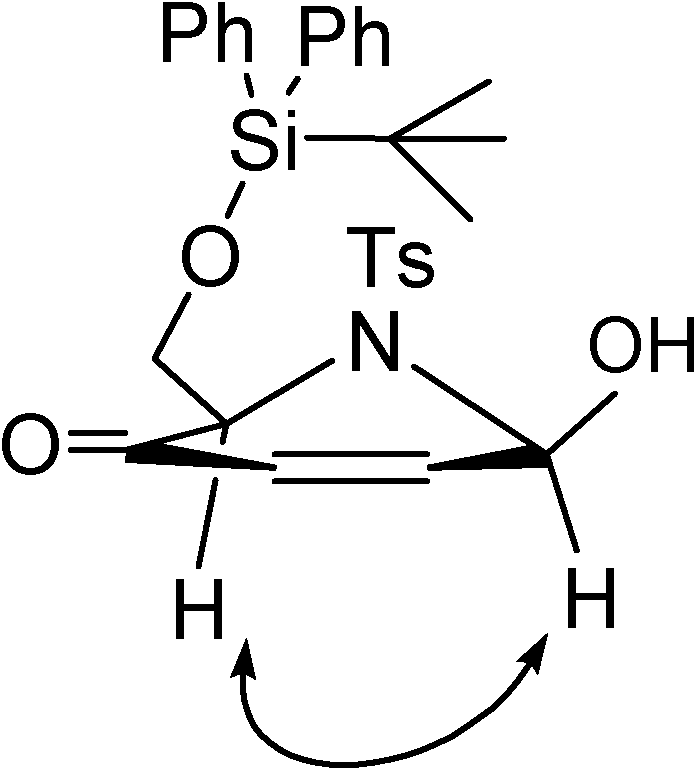
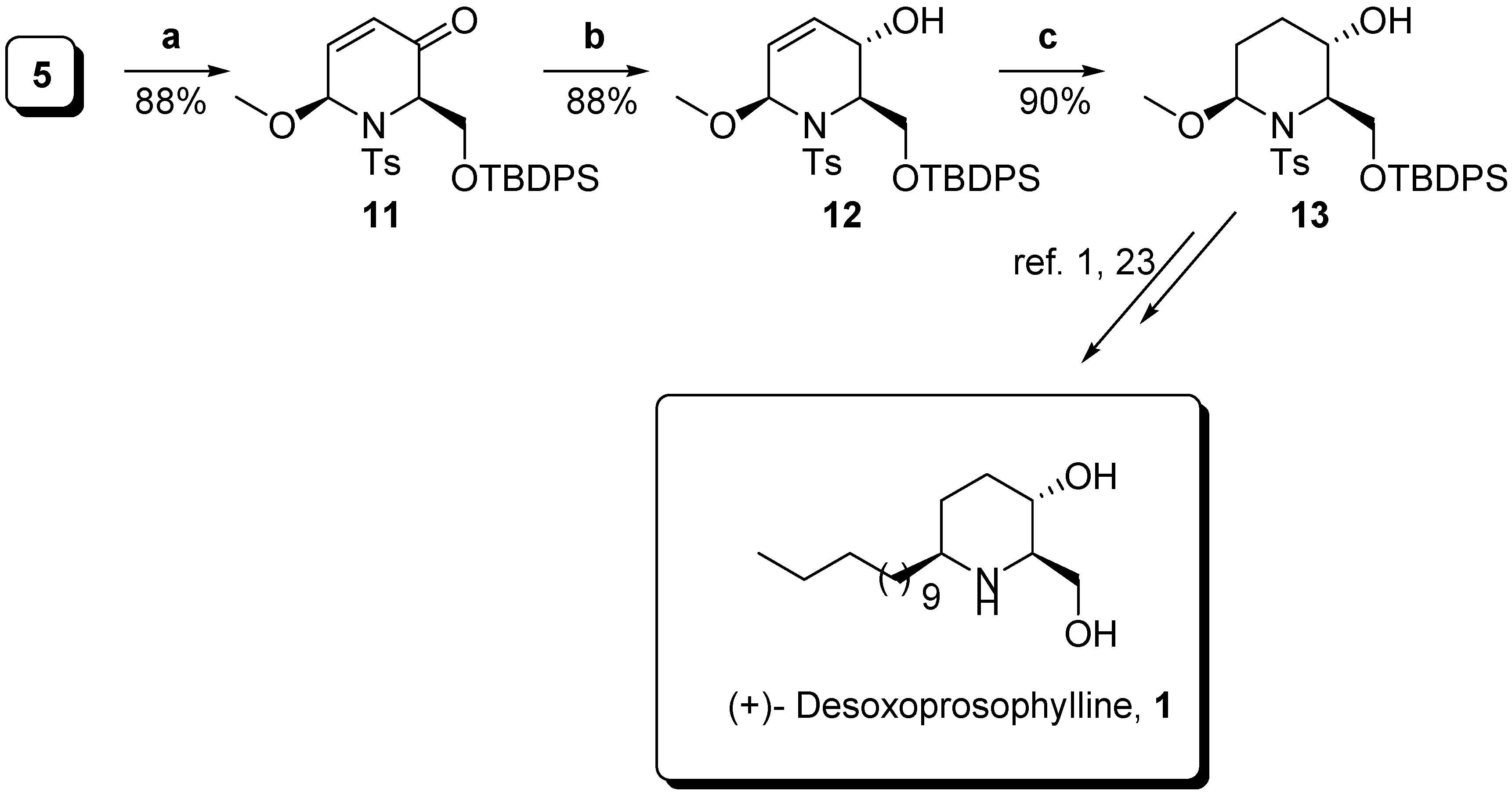
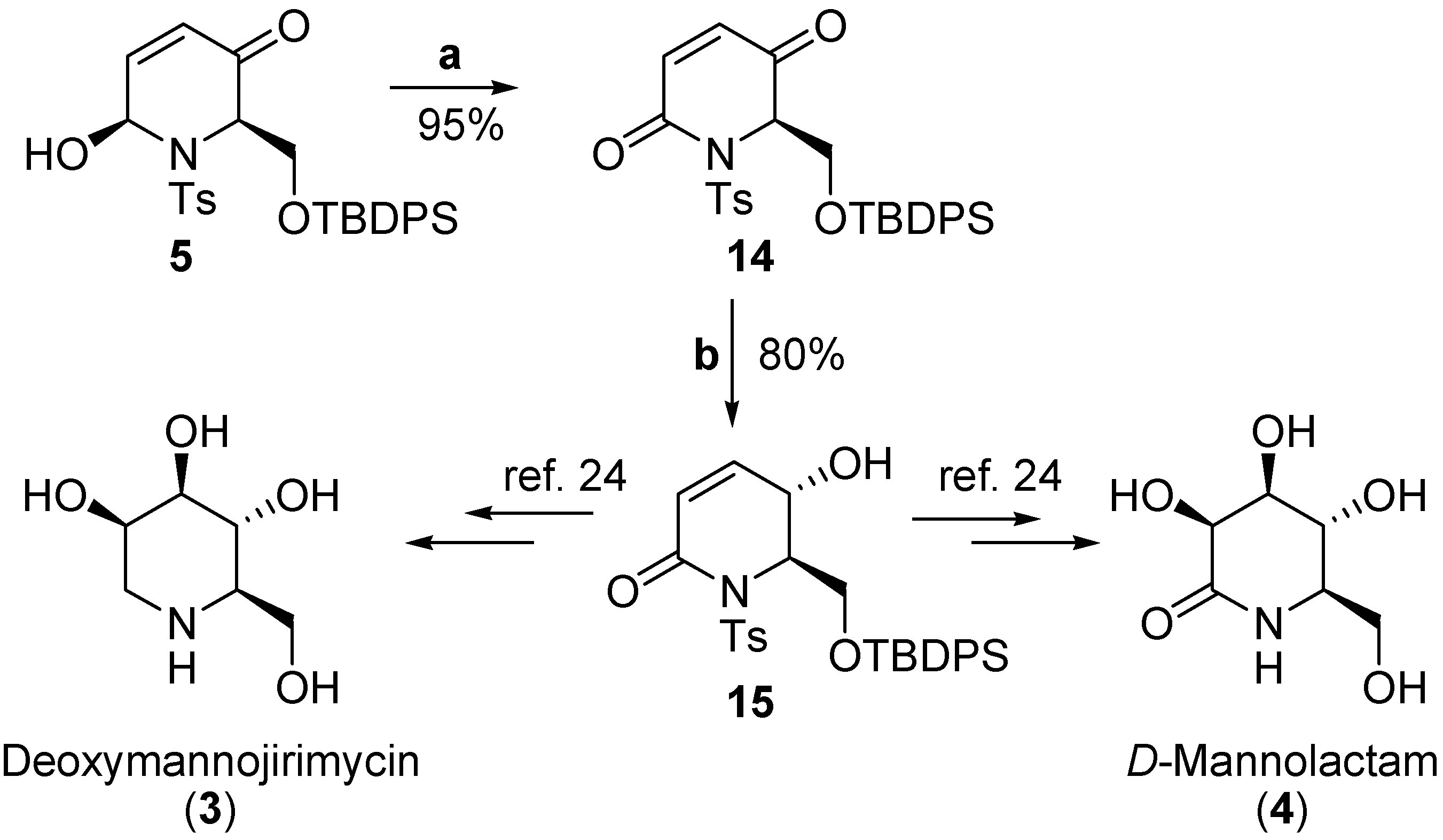
Conclusions
Experimental
General
+28.7, c 2.03, MeOH) was prepared according to a literature procedure [22]. All reactions were monitored by thin-layer chromatography using TLC sheets coated with silica gel 60 F254 (Merck); spots were visualized with UV light or/and an alcohol solution of anisaldehyde. Products were purified by flash chromatography on Merck silica gel 60 (230-400 mesh ASTM). Melting points (uncorrected): Büchi melting point apparatus. FT-IR: Nicolet Magna 750, series II. Samples were recorded as KBr pellets, unless otherwise stated. Optical rotations were measured with a Perkin-Elmer-241 polarimeter. 1H-NMR spectra were recorded on a Bruker DRX-400 (400 MHz) spectrometer, in CDCl3. Chemical shifts are referenced to internal TMS. Coupling constants (J) are expressed in Hz. HPLC: Hewlett Packard 1100 series instrument with a variable wavelength UV detector and coupled to HP Chem-Station utilizing the manufacturer’s 5.01 software package. −5.7 (c 1.00, EtOAc); 1H-NMR δ: 1.42 (s, 9H, C-CH3), 4.53 (dd, J = 10.7, 4.8, 1H, CH2), 4.65 (dd, J = 10.7, 4.8, 1H, CH2), 6.78 (dd, J = 7.2, 4.9, 1H, CH), 6.88 (d, J = 6.8, 1H, H-3), 7.70-8.10 (m, 15H, Ph-H, H-4, H-5), 8.50 (d, J = 6.8, 2H, Ph-H); Anal. Calcd. for C29H30O4Si (470.63) C, 74.01; H, 6.43. Found: C, 74.19; H, 6.30. –4.2 (c 1.03, EtOAc); IR (neat): ṽ= 3350 (OH), 740, 1020 (furan) cm-1; 1H-NMR δ: 1.07 (s, 9H, C-CH3), 3.95 (d, J = 1.4, 2H, CH2), 4.83 (m, 1H, CH), 6.27 (d, J = 3.2, 1H, H-3), 6.3 (dd, J = 5.0, 1.8, 1H, H-4), 7.3-7.6 (m, 11H, Ph-H, H-5); Anal. Calcd. for C22H26O3Si (366.53): C, 72.09; H, 7.15. Found: C, 72.31; H, 7.02. +50.8 (c 1.01, EtOAc); IR (neat): ṽ= 2110 (N3), 742, 1020 (furan) cm-1; 1H-NMR