Determination of the Three-dimensional Structure of Gynoside A in Solution using NMR and Molecular Modeling

Abstract
:Introduction
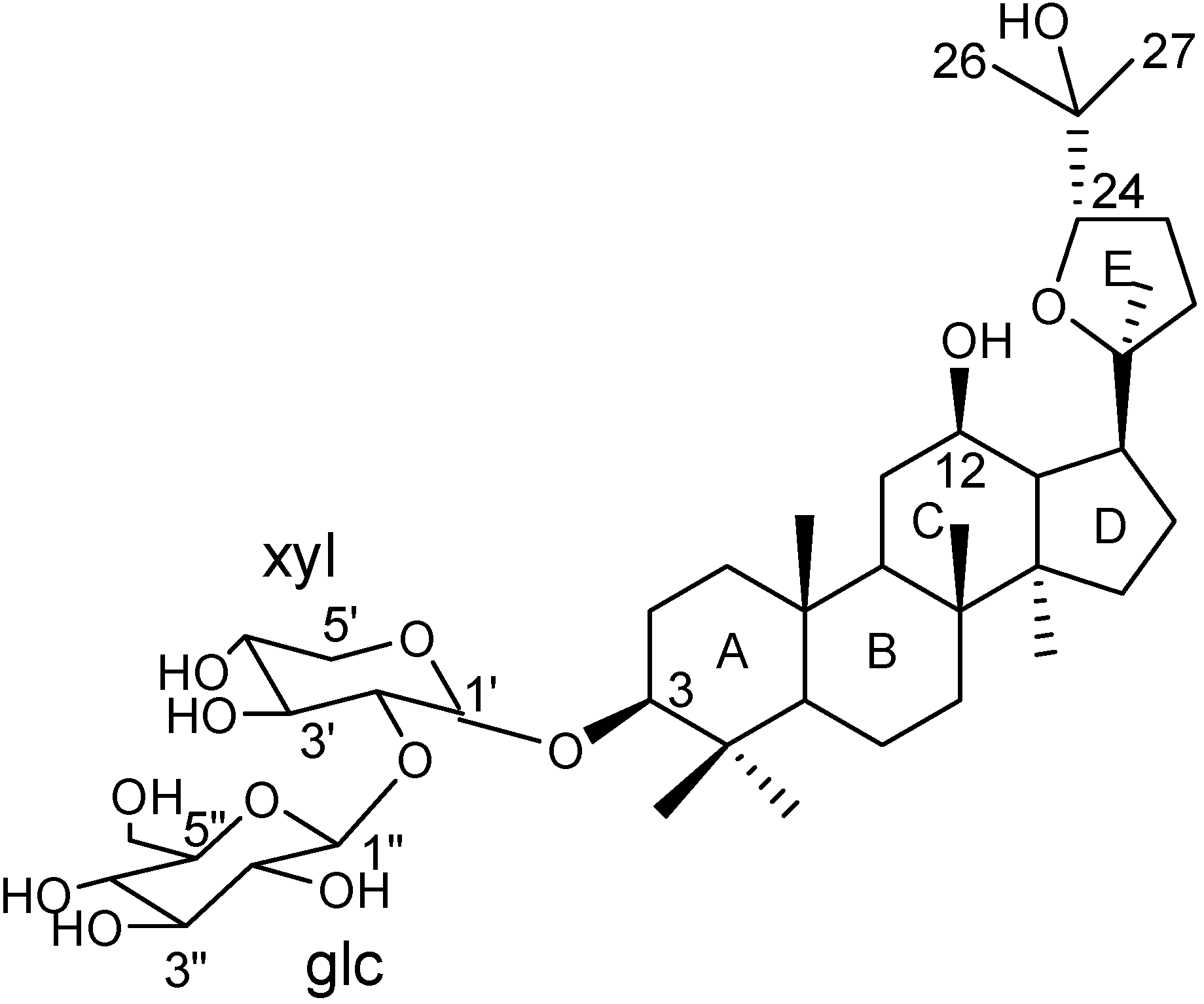
Results and Discussion
NMR assignments

{kind=link}
{kind=link}
{kind=link}
| No. | Pyridine-d5 | DMSO-d6 | ||
|---|---|---|---|---|
| δ (13C) | δ (1H) | δ (13C) | δ (1H) | |
| 1 | 39.8 | 1.68 | 38.5 | 1.60 (He), 0.93 (Ha) |
NMR constraints
| Coupled protons (A-X-Y-B) | 3J(A,B) | |
|---|---|---|
| in pyridine-d5 | in DMSO-d6 | |
| 1'H-1'C-2'C-2'H | 6.7 | 6.1 |
| 4'H-4'C-5'C-5'He | 4.9 | 4.5 |
| 4'H-4'C-5'C-5'Ha | 10.5 | 9.6 |
| 1''H-1''C-2''C-2''H | 7.6 | 8.5 |
| 2''H-2''C-3''C-3''H | 8.2 | 8.0 |
| 3''H-3''C-4''C-4''H | 8.9 | 8.4 |
| 4''H-4''C-5''C-5''H | 9.4 | 9.4 |
| 5''H-5''C-6''C-6''Ha | 3.5 | 4.6 |
| 5''H-5''C-6''C-6''He | 3.5 | 4.6 |
Structural results
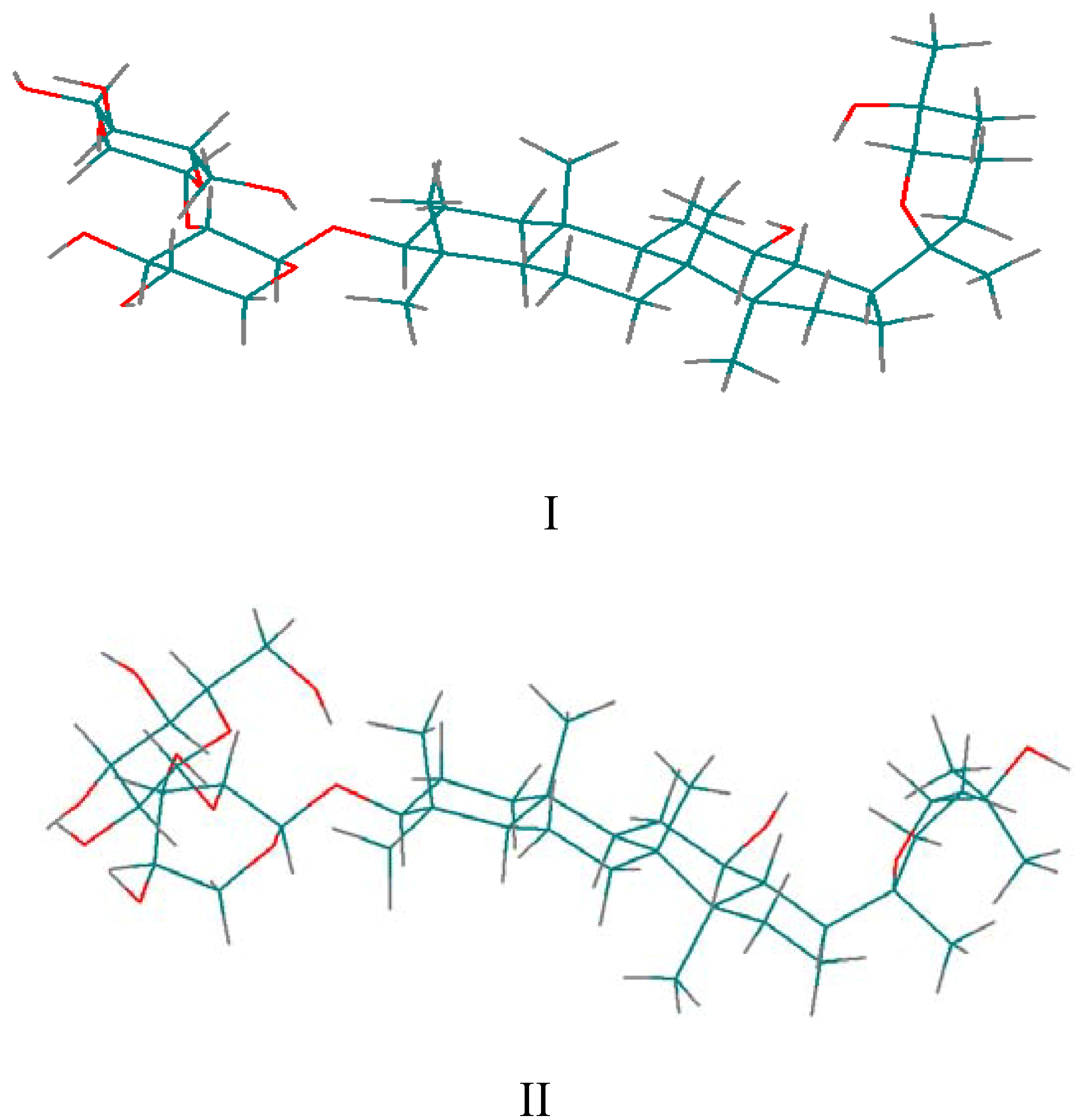
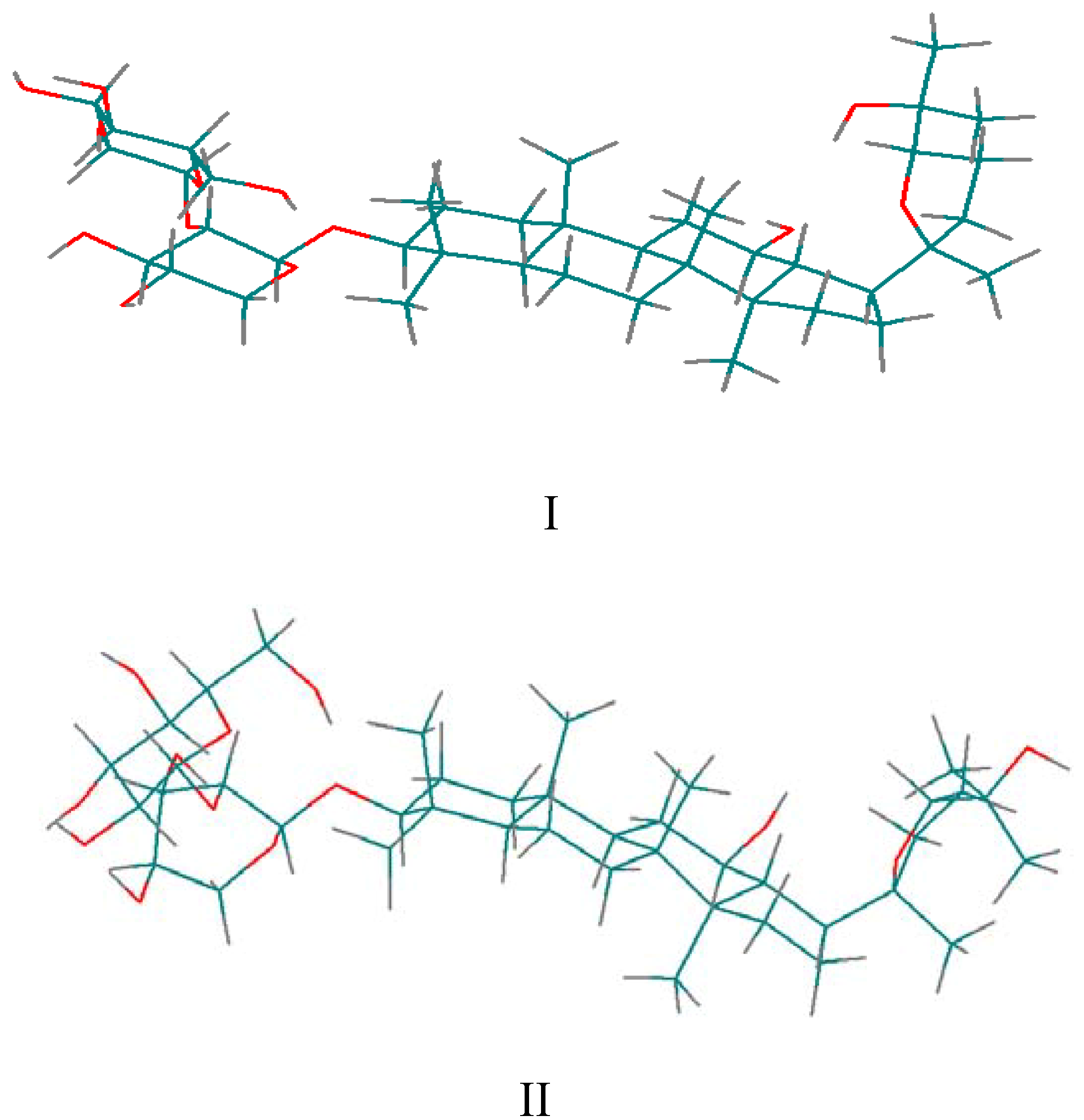
 for the pyridine-d5 solution work. due to the fact that the hydroxyl proton signals of sugar rings can be distinguished in DMSO-d6 solution (Figure 2). After calculation, 10 minimum energy structures among the last 100 ps were extracted and optimized by the conjugate-gradient method. The results are given in Table 3. All of the structures satisfied the distance constraint criteria with energies in the 186.2-191.5 kcal mol-1 range (1 kcal=4.184 kJ) in pyridine-d5 and 203.8-207.9 kcal mol-1 for DMSO-d6. The average root mean square deviation (rmsd) values for the mean structure in pyridine-d5 and DMSO-d6 were 0.837 and 0.521 Å, respectively. Obviously, the structure in pyridine-d5 had lower energies than that in DMSO-d6. The two global minimum energy conformations are displayed in Figure 3.
