Preparation of Second Generation Ionic Liquids by Efficient Solvent-Free Alkylation of N-Heterocycles with Chloroalkanes

 ,
,  , ,
, ,
Abstract
:Introduction
Results and Discussion

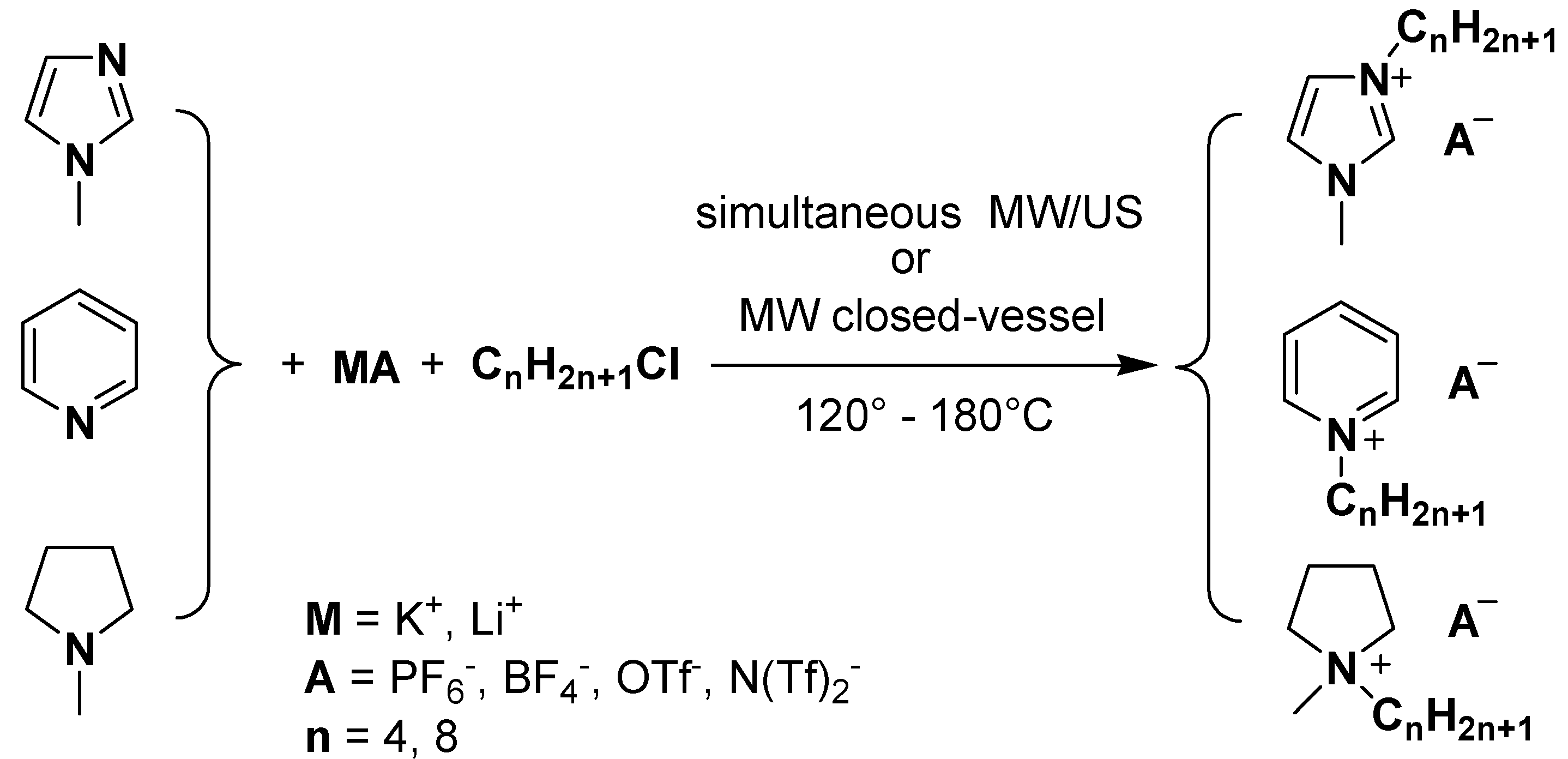{kind=link}
| Entry | Anion source | Irradiation time (min) | MW power (W) | US power (W) | Temperature (°C)* | Yield (%) |
|---|---|---|---|---|---|---|
| 1a | KPF6 | **10/20 | 90 | 40 | 120/140 | 98 |
| 1b | KPF6 | 60 | 60 | - | 140 | 88 |
| 1c | KPF6 | 60 (oil bath) | - | 40 | 140 | 62 |
| 1d | KPF6 | 60 (oil bath) | - | - | 140 | 47 |
| 1e | KPF6 | 240 (oil bath) | - | - | 140 | 84 |
| 2 | KBF4 | **10/20 | 75 | 40 | 120/140 | 95 |
| 3 | KOTf | **10/45 | 85 | 45 | 120/140 | 65 |
| 4 | LiN(Tf)2 | **10/90 | 110 | 45 | 120/140 | 72 |
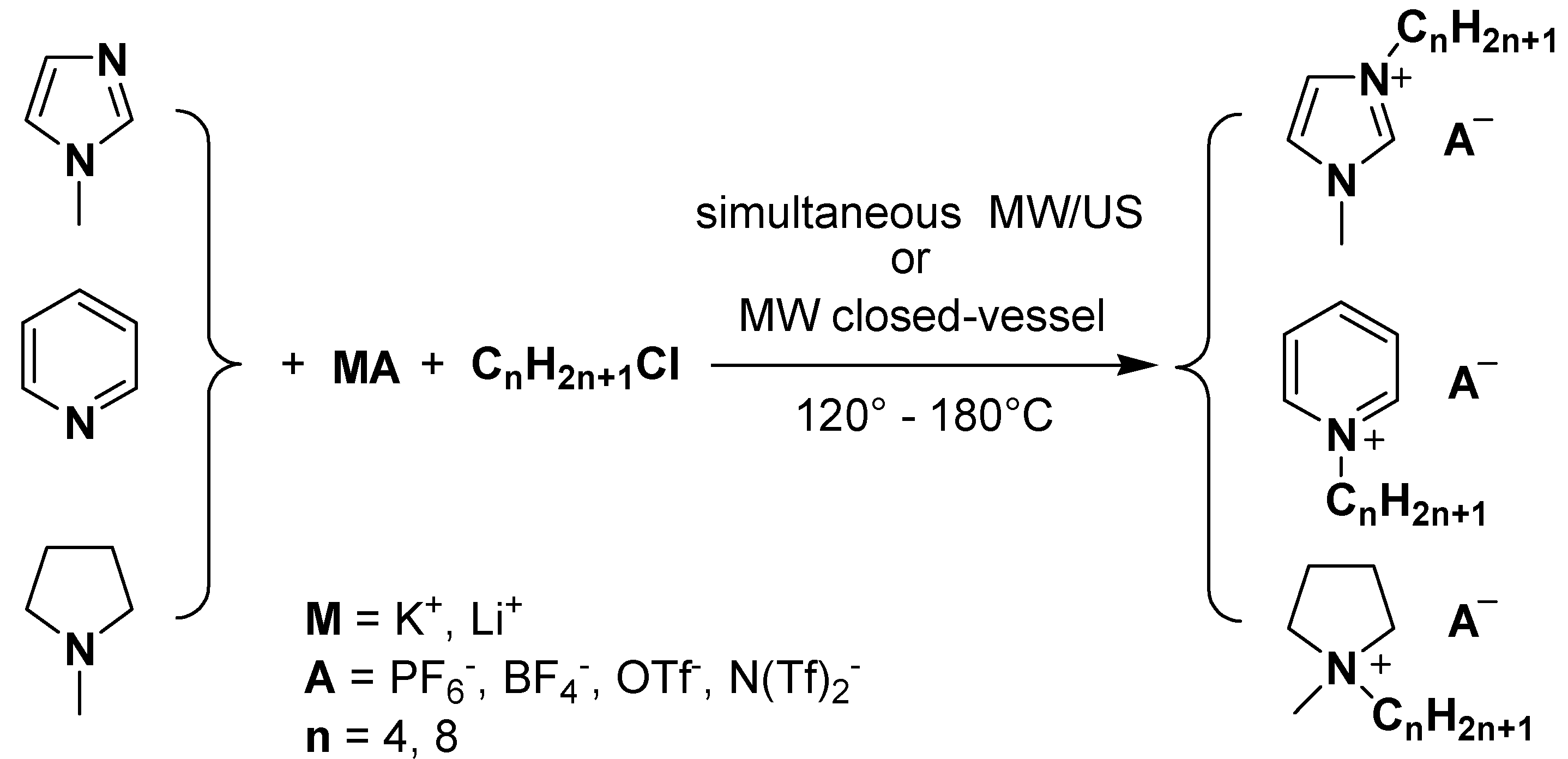
| Entry | Heterocycle* | Alkyl halide | Irradiation time (min) (1 or 2 steps) | MW power (W) | US power (W) | Temperature (°C) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | 1-mim | 1-ClC4 | **60/240 | - | - | 140/180 | 95 |