δ: 1.10 (s, 9H, C-CH3), 4.01 (d, J = 6.7, 2H, CH2), 4.60 (t, J = 5.5, 1H, CH), 6.37 (d, J = 1.8, 2H, H-3, H-4), 7.45 (m, 7H, H-5, Ph-H), 7.70 (m, 4H, Ph-H); Anal. Calcd. for C22H25N3O2Si (391.54): C, 67.49; H, 6.44; N, 10.73. Found: C, 67.27; H, 6.52; N, 10.59. +5.5 (c 1.01, EtOAc); IR (neat): ṽ= 3285 (N-H), 740, 1030 (furan), cm-1; 1H-NMR δ: 0.99 (s, 9H, C-CH3), 2.41 (s, 3H, PhCH3), 3.72 (dd, J = 10.1, 4.9, 1H, CH2), 3.85 (dd, J = 10.1, 4.9, 1H, CH2), 4.43 (m, 1H, CH), 5.2 (d, J = 7.6, 1H, NH), 6.13 (d, J = 3.11, 1H, H-3), 6.24 (dd, J = 3.1, 1.9, 1H, H-4), 7.20-7.52 (m, 13H, Ph-H, H-5), 7.22 (d, J = 8.3, 1H), 7.66 (d, J = 8.0, 2H, Ph-H); Anal. Calcd. for C29H33NO4SSi (519.73): C, 67.02; H, 6.40; N, 2.70. Found: C, 66.78; H, 6.30; N, 2.81. –25.5 (c 1.00, MeOH); IR (neat): ṽ= 3397 (OH), 1692 (C=O), 1595 (C=C) cm-1; 1H-NMR δ: 0.95 (s, 9H, C-CH3), 2.45 (s, 3H, PhCH3), 3.60 (dd, J = 10.7, 2.4, 1H, CH2), 3.90 (dd, J =10.7, 2.4, 1H, CH2 ), 4.55 (m, 1H, H-2), 4.96 (d, J = 11.5, 1H, OH), 6.10 (m, 1H, H-6), 6.22 (d, J = 10.4, 1H, H-4), 7.08 (dd, J = 10.4, 4.8, 1H, H-5), 7.3-7.5 (m, 12H, Ph-H), 7.79 (d, J = 8.0, 2H, Ph-H); Anal. Calcd. for C29H33NO5SSi (535.73): C, 65.02; H, 6.21; N, 2.61. Found: C, 65.17; H, 6.28; N, 2.68. −45 (c 1.02, EtOAc); IR (neat): ṽ= 1694 (C=O), 1596 (C=C) cm-1; 1H-NMR δ: 1.07 (s, 9H, C-CH3), 2.39 (s, 3H, PhCH3), 3.54 (s, 3H, CH3), 3.97 (dd, J = 10.2, 6.6, 1H, CH2), 4.07 (dd, J = 10.2, 6.6, 1H, CH2), 4.47 (t, J = 6.9, 1H, H-2), 5.51 (d, J = 4.3, 1H, H-6), 5.74 (d, J = 10.36, 1H, H-4), 6.68 (dd, J = 10.3, 4.4, 1H, H-5), 7.24 (d, J = 7.4, 2H, Ph-H), 7.44 (m, 6H, Ph-H), 7.55 (d, J = 8.2, 2H, Ph-H), 7.67 d, J = 7.4, 4H, Ph-H); Anal. Calcd. for C30H35NO5SSi (549.75): C, 65.54; H, 6.42; N, 2.55. Found: C, 65.69; H, 6.52; N, 2.44. −32.5 (c 0.98, EtOAc); IR (neat): ṽ= 3460 (OH), 1650 (C=C) cm-1; 1H-NMR δ: 1.05 (s, 9H, C-CH3), 2.42 (s, 3H, PhCH3), 3.31 (s, 3H, CH3), 3.77 (dd, J = 10.6, 4.2, 1H, CH2), 3.95 (m, 1H, H-3), 4.17 (d, J = 6.85, 1H, OH), 4.2 (m, 1H, H-2), 4.4 (t, J = 10.3, 1H, CH2), 5.24 (m, 1H, H-6), 5.69 (m, 1H, H-5), 5.84 (m, 1H, H-4), 7.25 (t, J = 9.01, 2H, Ph-H), 7.4-7.5 (m, 6H, Ph-H), 7.6-7.7 (m, 6H, Ph-H); Anal. Calcd. for C30H37NO5SSi (551.8): C, 65.30; H, 6.76; N, 2.54. Found: C, 65.55; H, 6.90; N, 2.63. −15.5 (c 1.02, EtOAc); IR (neat): ṽ= 3500 (OH) cm-1; 1H-NMR δ: 1.07 (s, 9H, C-CH3), 1.65 (m, 2H, H-4), 1.9 (m, 2H, H-5), 2.4 (s, 3H, PhCH3), 3.1 (s, 3H, CH3), 3.44 (m, 1H, H-3), 3.66 (m, 1H, CH2), 3.88 (dd, J = 10.2, 4.1, 1H, H-2), 4.35 (d, J = 6.26, 1H, OH), 4.5 (t, J = 10.4, 1H, CH2), 5.01 (m, 1H, H-6), 7.27 (t, J = 8.15, 2H, Ph-H), 7.3-7.5 (m, 6H, Ph-H), 7.6-7.7 (m, 6H, Ph-H); Anal. Calcd. for C30H39NO5SSi (553.8): C, 65.07; H, 7.10; N, 2.53. Found: C, 65.31; H, 7.23; N, 2.42. −10.7 (c 1.00, EtOAc); IR (neat): ṽ= 1725 (C=O), 1692 (N-C=O) cm-1; 1H-NMR δ: 0.92 (s, 9H, C-CH3), 2.44 (s, 3H, PhCH3), 4.11 (dd, J = 10.6, 1.6, 1H, CH2), 4.42 (dd, J =10.6, 1.6, 1H, CH2), 5.02 (s, 1H, H-6), 6.72 (d, J = 10.1, 1H, H-3), 6.8 (d, J = 10.1, 1H, H-4), 7.27 (d, J = 8.1, 2H, Ph-H), 7.3-7.55 (m, 10H, Ph-H), 7.94 (d, J = 8.3, 2H, Ph-H); Anal. Calcd. for C29H31NO5SSi (533.7): C, 65.26; H, 5.85; N, 2.62. Found. C, 65.01; H, 5.98; N, 2.49. −2.7 (c 1.02, EtOAc); IR (neat): ṽ= 1450 (C=O), 1670 (N-C=O) cm-1; 1H-NMR δ: 0.92 (s, 9H, C-CH3), 2.44 (s, 3H, PhCH3), 3.82 (dd, J = 10.6, 4.2, 1H, CH2), 4.02 (dd, J = 10.6, 4.2, 1H, CH2), 4.17 (d, J = 6.85, 1H, OH), 5.04 (m, 1H, H-5), 5.04 (m, 1H, H-5), 5.07 (m, 1H, H-6), 5.60 (dd, J = 10.0, 4.2, 1H, H-4), 7.01 (dt, J = 10.2, 1.7, 1H, H-3), 7.2 (d, J = 7.2, 2H, Ph-H), 7.40-7.52 (m, 6H, Ph-H), 7.60 (dd, J = 7.5, 1.5, 2H, Ph-H), 7.7 (d, J = 8.4, 4H, Ph-H); Anal. Calcd. for C29H31NO5SSi (533.7): C, 65.26; H, 5.85; N, 2.62. Found: C, 65.09; H, 5.71; N, 2.72.References