for the pyridine-d5 solution work. due to the fact that the hydroxyl proton signals of sugar rings can be distinguished in DMSO-d6 solution (Figure 2). After calculation, 10 minimum energy structures among the last 100 ps were extracted and optimized by the conjugate-gradient method. The results are given in Table 3. All of the structures satisfied the distance constraint criteria with energies in the 186.2-191.5 kcal mol-1 range (1 kcal=4.184 kJ) in pyridine-d5 and 203.8-207.9 kcal mol-1 for DMSO-d6. The average root mean square deviation (rmsd) values for the mean structure in pyridine-d5 and DMSO-d6 were 0.837 and 0.521 Å, respectively. Obviously, the structure in pyridine-d5 had lower energies than that in DMSO-d6. The two global minimum energy conformations are displayed in Figure 3.
| Atom A-Atom B | Pyridine-d5 | DMSO-d6 | Atom A-Atom B | Pyridine-d5 | DMSO-d6 |
|---|---|---|---|---|---|
| H5’’—H6’’ | 3 | - | H3—C28 | 4 | 4 |
| H5’’—H1’’ | 4 | - | H3—C29 | 5 | - |
| H1’’—C29 | 5 | 5 | H5—C28 | 5 | - |
| H1’—H2 | 4 | - | H5’’—C29 | 5 | - |
| H1’—H3 | 4 | - | H6’’—C29 | 5 | - |
| H1’—H3’ | 4 | H12—H11 | 3 | 3 | |
| H1’—H5’ | 4 | - | H12—H17 | 3 | 3 |
| H1’—C28 | 5 | 5 | H12—C30 | 5 | 5 |
| H3—H5 | 3 | - | OH12—H13 | 3 | 3 |
| H3—H19 | 3 | - | OH12—H12 | 3 | 3 |
| OH12—H17 | 4 | 3 | OH12—H11 | 4 | |
| OH12—H24 | 3 | 3 | OH12—C26 | 5 | - |
| OH12—C21 | - | 5 | OH12—C27 | - | 5 |
| OH25—C26 | 4 | 5 | OH25—C27 | 5 | - |
