Antiparkinson Prodrugs


Department of Drug Sciences, “G. d'Annunzio” University, School of Pharmacy, Via dei Vestini 31, 66100 Chieti, Italy
*
Author to whom correspondence should be addressed.
Molecules 2008, 13(1), 46-68; https://doi.org/10.3390/molecules13010046
Submission received: 4 December 2007
/
Revised: 11 January 2008
/
Accepted: 11 January 2008
/
Published: 16 January 2008
(This article belongs to the Section Medicinal Chemistry)
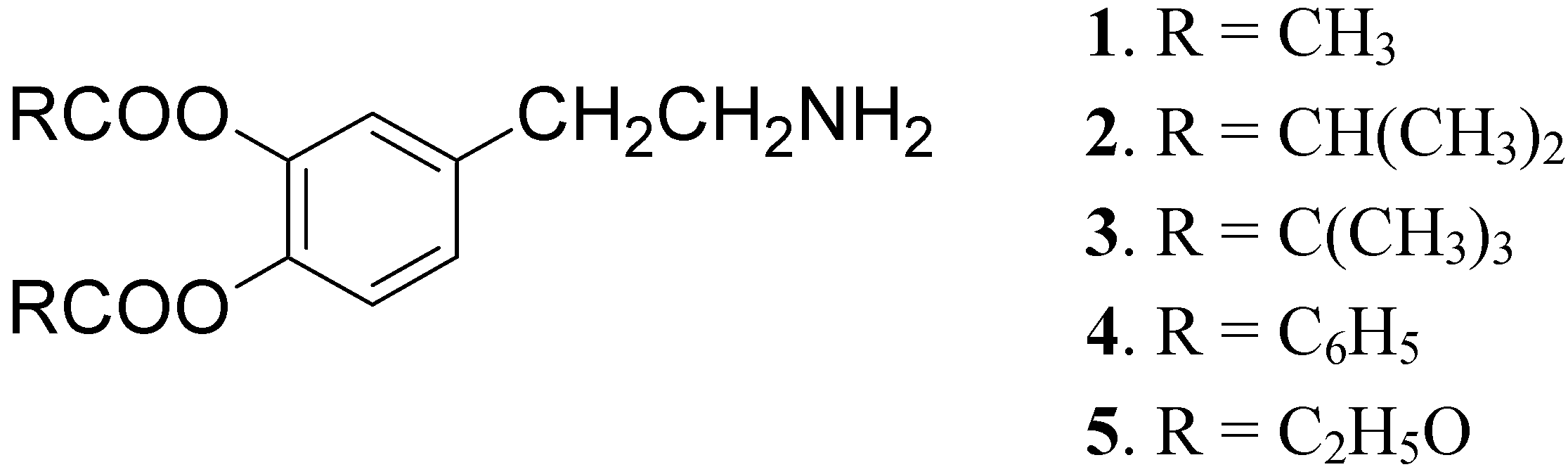{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
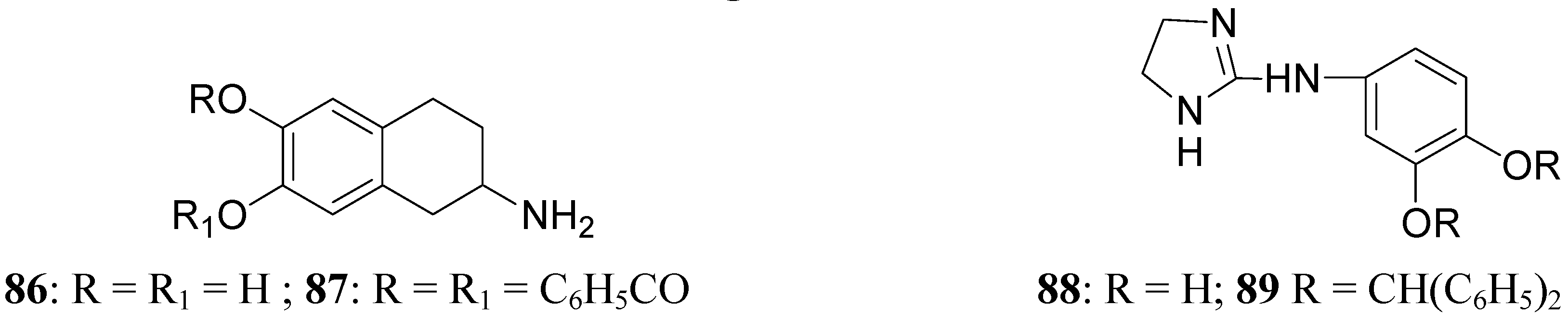{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Parkinson’s disease (PD) is a progressive, neurodegenerative disorder which involves the loss of dopaminergic neurons of the substantia nigra pars compacta. Current therapy is essentially symptomatic, and L-Dopa (LD), the direct precursor of dopamine (DA), is the treatment of choice in more advanced stages of the disease. Substitution therapy with LD is, however, associated with a number of acute problems. The peripheral conversion of LD by amino acid decarboxylase (AADC) to DA is responsible for the typical gastrointestinal (nausea, emesis) and cardiovascular (arrhythmia, hypotension) side effects. To minimize the conversion to DA outside the central nervous system (CNS) LD is usually given in combination with peripheral inhibitors of AADC (carbidopa and benserazide). In spite of that, other central nervous side effects such as dyskinesia, on-off phenomenon and end-of-dose deterioration still remain. The main factors responsible for the poor bioavailability and the wide range of inter- and intra-patient variations of plasma levels are the drug’s physical-chemical properties: low water and lipid solubility, resulting in unfavourable partition, and the high susceptibility to chemical and enzymatic degradation. In order to improve the bioavailability, the prodrug approach appeared to be the most promising and some LD prodrugs have been prepared in an effort to solve these problems. We report here a review of progress in antiparkinson prodrugs, focusing on chemical structures mainly related to LD, DA and dopaminergic agonists.