| 2 | 1-mim | 1-ClC4 | 150 | 120*** | 30 | 75 | <5 |
| 3 | 1-mim | 1-ClC4 | 10/30 | 85 | - | 120/180 | 98 |
| 4 | pyr | 1-ClC8 | **60/240 | - | - | 140/180 | 80 |
| 5 | pyr | 1-ClC8 | 180 | 60*** | 25 | 100 | <5 |
| 6 | pyr | 1-ClC8 | 10/30 | 85 | - | 120/180 | 85 |
| 7 | 1-mpyr | 1-ClC8 | **60/240 | - | - | 140/180 | 88 |
| 8 | 1-mpyr | 1-ClC8 | 10/60 | 40 | - | 80/120 | 0 |
| 9 | 1-mpyr | 1-ClC8 | 15/50 | 80 | - | 120/160 | 20 |
| 10 | 1-mpyr | 1-ClC8 | 15/50 | 90 | - | 120/180 | 99 |
| Entry | Heterocycle** | Alkyl halide | Anion source | Irradiation steps (min) | MW power (W) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 1-mim | 1-ClC4 | KPF6 | 15/30 | 60 | 90 |
| 2 | 1-mim | 1-ClC4 | KBF4 | 10/30 | 55 | 72 |
| 3 | 1-mim | 1-ClC4 | KOTf | 15/40 | 65 | 75 |
| 4 | 1-mim | 1-ClC4 | LiN(Tf)2 | 10/30 | 65 | 62 |
| 5 | pyr | 1-ClC8 | KPF6 | 15/50 | 65 | 83 |