| OH25—C21 | - | 5 | OH25—C24 | 4 | 4 |
| OH2’’—H1’’ | - | 4 | H1’’—H4’’ | - | 4 |
| OH2’’—H3’’ | 4 | 4 | OH2’’—H2’’ | - | 3 |
| OH3’’—H2’’ | 4 | OH2’’—OH3’’ | - | 4 | |
| OH4’’—H3’’ | 4 | OH3’’—H3’’ | 3 | 3 | |
| H3—H2 | - | 3 | H1’’—H5’’ | 4 | |
| OH3’—H5’ | - | 4 | OH4’’—H6’’a | - | 4 |
| OH3’—H2’ | 4 | OH4’’—H6’’e | - | 4 | |
| OH3’—H3’ | 3 | H1’’—H2’ | 4 | ||
| OH4’—H1’’ | 5 | 4 | OH6’’—H6’’a | - | 3 |
| OH4’—OH3’ | - | 4 | OH6’’—H6’’e | - | 3 |
| H6’’a—H4’’ | - | 4 | H12—H9 | - | 3 |
| H6’’e—H4’’ | - | 4 | H17—C21 | 5 | 5 |
| H17—H22 | - | 3 | H17—C30 | 5 | 5 |
| H22a—C21 | - | 4 | H22e—C21 | - | 4 |
| H24—H13 | - | 3 | H24—H22e | - | 3 |
| H24—C27 | - | 4 | H24—C26 | 5 | 5 |
| Torsional angle | Pyridine-d5 | DMSO-d6 |
|---|---|---|
| C3-C4-C5-C10 | 50.4±0.9 | 53.1±1.6 |
| C3-C4-C5-C6 | 183.5±1.0 | 188.4±1.4 |
| C4-C5-C6-C7 | 163.8±1.7 | 161.2±2.2 |
| C4-C5-C10-C9 | 191.47±1.3 | 193.3±1.5 |
| C6-C7-C8-C14 | 193.0±1.9 | 192.3±1.6 |
| C6-C7-C8-C9 | 312.0±1.7 | 311.3±1.7 |
| C7-C8-C9-C10 | 47.3±2.0 | 47.9±1.7 |
| C7-C8-C9-C11 | 185.9±2.3 | 185.9±0.6 |
| C7-C8-C14-C13 | 177.1±1.5 | 176.9±0.6 |
| C7-C8-C14-C15 | 289.7±2.5 | 286.8±1.2 |
| C8-C9-C11-C12 | 50.7±2.6 | 52.7±0.5 |
| C8-C14-C13-C17 | 165.4±2.0 | 161.9±0.8 |
| C8-C14-C15-C16 | 201.0±3.0 | 199.5±0.5 |
| C16-C17-C20-O | 194.5±2.2 | 190.3±1.0 |
| C16-C17-C20-C22 | 306.2±2.8 | 306.7±1.1 |
| C15-C16-C17-C20 | 120.1±2.9 | 117.1±1.1 |
| C2-C3-O-C1' | 135.9±3.5 | 145.0±1.5 |
| C3-O-C1'-C2" | 187.4±2.4 | 146.1±2.0 |
| C1'-C2'-O-C1" | 113.5±2.3 | 154.1±1.2 |
| C2'-O-C1"-C2" | 91.2±1.5 | 155.7±2.4 |
Experimental
General
Acknowledgments