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder, characterized by a specific loss of dopamine (DA) neurons in the substantia nigra pars compacta (SNpc) and affecting about 1% of the population above the age of 60 [1]. The major clinical symptoms of PD include bradykinesia, postural instability, rigidity and tremor; furthermore, a number of patients also suffer from anxiety, depression, autonomic disturbances and dementia. These are the result of the degeneration of DA-containing neurons in the SNpc, which causes a consequent reduction of DA levels in the striatum. However, PD is more than a disorder of DA neurotransmitter system; additional neuronal fields and neurotransmitter systems are also involved, including the amygdala, hippocampus, the autonomic nervous system and the cerebral cortex leading to depletion of noradrenaline, serotonine and acetylcholine. This loss results in symptoms that include cognitive decline, sleep abnormalities and depression, as well as gastrointestinal and genitourinary disturbances [2]. DA deficiency appears to be responsible for the motor deficits of the disorder but PD cannot be treated directly with DA or related catecholamines due to their inability to cross the blood brain barrier (BBB). LD, which could be considered as a prodrug of DA, still remains the most clinically useful drug for treatment of PD [3,4,5]. When administered orally LD is adsorbed by a specific carrier mediate transport system and transported through the BBB where it undergoes decarboxylation to DA within the brain. However, during chronic treatment with LD, a variety of problems may emerge: patients experience a decrease in the duration of drug effect (‘wearing-off’ phenomenon) and, as the number of functioning DA neurons decreases in the central nervous system (CNS), the patient becomes more sensitive to LD plasma level fluctuations (on/off effects). LD is usually administered orally but the clinical response is variable because of its erratic oral absorption and gastrointestinal tract metabolism, so that relatively little arrives in the bloodstream as intact drug. The oral bioavailability of LD alone is estimated to be about 10% and less than 1% of the administered oral dose reaches the brain unchanged [6]. The major peripheral side effects such as cardiac arrhythmias, vomiting and hypotension resulting from the oral administration of LD appear due to the formation of large amounts of DA during first-pass metabolism in the gastrointestinal tract [6,7]. Variability in the degree of this first-pass effect is the main cause of the common difficulty of maintaining an effective therapeutic regimen with LD. Decarboxylase inhibitors are co-administered with LD to decrease its gastrointestinal tract metabolism; the most notable effects of this are enhanced bioavailability, reduction in total daily LD dose and a decrease of peripheral side effects [9,10,11]. However, the on-off fluctuation remains because the oral absorption is still erratic and plasma concentrations still fluctuate [12,13,14]. Intravenous (i.v.) application of LD was found to increase not only the plasma levels but led at the same time to an improvement of the kinetic behaviour: the duration of mobility was enhanced and the frequency of fluctuation was reduced with significant mobility improvement [15]. Furthermore i.v. coadministration of LD with carbidopa, a decarboxylase inhibitor, resulted in significant increases in both the area under the plasma (AUC) LD concentration versus time profile and the plasma LD half-life [16]. Since i.v. infusion is inconvenient for routine clinical use, several approaches have been attempted to enhance the bioavailability and minimize the side effects of LD but it has not been easy to produce a controlled release preparation of LD capable of more effectively maintaining adequate plasma levels [17,18,19,20]. For this reason, attempts were made to ameliorate the dissolution, absorption and metabolism problems of LD and great interest has been addressed toward the production of prodrugs with improved pharmacological and pharmacokinetic properties compared with LD. Several derivatives were studied with the aim of enhancing its chemical stability, water or lipid solubility, as well as diminishing the susceptibility to enzymatic degradation [21]. We report here the progresses in antiparkinson prodrugs, focusing on chemical structures mainly related to LD, DA and dopaminergic agonists.
Dopamine prodrugs
DA is synthesized in sympathetic neurons and chromaffin cells, the biosynthesis starting from the amino acid tyrosine in a two step process. The DA precursor is LD which, in turn, is formed from tyrosine (Scheme 1).
Scheme 1.
Dopamine biosynthesis.
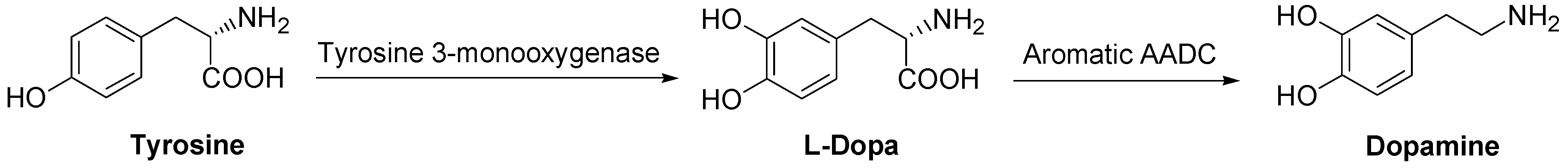
DA is subject to extensive hepatic metabolism following oral administration. Due to the presence of the catechol moiety it is essentially completely ionized at physiological pH, which results in its poor permeation across the BBB and other cell membranes. For these reasons the use of DA itself in PD treatment is precluded [22]. To overcome these problems a series of lipophilic 3,4-O-diesters 1-5 (Figure 1) were proposed as latent lipophilic derivatives of DA usable in therapy of parkinsonism, hypertension and renal failure [23,24].
Figure 1.
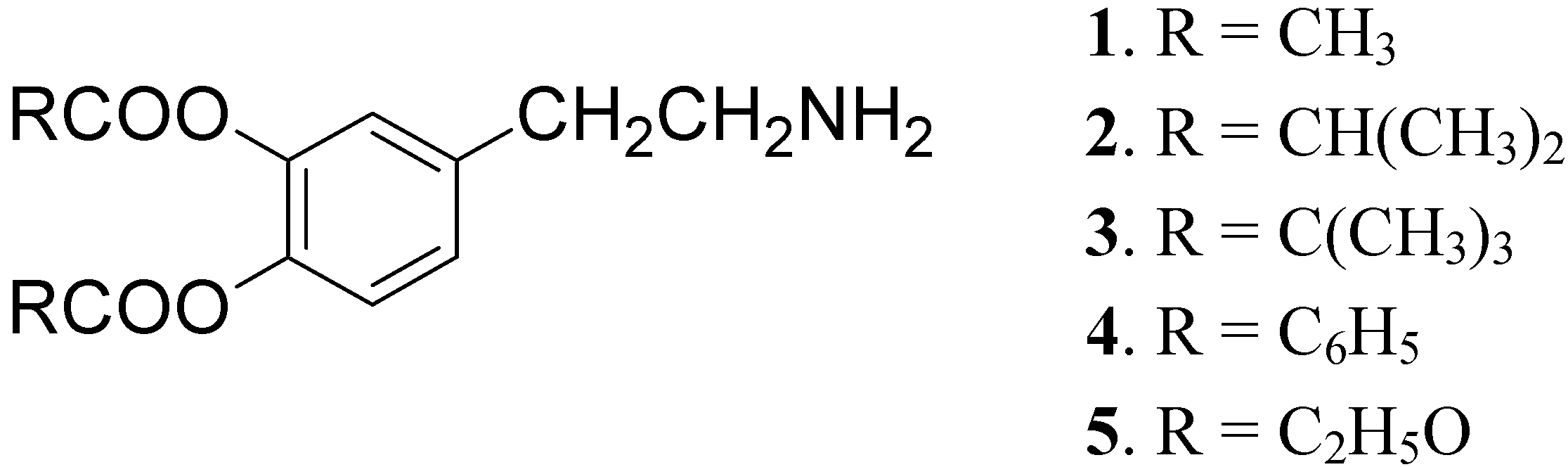
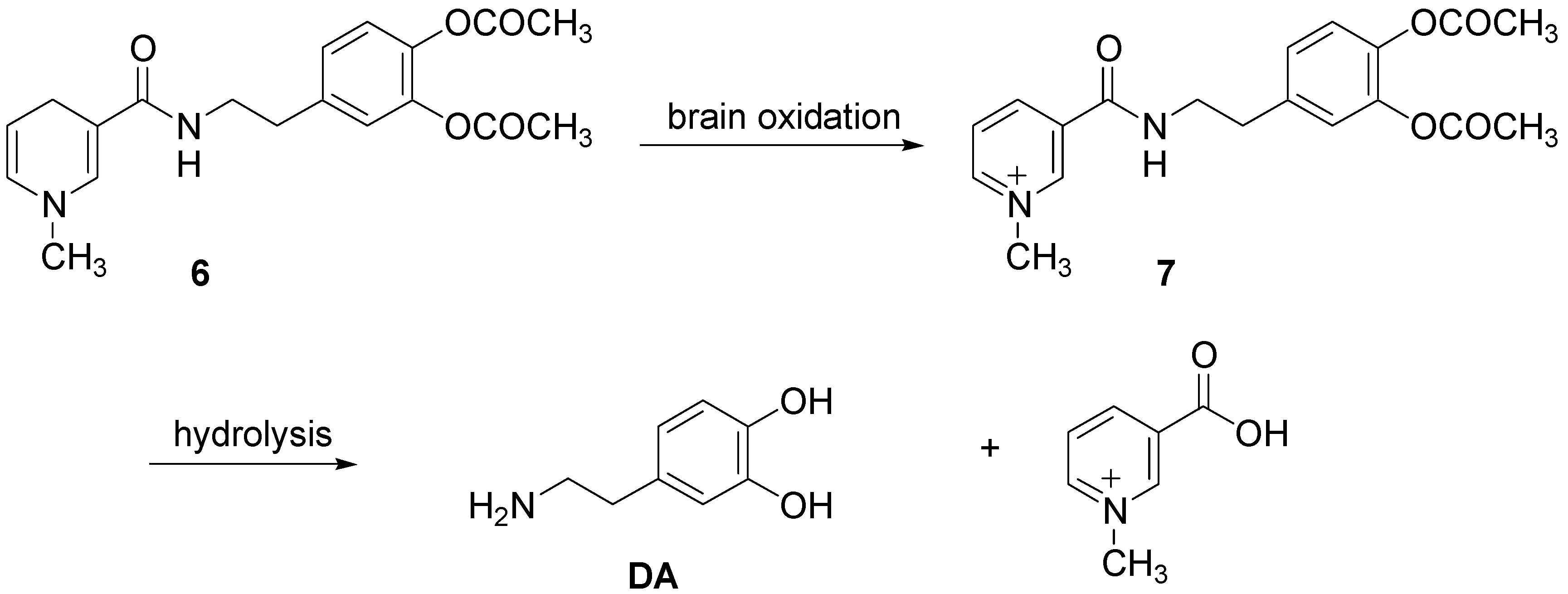
Furthermore Bodor et al. [4] proposed the application of chemical delivery system based on a pyridinium/dihydropyridine redox carrier for brain specific delivery of DA [25]. This redox delivery system was successfully applied for brain specific delivery of DA by using the dihydro derivative 6 to deliver the quaternary precursor 7, which while “locked in” the brain provided a sustained release form of DA (Scheme 2).