- Dransfield, J. R.; Gore, P. M.; Shipman, M.; Slawin, A. M. Z. Divergent approach to imino sugar C-glycosides using imino glycals: application to the stereocontrolled synthesis of (+)-deoxoprosophylline. Chem. Commun. 2002, 2, 150–151. [Google Scholar] Yang, C.-F.; Xu, Y.-M.; Liao, L.-X.; Zhou, W.-S. Asymmetric total synthesis of (+)-desoxoprosophylline. Tetrahedron Lett. 1998, 39, 9227–9228. [Google Scholar]
- Schneider, M. J. Alkaloids: Chemical and Biological Perspectives; Pelletier, S. W., Ed.; Pergamon: Oxford, 1996; Vol. 10, pp. 155–299. [Google Scholar]
- Stutz, A. E. Iminosugars as Glycosidase Inhibitors; Wiley-VCH: Weinheim, 1998. [Google Scholar]
- Hughes, A. B.; Rudge, A. J. Deoxynojirimycin – synthesis and biological activity. Nat. Prod. Rep. 1994, 11, 135–162. [Google Scholar]
- Butters, T. D.; Dwek, R. A.; Platt, F. M. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology 2005, 10, R-43–R52. [Google Scholar] Butters, T. D.; Dwek, R. A.; Platt, F. M. Therapeutic applications of imino sugars in lysosomal storage disorders. Curr. Top. Med. Chem. 2003, 3, 561–574. [Google Scholar]
- Ye, X.-S.; Sun, F.; Liu, M.; Li, Q.; Wang, Y.; Zhang, G.; Zhang, L.-H.; Zhang, X.-L. Synthetic iminosugar derivatives as new potential immunosuppressive agents. J. Med. Chem. 2005, 48, 3688–3691. [Google Scholar] [CrossRef]
- Greimel, P.; Spreitz, J.; Stütz, A. E.; Wrodnigg, T. M. Iminosugars and relatives as antiviral and potential anti-infective agents. Curr. Topics Med. Chem. 2003, 3, 513–523. [Google Scholar] Robina, I.; Moreno-Vargas, A. J.; Carmona, A. T.; Vogel, P. Glycosidase inhibitors as potential HIV entry inhibitors? Curr. Drug. Metab. 2004, 5, 329–361. [Google Scholar]