References and Notes
- Cho, J.Y.; Yoo, E.S.; Cha, B.C.; Park, H.J; Rhee, M.H.; Han, Y.N. The inhibitory effect of triterpenoid glycosides originating from Sanguisorba officinalis on tissue factor activity and the production of TNF-α. Planta Medica 2006, 72, 1279–1284. [Google Scholar] [CrossRef]
- Popovich, D.G.; Kitts, D.D. Anticancer activity of ginseng and soy saponins. In Nutrition and Cancer Prevention; Awad, A.B., Bradford, P.G., Eds.; Taylor and Francis: Boca Raton, FL, 2006; pp. 457–483. [Google Scholar]
- Sugishita, E.; Amagaya, S.; Ogihara, Y. Structure-activity studies of some oleanane triterpenoid glycosides and their related compounds from the leaves of Tetrapanax papyriferum on antiinflammatory activities. J. Pharmacobio-Dynam. 1982, 5, 379–87. [Google Scholar] [CrossRef]
- Rao, A.V.; Gurfinkel, D.M. The bioactivity of saponins: triterpenoid and steroidal glycosides. Drug Metab. Drug Interact. 2000, 17, 211–235. [Google Scholar]
- Liu, X.; Ye, W.C.; Mo, Z.Y.; Hsiao, W.L. Five New Ocotillone-Type Saponins from Gynostemma pentaphyllum. J. Nat. Prod. 2004, 67, 1147–1151. [Google Scholar] [CrossRef]
- Gao, J.H.; Shi, G.B.; Song, G.Q.; Shao, Y.; Zhou, B.N. Further NMR Investigation and Conformational Analysis of an Acylated Flavonol Glucorhamnoside. Magn. Reson. Chem. 1996, 34, 249–254. [Google Scholar] [CrossRef]
- Hudson, B.P.; Hudson; Barton, J.K. Solution Structure of a Metallointercalator Bound Site Specifically to DNA. J. Am. Chem. Soc. 1998, 120, 6877–6888. [Google Scholar] [CrossRef]
- Goddard, T.D.; Kneller, D.G. SPARKY 3; University of California: San Francisco, CA, 2006. [Google Scholar]