“In vivo” administration of the catechol protected DA resulted in brain-specific, high and sustained concentration of the 1-methyl-3-[N-(-(β-3,4-dihydroxyphenyl)ethyl)carbamoil]pyridinium salt (7), the direct DA precursor, locked in the brain for many hours, while systemic concentration decreased fast, with a t½ of less than 30 min. Significant dopaminergic activity was observed in the brain, which was sustained for hours.
Scheme 2.
DA delivery from pyridinium/dihydropyridine redox carrier system.

During the last years a new approach to deliver DA into the CNS, based on the use of D-glucose as transportable agent, has been studied. Glycosyl-DA derivatives bearing the sugar moiety linked to either the amino group or the catechol ring of DA through amide, ester or glycosidic bonds were synthesised as potential antiparkinsonian agents [26,27,28].
Figure 2.
Glycosyl DA derivatives.
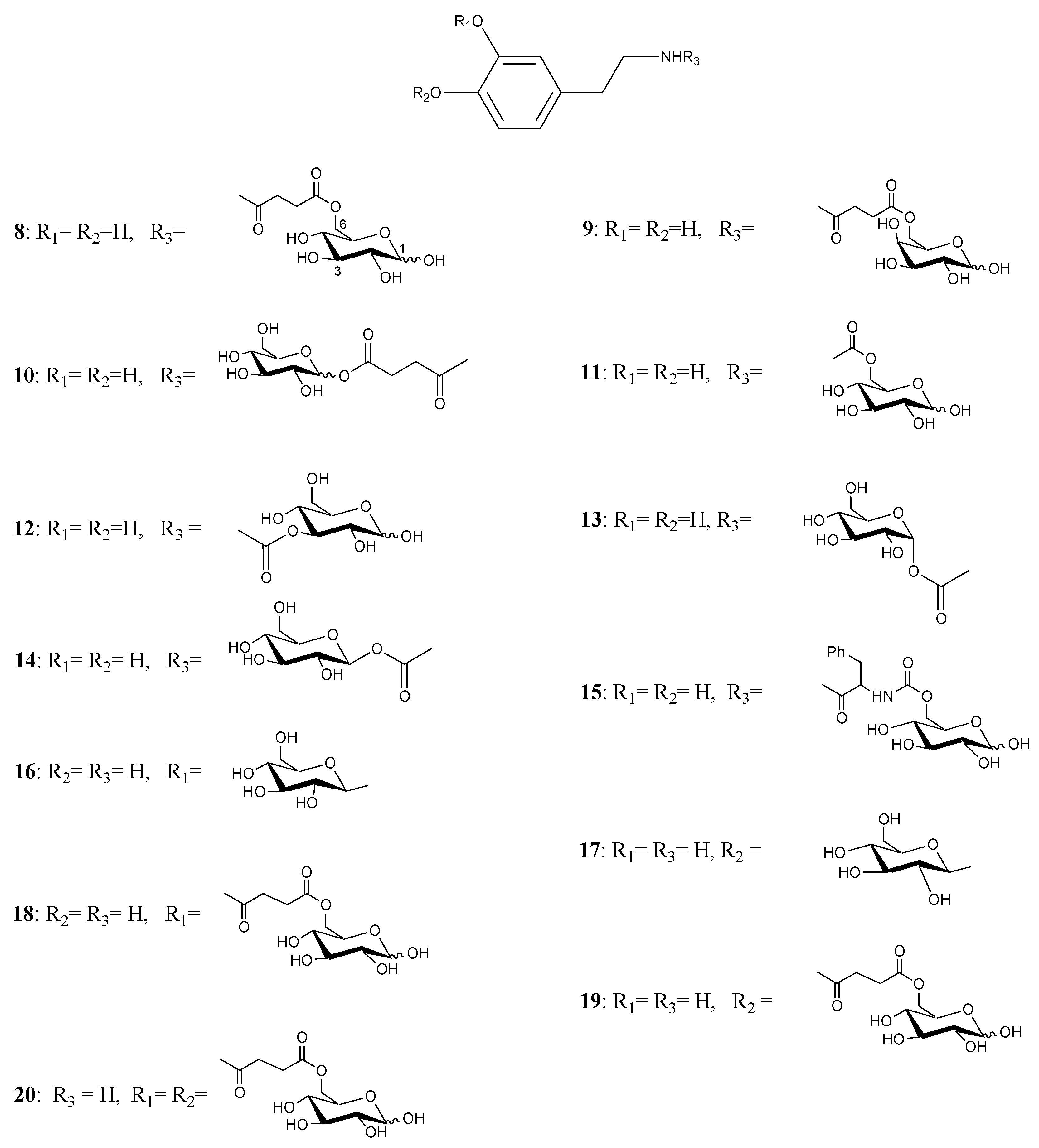
These DA-derivatives might be able to penetrate the BBB, by making use of the specific glucose transport system named GLUT-1 [29,30]. This proteic transporter is located in the membrane of brain capillary endothelial cells composing the BBB. Thus, one could envisage a new approach to delivering DA into the CNS by linking the neurotransmitter to a sugar molecule so that the resulting glycoconjugate may cross the BBB using this carrier-mediated transport [31]. Upon reaching the CNS, the prodrug should be enzymatically cleaved to release the active compound.
Fernandez et al. [26,27] described the synthesis and biological activities of glycoconjugates in which DA was attached to different positions of the glucose molecule using a variety of linkages susceptible of enzymatic hydrolysis. A series of compounds had the amino group of DA linked to C-6, C-3 and C-1 of the sugar through a succinyl linker, carbamate bond, glycosidic and ester bonds (compounds 8-20, Figure 2). The affinity of the glycoconjugates for the glucose carrier GLUT-1 using human erythrocytes was also studied. Several compounds released DA when incubated with brain extracts and the rate was related to the bond linking DA with glucose. The glycosyl conjugates substituted at the C-6 position of the sugar were more potent inhibitors of glucose transport when compared to C-1 and C-3 substituted derivatives. From the studied compounds the carbamate derivatives 11, 13 and 14 were the prodrugs of choice, in particular compound 11, which showed the best affinity for GLUT-1, even higher than glucose itself.