| 6 | pyr | 1-ClC8 | KBF4 | 10/40 | 60 | 68 |
| 7 | pyr | 1-ClC8 | KOTf | 10/50 | 55 | 60 |
| 8 | pyr | 1-ClC8 | LiN(Tf)2 | 15/50 | 65 | traces |
| 9 | 1-mpyr | 1-ClC8 | KPF6 | 15/35 | 95 | 85 |
| 10 | 1-mpyr | 1-ClC8 | KBF4 | 10/45 | 113 | 81 |
| 11 | 1-mpyr | 1-ClC8 | KOTf | 10/45 | 104 | 79 |
Conclusions
Experimental
General
General procedure
Acknowledgements
References
- Wilkes, J.S. Introduction. In Ionic Liquids in Synthesis; Wassercheid, P., Welton, T., Eds.; Wiley-VCH: Weiheim, Germany, 2002; p. 1. [Google Scholar]
- Welton, T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2083. [Google Scholar]
- Lévêque, J.-M.; Estager, J.; Draye, M.; Boffa, L.; Cravotto, G.; Bonrath, W. Synthesis of ionic liquids using non-conventional activation methods: An Overview. Monatshe. Chem. 2007, 138, 1103–1113. [Google Scholar] [CrossRef]
- Loupy, A. (Ed.) Microwaves in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006.