- Wüthrich, K.; Billeter, M.; Braun, W. Pseudo-structures for the 20 common amino acids for use in studies of protein conformations by measurements of intramolecular proton-proton distance constraints with nuclear magnetic resonance. J. Mol. Biol. 1983, 169, 949–961. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley: New York, 1986. [Google Scholar]
- Steinmetz, W.E.; Sadowshy, J.D.; Rice, J.S.; Roberts, J.J.; Bui, Y.K. Determination of the aqueous-phase structure of 6-O-methylerythromycin from NMR constraints. Magn. Reson. Chem. 2001, 39, 163. [Google Scholar] [CrossRef]
- Steinmetz, W.E.; Sparrow, A.; Somsouk, M. Determination of the three-dimensional, solution-phase structure of the macrolide antibiotic oxolide in CD2Cl2 and D2O from NMR constraints. Magn. Reson. Chem. 2005, 43, 16–20. [Google Scholar] [CrossRef]
- Liu, S.B.; Shi, Y.H.; Zhang, Q.W.; Song, G.Q. Conformational study of fosinopril sodium in solution using NMR and molecular modeling. Magn. Reson. Chem. 2003, 41, 609–614. [Google Scholar] [CrossRef]
- Shi, YH; Song, YL; Li, Q; Song, GQ. Binding affinity difference induced by the stereochemistry of the sulfoxide bridge of the cyclic peptide inhibitors of Grb2-SH2 domain: NMR studies for the structural origin. Biochim. Biophys. Res. Commu. 2005, 330, 1254–1261. [Google Scholar] [CrossRef]
- Hyperchem 7.5 Master manual: Release 7 for Windows; Hypercube Inc.: Florida, USA, 2002.