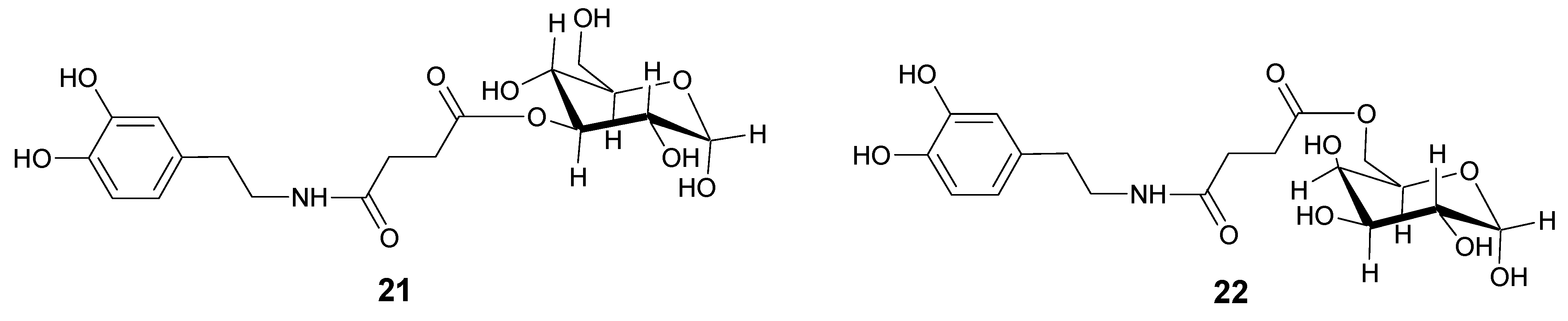
In order to overcome the problem of BBB low permeability of DA, two new glycosyl derivatives of DA were synthesized (Figure 3) [28]. The new esters link DA by a succinyl spacer to C-3 position of glucose (21) and to C-6 of galactose (22). Pharmacological studies showed that compounds 21 and 22 were equiactive in reversing reserpine-induced hypolocomotion in rats, and both were more active than LD. The minimal vascular effects of these derivatives allow underlining the possibility to use the glycosuccinyl DA derivatives in pathologies such as PD.
Figure 3.
Chemical structures of glycosuccinyl-derivatives of DA.
L-Dopa prodrugs
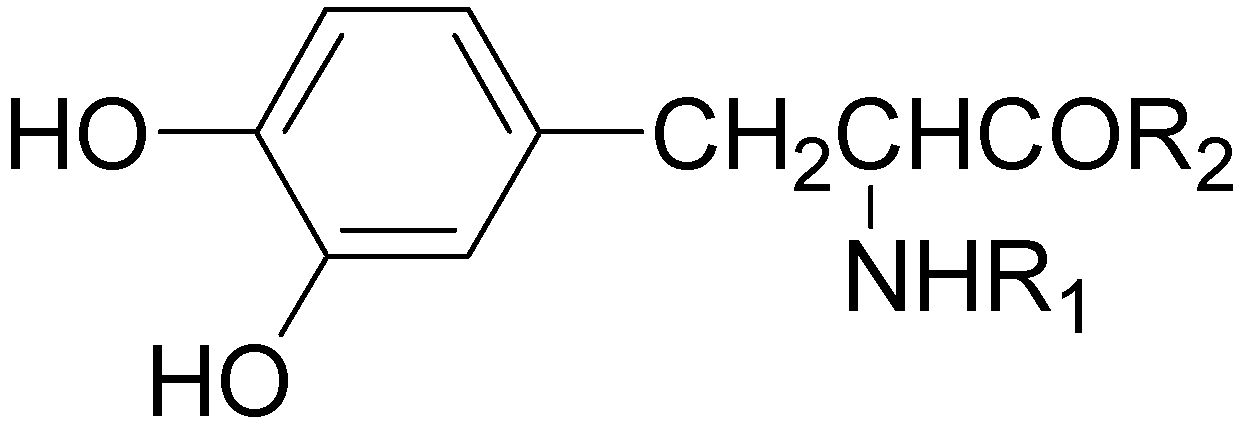
LD is the key compound in the treatment of PD, acting as a precursor of neurotransmitter DA [32,33]. Despite the good symptoms improvement during the LD therapy in PD patients, the drug however has a number of disadvantages. This has prompted the search for compounds which could replace LD in the treatment of this neurodegenerative disease. LD offers a numbers of possibilities for derivative formation containing biodegradable linkage and useful for prodrug approach. Starting from these considerations a series of di- and tripeptides 23-46 containing LD were proposed and examined for their antiparkinson activity in mice by Felix et al. [34] (Figure 4).
Figure 4.
Chemical structures of di- and tripeptides of LD.
Some of the peptides were more effective in reversing reserpine-induced catatonia than LD. In general, the peptides were relatively non-toxic and resulted in a low degree of stereotypic behaviour than LD. More recently a tripeptide mimetic DA prodrug in which D-p-hydroxyphenylglycine-L-proline was attached to LD (47, Figure 5) was synthesized as delivery system for improving the oral absorption [35].
Figure 5.
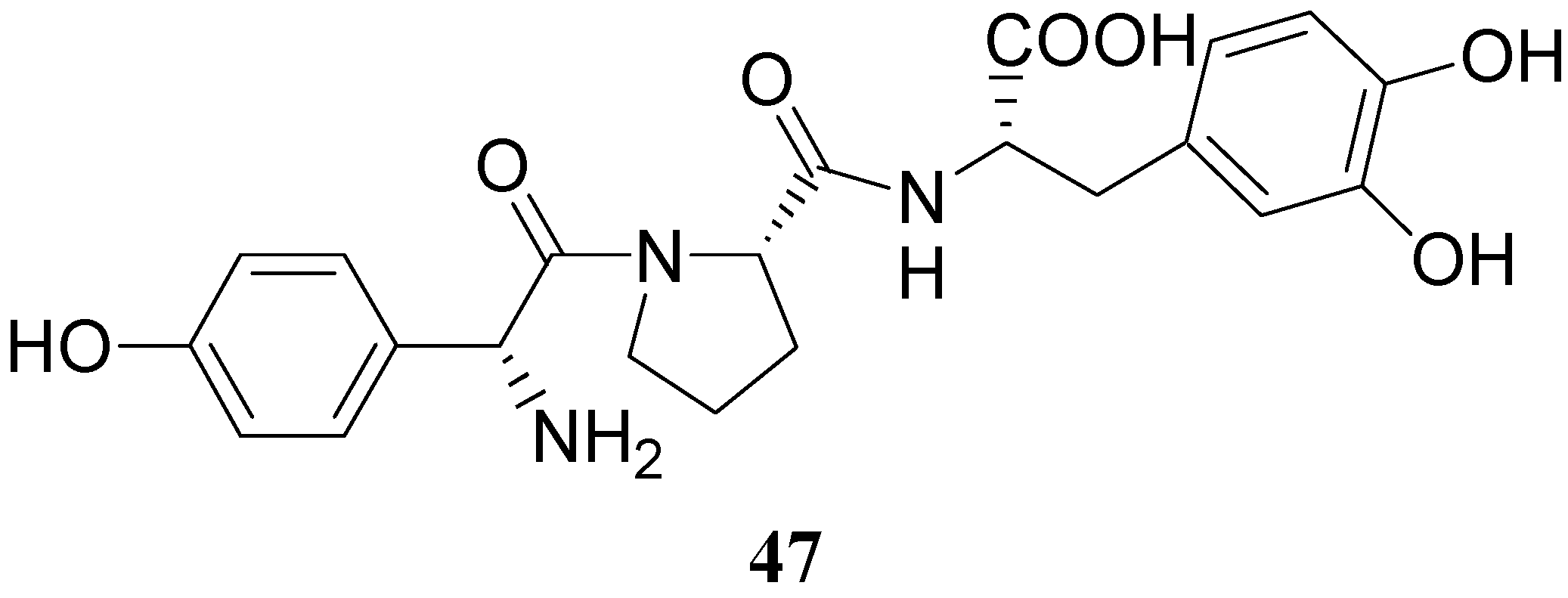
This tripeptide was well absorbed in a perfusion study in rat intestine, devoid of DA-like side effect on isolate muscles and significantly decreased the metamphetamine-induced rotational behaviour in nigrostriatal-lesioned rats, suggesting that it might be an effective agent for PD disease.