- Somsak, L.; Nagy, V.; Hadazy, Z.; Dosca, T.; Gergely, P. Glucose analog inhibitors of glycogen phosphorylases as potential antidiabetic agents: Recent developments. Curr. Pharm. Design 2003, 9, 1177–1189. [Google Scholar]
- Lu, Y.; Xu, Y.-Y.; Fan, K.-Y.; Shen, Z.-H. 1-deoxymannojirimycin, the alpha 1,2-mannosidase inhibitor, induced cellular endoplasmic reticulum stress in human hepatocarcinoma cell 7721. Biochem. Biophys. Res. Commun. 2006, 344, 221–225. [Google Scholar] Martin, O. R.; Compain, P. Imino-sugars: Recent insights into their bioactivity and potential as therapeutic agents – Preface. Curr. Topics Med. Chem. 2003, 3, U2–U5. [Google Scholar]
- Fuhrmann, U.; Bause, E.; Legler, G.; Ploegh, H. Novel Mannosidase Inhibitor Blocking Conversion of High Mannose to Complex Oligosaccharides. Nature 1984, 307, 755–758. [Google Scholar] [CrossRef]
- Kato, A.; Kato, N.; Kano, E.; Adachi, I.; Ikeda, K.; Yu, L.; Okamoto, T.; Banba, Y.; Ouchi, H.; Takahata, H.; Asano, N. Biological properties of D- and L-1-deoxyazasugars. J. Med. Chem. 2005, 48, 2036–2044. [Google Scholar] [CrossRef]
- Spreitz, J.; Stütz, A. E. Golgi endomannosidase inhibitor, alpha-D-glucopyranosyl-(1->3)-1-deoxymannojirimycin: a five-step synthesis from maltulose and examples of N-modified derivatives. Carbohydr. Res. 2004, 339, 1823–1827. [Google Scholar] [CrossRef]
- Boucheron, C.; Compain, P.; Martin, O. R. A stereodivergent approach to 1-deoxynojirimycin, 1-deoxygalactonojirimycin and 1-deoxymannojirimycin derivatives. Tetrahedron Lett. 2006, 47, 3081–3084. [Google Scholar] [CrossRef]
- Kennedy, A.; Nelson, A.; Perry, A. Methods for the synthesis of polyhydroxylated piperidine by diastereoselective dihydroxylation. Exploitation in the two-directional synthesis of aza-C-linked disaccharide derivatives. Beilstein J. Org. Chem. 2005, 1, 2. [Google Scholar] [CrossRef]