- Lins, L.; Brasseur, R.; Malaisse, W.J.; Biesemans, M.; Verheyden, P.; Willem, R. Importance of the hydrophobic energy: structural determination of a hypoglycemic drug of the meglitinide family by nuclear magnetic resonance and molecular modeling. Biochem. Pharmacol. 1996, 52, 1155–1168. [Google Scholar] [CrossRef]
- Gao, J.H.; Shi, G.B.; Song, G.Q.; Chen, K.X.; Ji, R.Y. Conformational studies of Asterin B and C in solution by NMR. II. Conformational analysis by NMR and molecular dynamic simulations. 1996, 54, 702–708. [Google Scholar]
- Crystallographic data for the compound Gynoside A reported in this paper have been deposited with the Cambridge Crystallographic Data Center and allocated the deposition number CCDC 200550. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0)-1223-336033 or e-mail: [email protected]]
- Sample Availability: Small samples (a few milligrams) of Gynoside A are available from the authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Li, Q.; Yao, Z.-H.; Shi, Y.-H.; Liu, X.; Yao, X.-S.; Ye, W.-C. Determination of the Three-dimensional Structure of Gynoside A in Solution using NMR and Molecular Modeling. Molecules 2007, 12, 907-916. https://doi.org/10.3390/12040907
Li Q, Yao Z-H, Shi Y-H, Liu X, Yao X-S, Ye W-C. Determination of the Three-dimensional Structure of Gynoside A in Solution using NMR and Molecular Modeling. Molecules. 2007; 12(4):907-916. https://doi.org/10.3390/12040907
Chicago/Turabian StyleLi, Qian, Zhi-Hong Yao, Yan-Hong Shi, Xin Liu, Xin-Sheng Yao, and Wen-Cai Ye. 2007. "Determination of the Three-dimensional Structure of Gynoside A in Solution using NMR and Molecular Modeling" Molecules 12, no. 4: 907-916. https://doi.org/10.3390/12040907