During the last 30 years various classes of transient derivatives of LD have been synthesized, systematically protecting on or more of the main sites of metabolism in the molecule: the carboxy function, the amino and/or the catechol system. The derivatives evaluated include carboxy ester, phenol esters, amides, and various combinations of these functions [5,19,36,37,38,39,40,41,42]. In particular compounds 48-57 (Figure 6) seems to effectively prevent the metabolism of LD prior to or during the absorption process, resulting in a significantly better bioavailability of the drug.
Figure 6.
Chemical structures of LD esters.
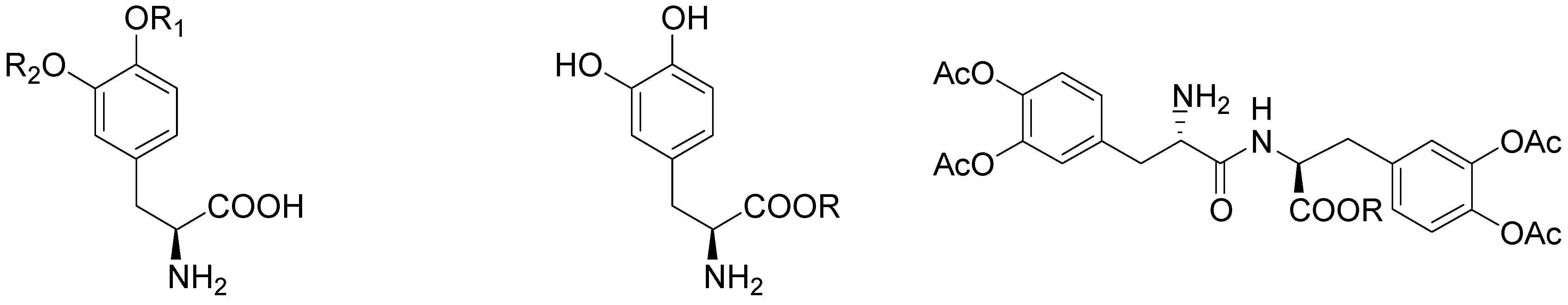
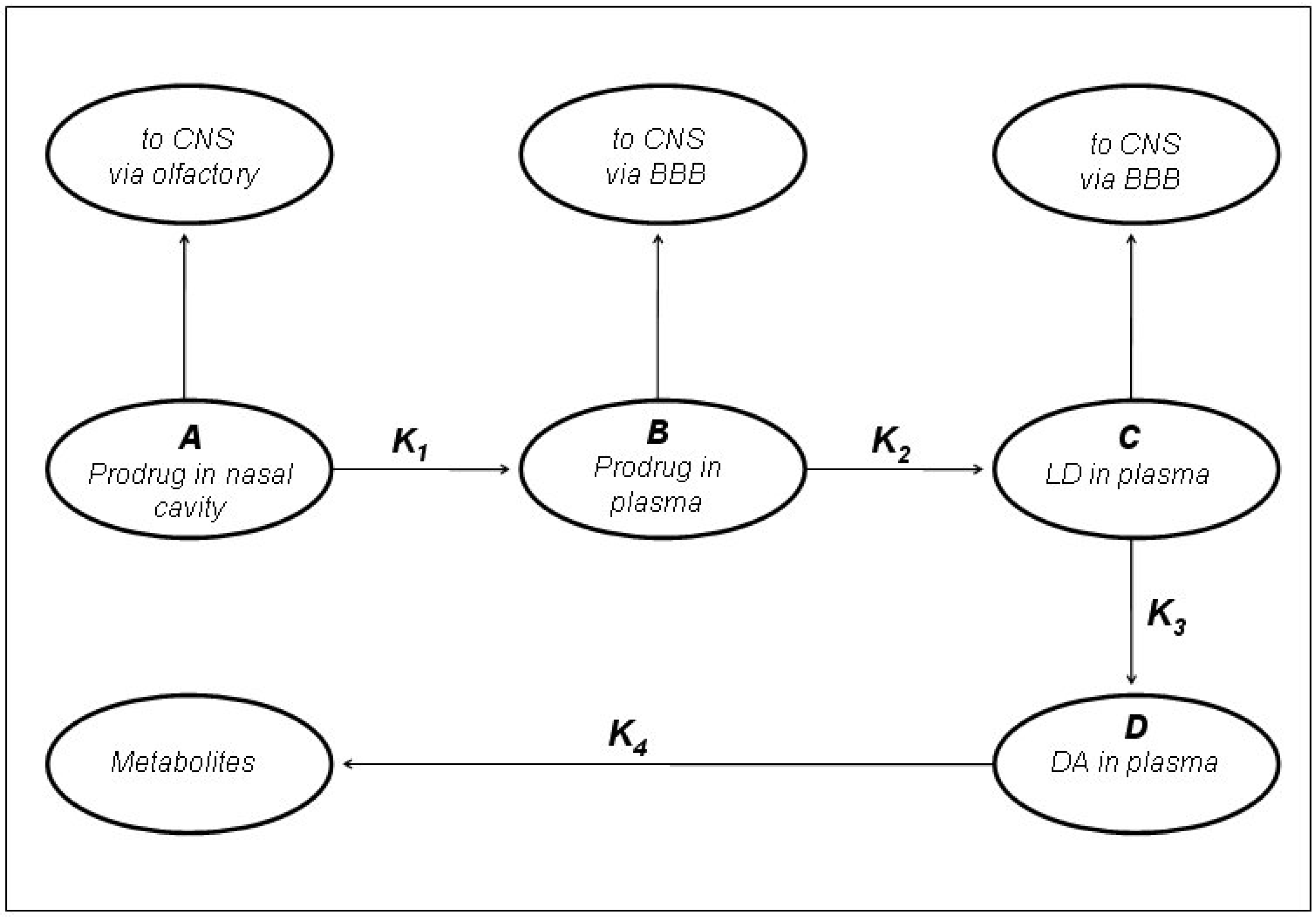
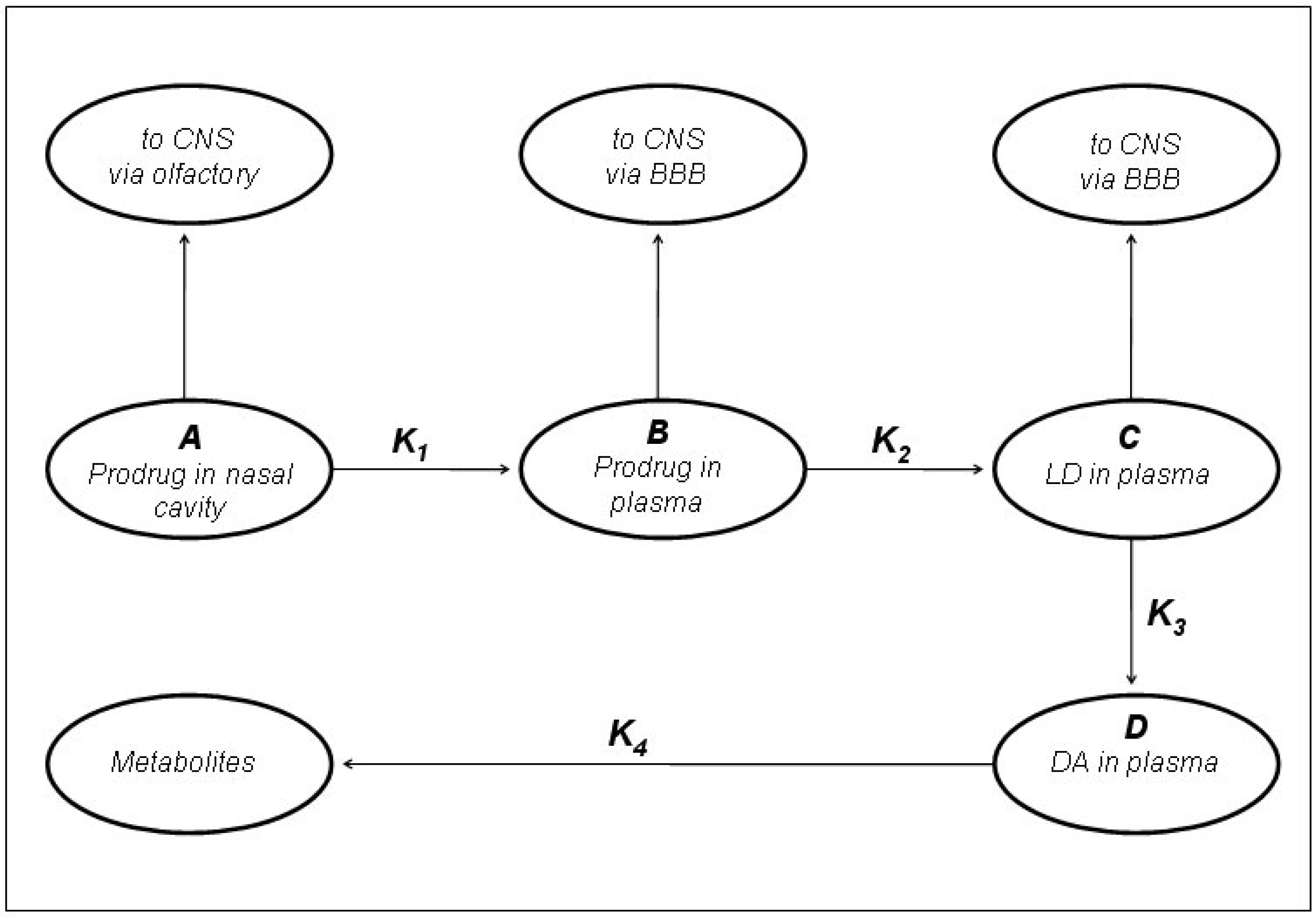
In preliminary “in vivo” studies on dogs compound 54 showed up to 2.5-fold increase in LD blood levels [19]. The ester prodrugs were effective as LD itself in producing motor activity but overall none of the compounds was markedly more potent or of long duration than LD itself [40]. The pivaloyl ester of LD (48, NB-355) was proposed in long-lasting