- Cravotto, G.; Cintas, P. Power ultrasound in organic synthesis: moving cavitational chemistry from academia to innovative and large-scale applications. Chem. Soc. Rev. 2006, 35, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Lévêque, J.-M.; Cravotto, G. Microwaves, Power Ultrasound and Ionic Liquids. A new Synergy in Green Organic Synthesis. Chimia 2006, 60, 313–320. [Google Scholar]
- Cravotto, G.; Cintas, P. The Combined Use of Microwaves and Ultrasound: New Tools in Process Chemistry and Organic Synthesis. Chem. Eur. J. 2007, 13, 1902–1909. [Google Scholar]
- Varma, R.S.; Namboodiri, V.V. An expeditious solvent-free route to ionic liquids using microwaves. Chem. Commun. 2001, 643–644. [Google Scholar] [CrossRef]
- Law, M.C.; Wong, K.Y.; Chan, T.H. Solvent-free route to ionic liquid precursors using a water-moderated microwave process. Green Chem. 2002, 4, 328–330. [Google Scholar] [CrossRef]
- Khadilkar, B.M.; Rebeiro, G.L. Microwave-Assisted Synthesis of Room-Temperature Ionic Liquid Precursor in Closed Vessel. Org. Proc. Res. Dev. 2002, 6, 826–828. [Google Scholar]
- Deetlefs, M.; Seddon, K.R. Improved preparations of ionic liquids using microwave irradiation. Green Chem. 2003, 5, 181–186. [Google Scholar]
- Namboodiri, V.V.; Varma, R.S. Solvent-Free Sonochemical Preparation of Ionic Liquids. Org. Lett. 2002, 4, 3161–3163. [Google Scholar] [CrossRef] [PubMed]
- Lévêque, J.-M.; Luche, J.L.; Pétrier, C.; Roux, R.; Bonrath, W. An improved preparation of ionic liquids by ultrasound. Green Chem. 2002, 4, 357–360. [Google Scholar] [CrossRef]
- Lévêque, J.-M.; Desset, S.; Suptil, J.; Fachinger, C.; Draye, M.; Bonrath, W.; Cravotto, G. A general ultrasound-assisted access to room temperature ionic liquids. Ultrason. Sonochem. 2006, 13, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Estager, J.; Lévêque, J.-M.; Cravotto, G.; Boffa, L.; Bonrath, W. One-pot and Solventless Synthesis of Ionic Liquids under Ultrasonic Irradiation. Synlett 2007, 2065–2068. [Google Scholar]
- Cravotto, G.; Lévêque, J.-M.; Estager, J.; Draye, M.; Boffa, L.; Bonrath, W. Solvent-free one-pot synthesis of ionic liquids using ultrasound irradiation. US Patent Appl. 60/880,011; filed by DSM Nutritional Products Ltd.,
- Cravotto, G.; Boffa, L.; Lévêque, J.-M.; Estager, J.; Draye, M.; Bonrath, W. A Speedy One-Pot Synthesis of Ionic Liquids under Microwave/Ultrasound Irradiation. Austr. J. Chem. 2007, 60, 946–950. [Google Scholar] [CrossRef]
- Palmisano, G.; Tagliapietra, S.; Barge, A.; Binello, A.; Boffa, L.; Cravotto, G. Efficient Regioselective Opening of Epoxides by Nucleophiles in Water under Simultaneous Ultrasound/Microwave Irradiation. Synlett 2007, 13, 2041–2044. [Google Scholar]
- Cassol, C.C.; Ebeling, G.; Ferrera, B.; Dupont, J. A Simple and Practical Method for the Preparation and Purity Determination of Halide-Free Imidazolium Ionic Liquids. Adv. Synth. Catal. 2006, 348, 243–248. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all the compounds described herein are available from the authors.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Cravotto, G.; Gaudino, E.C.; Boffa, L.; Lévêque, J.-M.; Estager, J.; Bonrath, W. Preparation of Second Generation Ionic Liquids by Efficient Solvent-Free Alkylation of N-Heterocycles with Chloroalkanes. Molecules 2008, 13, 149-156. https://doi.org/10.3390/molecules13010149
Cravotto G, Gaudino EC, Boffa L, Lévêque J-M, Estager J, Bonrath W. Preparation of Second Generation Ionic Liquids by Efficient Solvent-Free Alkylation of N-Heterocycles with Chloroalkanes. Molecules. 2008; 13(1):149-156. https://doi.org/10.3390/molecules13010149
Chicago/Turabian StyleCravotto, Giancarlo, Emanuela Calcio Gaudino, Luisa Boffa, Jean-Marc Lévêque, Julien Estager, and Werner Bonrath. 2008. "Preparation of Second Generation Ionic Liquids by Efficient Solvent-Free Alkylation of N-Heterocycles with Chloroalkanes" Molecules 13, no. 1: 149-156. https://doi.org/10.3390/molecules13010149