- Singh, O. V.; Han, H. A general methodology for the asymmetric synthesis of 1-deoxy-iminosugars. Tetrahedron Lett. 2003, 44, 2387–2391. [Google Scholar]
- Boglio, C.; Stahlke, S.; Thorimbert, S.; Malacria, M. A stereoselective route toward poly-hydroxylated piperidines. A total synthesis of (+/-)-deoxymannojirimycin. Org. Lett. 2005, 7, 4851–4854. [Google Scholar] [CrossRef]
- Pyun, S.-J.; Lee, K.-Y.; Oh, C.-Y.; Joo, J.-E.; Cheon, S.-H.; Ham, W.-H. Total synthesis of 1-deoxygulonojirimycin. Revision of the absolute configuration of the natural product. Tetrahedron 2005, 61, 1413–1416. [Google Scholar]
- Takahata, H.; Banba, Y.; Sasatani, M.; Nemoto, H.; Kato, A.; Adachi, I. Asymmetric synthesis of 1-deoxynojirimycin and its congeners from a common chiral building block. Tetrahedron 2004, 60, 8199–8205. [Google Scholar] [CrossRef]
- Kirhara, M.; Nishio, T.; Yokohama, S.; Kakuda, H.; Momose, T. Hypervalent lambda(n)-iodane-mediated fragmentation of tertiary cyclopropanol systems - II: Application to asymmetric syntheses of piperidine and indolizidine alkaloids. Tetrahedron 1999, 55, 2911–2926. [Google Scholar] [CrossRef]
- Zhou, W. S.; Lu, Z. H.; Wang, Z. M. An efficient preparation of optically-active alpha-furfuryl amide by kinetic resolution using the modified sharpless asymmetric epoxidation reagents. Tetrahedron 1993, 49, 2641–2654. [Google Scholar] Koulocheri, S. D.; Magiatis, P.; Skaltsounis, A.-L.; Haroutounian, S. A. Stereoselective Michael addition of thiophenols, amino acids and hydrazoic acid to (2S)-hydroxymethyl-dihydropyridone as a convenient route to novel azasugar derivatives. Tetrahedron 2000, 56, 6135–6141. [Google Scholar]