LD therapy for the treatment of PD with little concern about toxicity. Following i.v. administration of 48, the elimination half-life and AUC of plasma LD were the same as those observed following LD treatment [41]. The hydrochlorides of compounds 50-54 were proposed as water soluble prodrugs for specific delivery of LD via the nasal route in the treatment of PD. Studied compounds may have therapeutic advantages such as improved bioavailability, decreased side effects, and potentially enhanced CNS delivery; in particular the nasal administration in rats of the butyl ester prodrug of LD 51 resulted in an improved CNS bioavailability compared to that achieved from an equivalent i.v. dose (Figure 7) [5,43].
Figure 7.
Kinetic model of LD prodrugs following the nasal administration as proposed by Kao et al. [5].
Figure 7.
Kinetic model of LD prodrugs following the nasal administration as proposed by Kao et al. [5].
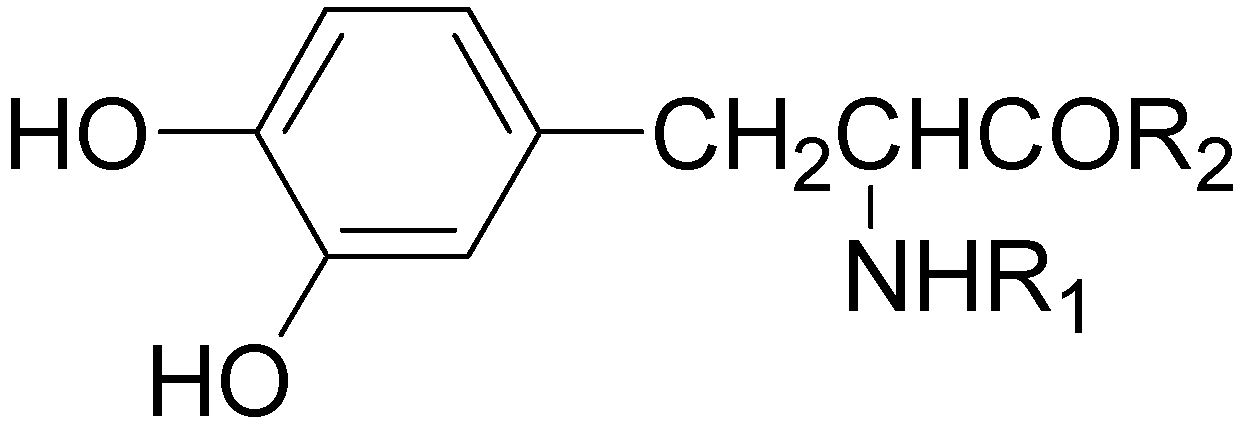
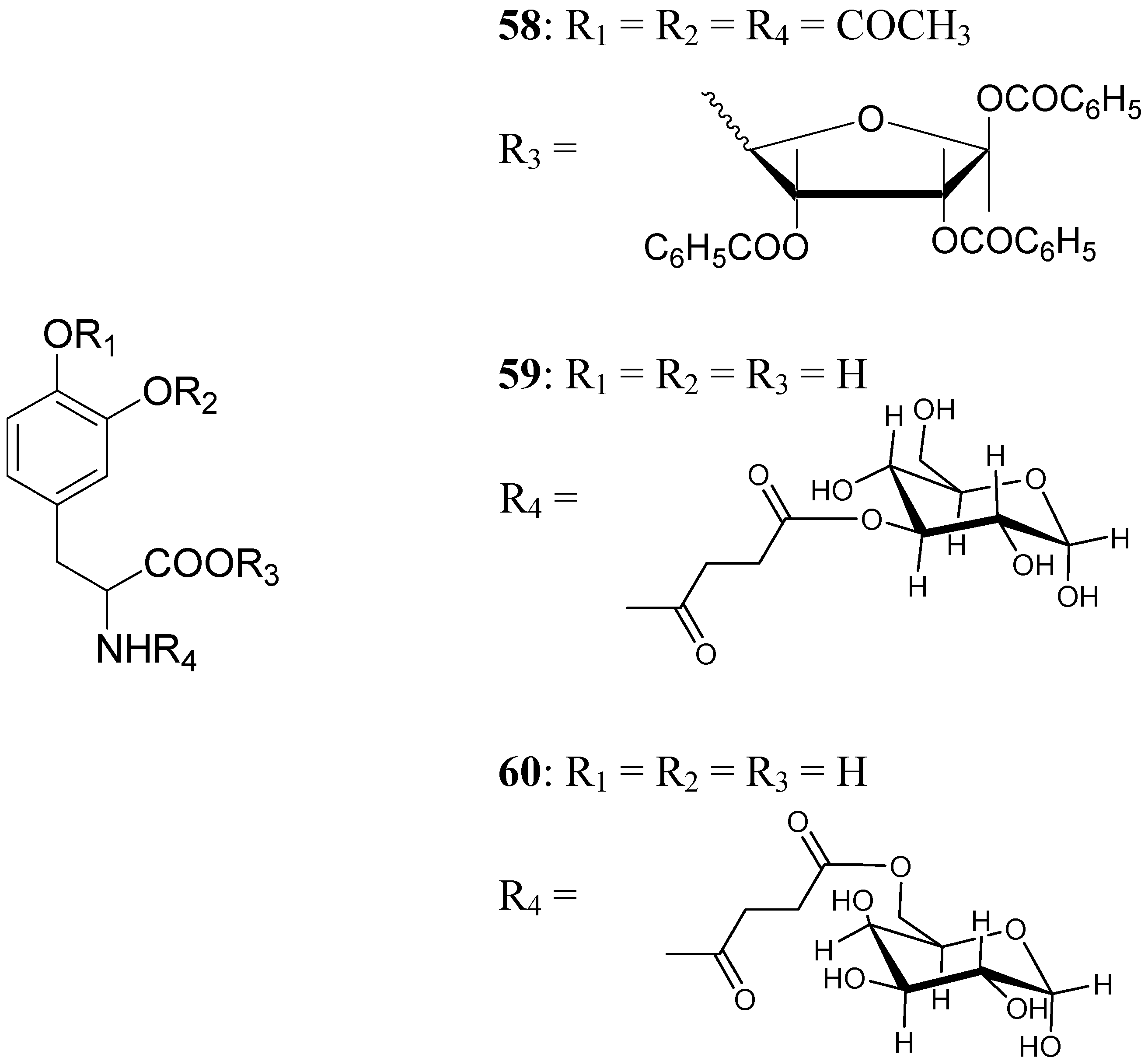
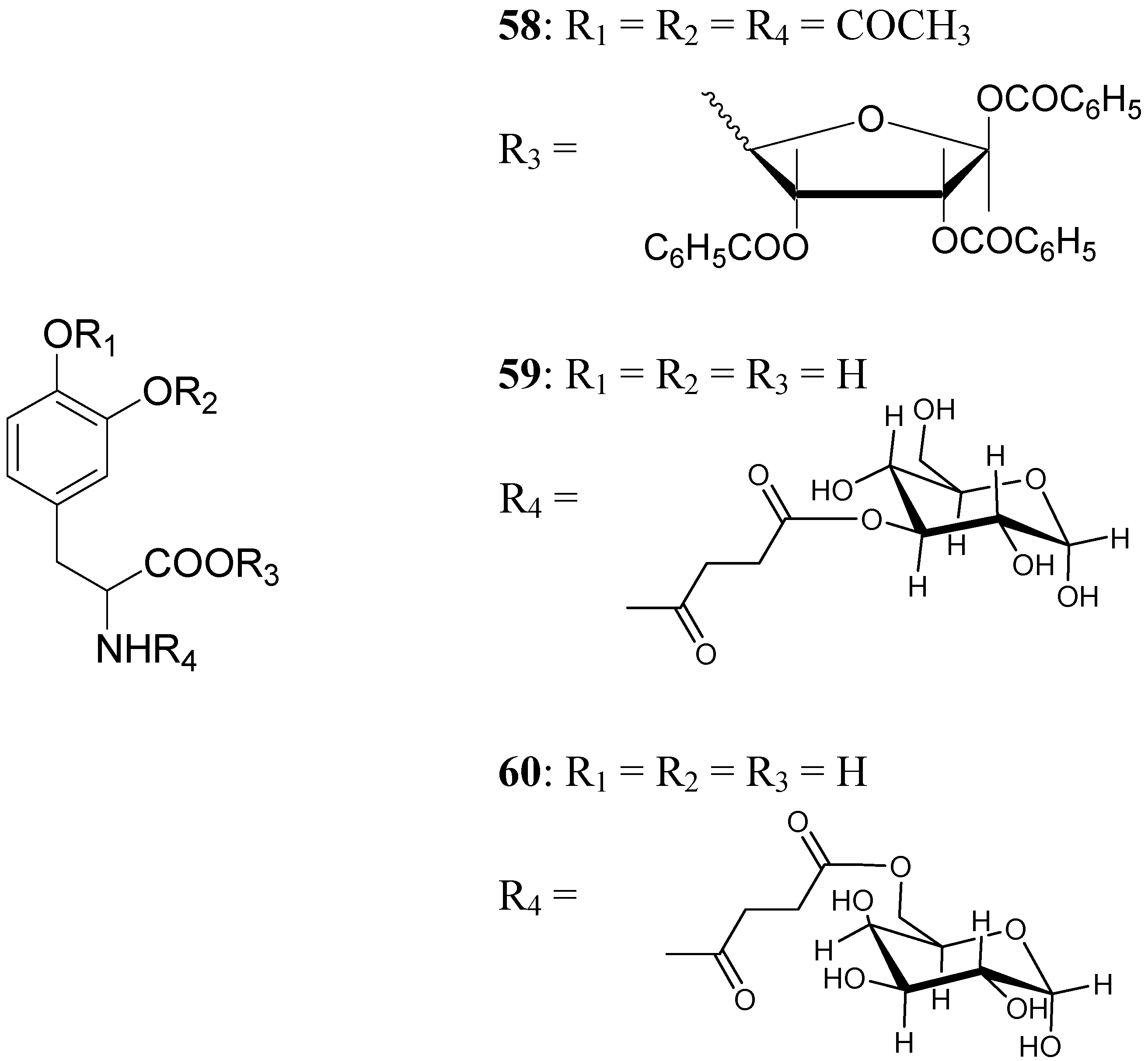
A series of glycosyl derivatives of LD were also proposed as prodrugs (Figure 8). The LD ribosides were prepared by direct condensation between sugar and LD, the latter both free or protected on the amino acyl group (58) [44]. The LD esters with C-3 position of glucose and C-6 position of galactose were synthesized (59 and 60 respectively) [28].
The results obtained indicate that compounds 59 and 60 appeared stable in solution at pH 7.4 and in rat plasma, but showed weak activity when compared with LD in reversing reserpine-induced hypolocomotion in rats. In order to obtain a lipophilic prodrug with enhanced absorption and to protect the LD aminoacidic moiety toward AADC l-3-(3-hydroxy-4-pivaloyloxybenzyl)-2,5-diketomorpholine (61) was synthesized as a LD prodrug [45].
Figure 8.
Chemical structures of glycosyl-derivatives of LD.
In this compound the carboxylic and amino groups were masked in the 2,5-diketomorpholine skeleton as amide and ester groups, respectively. The catechol was protected by the pivaloyl group considering that the NB-355 (48) produces a sustained LD plasma level after oral dosing in rats and dogs [41].
Figure 9.