- Koulocheri, S. D.; Haroutounian, S. A. Transformation of D-glucal to (2S)-hydroxymethyl-dihydropyridones as intermediates to piperidine alkaloids. Synthesis 1999, 11, 1889–1892. [Google Scholar] [CrossRef]
- Hauser, F. M.; Ellenberger, S. R.; Ellenberger, W. P. A simple procedure for the preparation of L-hexoses. Tetrahedron Lett. 1988, 29, 4939–4942. [Google Scholar] [CrossRef]
- Thompson, A. S.; Humphrey, G. R.; De Marco, A. M.; Mathre, D. J.; Grabowski, E. J. J. Direct conversion of activated alcohols to azides using diphenyl phosphorazidate – A practical alternative to Mitsunobu conditions. J. Org. Chem. 1993, 58, 5886–5888. [Google Scholar] [CrossRef]
- Yang, C.; Liao, L.; Xu, Y.; Zhang, H.; Xia, P.; Zhou, W. New concise asymmetric total synthesis of (+)-desoxoprosophylline and prosophylline. Tetrahedron: Asymmetry 1999, 10, 2311–2318. [Google Scholar] [CrossRef]
- Knight, J. G.; Tchabanenko, K. Total synthesis of deoxymannojirimycin and D-mannolactam via carbonylation of 5-vinyloxazolidin-2-ones. Tetrahedron 2003, 59, 281–286. [Google Scholar] [CrossRef]
- Sample availability: Available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Tzanetou, E.N.; Kasiotis, K.M.; Magiatis, P.; Haroutounian, S.A. Synthesis of (R)-Dihydropyridones as Key Intermediates for an Efficient Access to Piperidine Alkaloids. Molecules 2007, 12, 735-744. https://doi.org/10.3390/12040735
Tzanetou EN, Kasiotis KM, Magiatis P, Haroutounian SA. Synthesis of (R)-Dihydropyridones as Key Intermediates for an Efficient Access to Piperidine Alkaloids. Molecules. 2007; 12(4):735-744. https://doi.org/10.3390/12040735
Chicago/Turabian StyleTzanetou, Evangelia N, Konstantinos M Kasiotis, Prokopios Magiatis, and Serkos A Haroutounian. 2007. "Synthesis of (R)-Dihydropyridones as Key Intermediates for an Efficient Access to Piperidine Alkaloids" Molecules 12, no. 4: 735-744. https://doi.org/10.3390/12040735