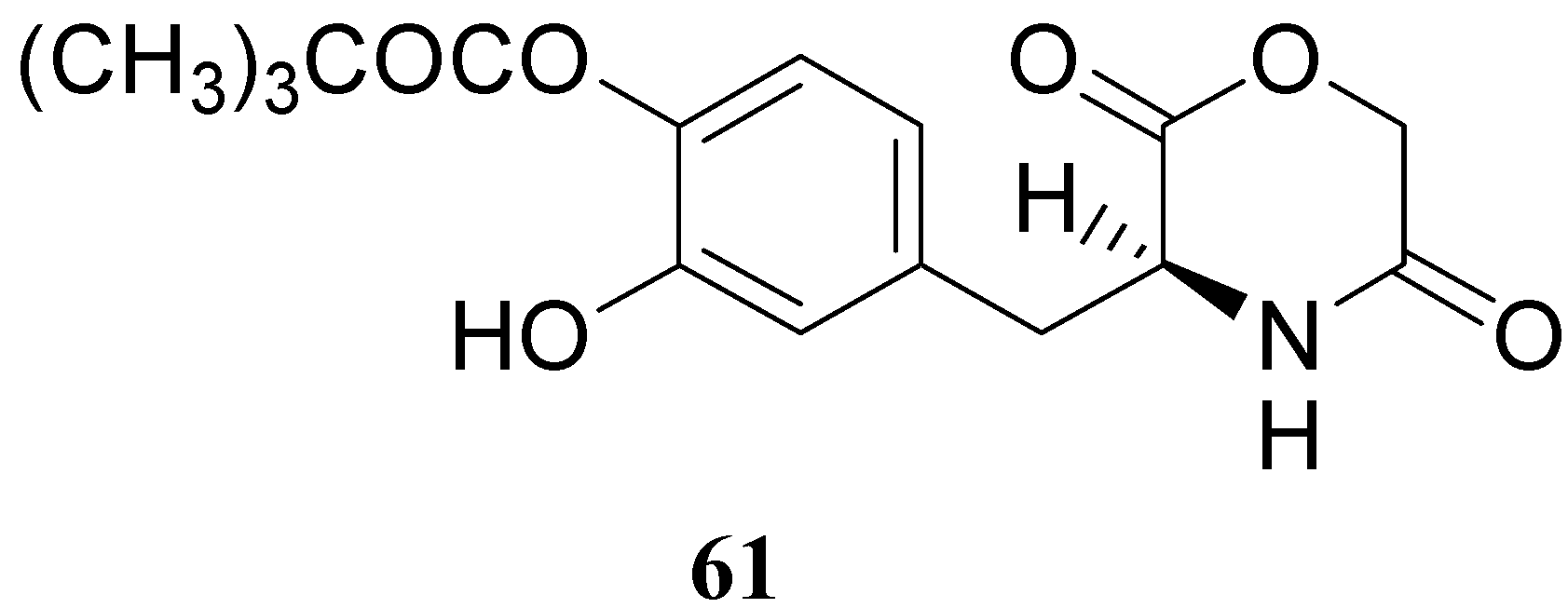
Compound 61 (Figure 9) showed good stability towards gastrointestinal hydrolysis and was able to release LD in human plasma after enzymatic hydrolysis, for these reasons the 2,5-diketomorpholine ring was proposed as useful system in the design of LD prodrugs. Dimeric derivatives have become a common strategy for the production of prodrug forms, in which two identical structural molecules are linked together through a spacer and after administration are metabolized into their two identical active components [46,47,48,49,50]. Starting from these studies a series of dimeric derivatives of LD diacetyl esters 62-69 (Figure 10) were synthesized and evaluated as potential LD prodrugs with improved physicochemical properties; furthermore, to improve the bioavailability of the synthesized dimeric drugs, they were encapsulated in unilamellar liposomes of dimiristoylphosphatidylcholine (DMPC) and cholesterol [21,51,52,53].
Figure 10.
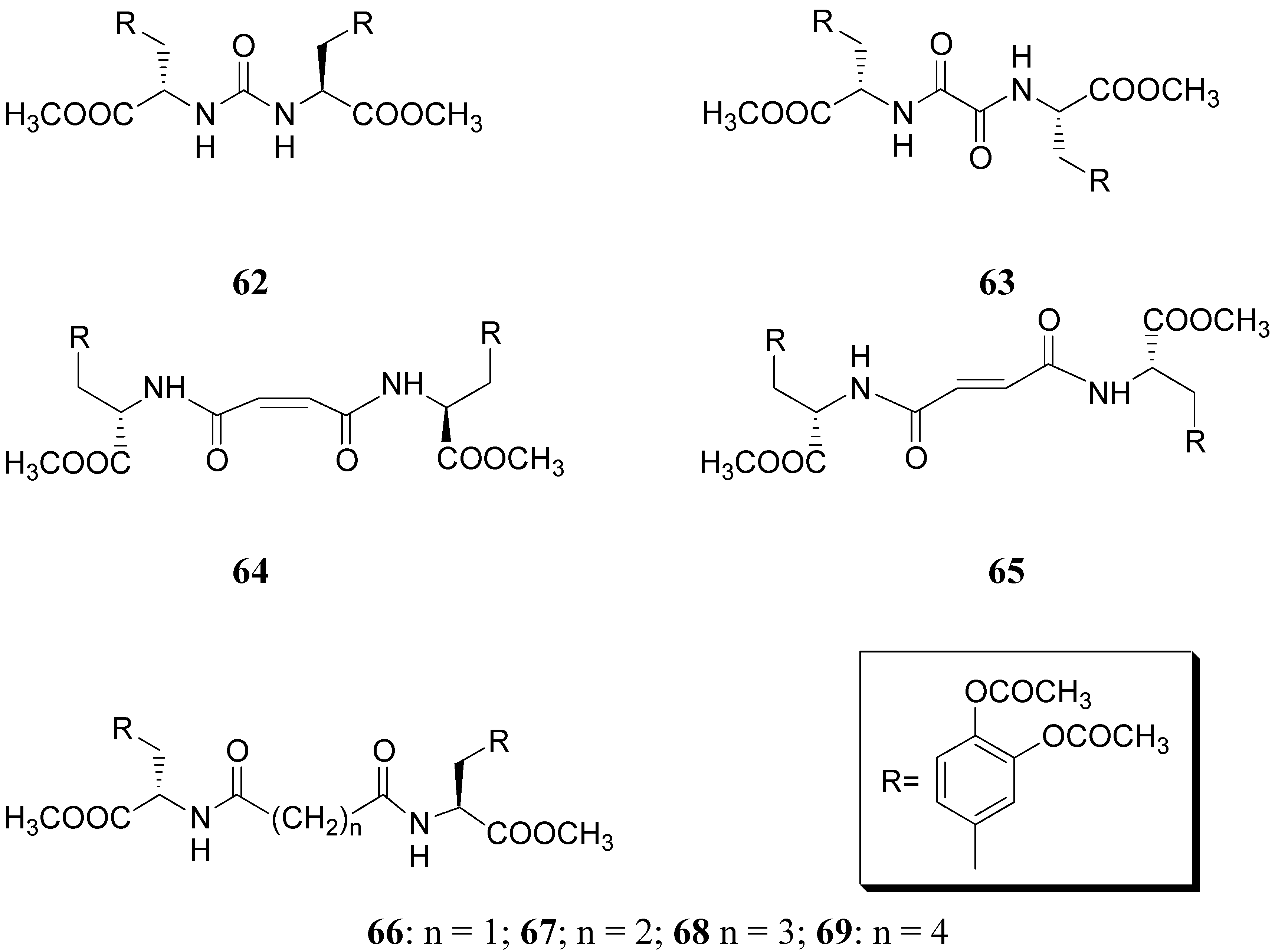
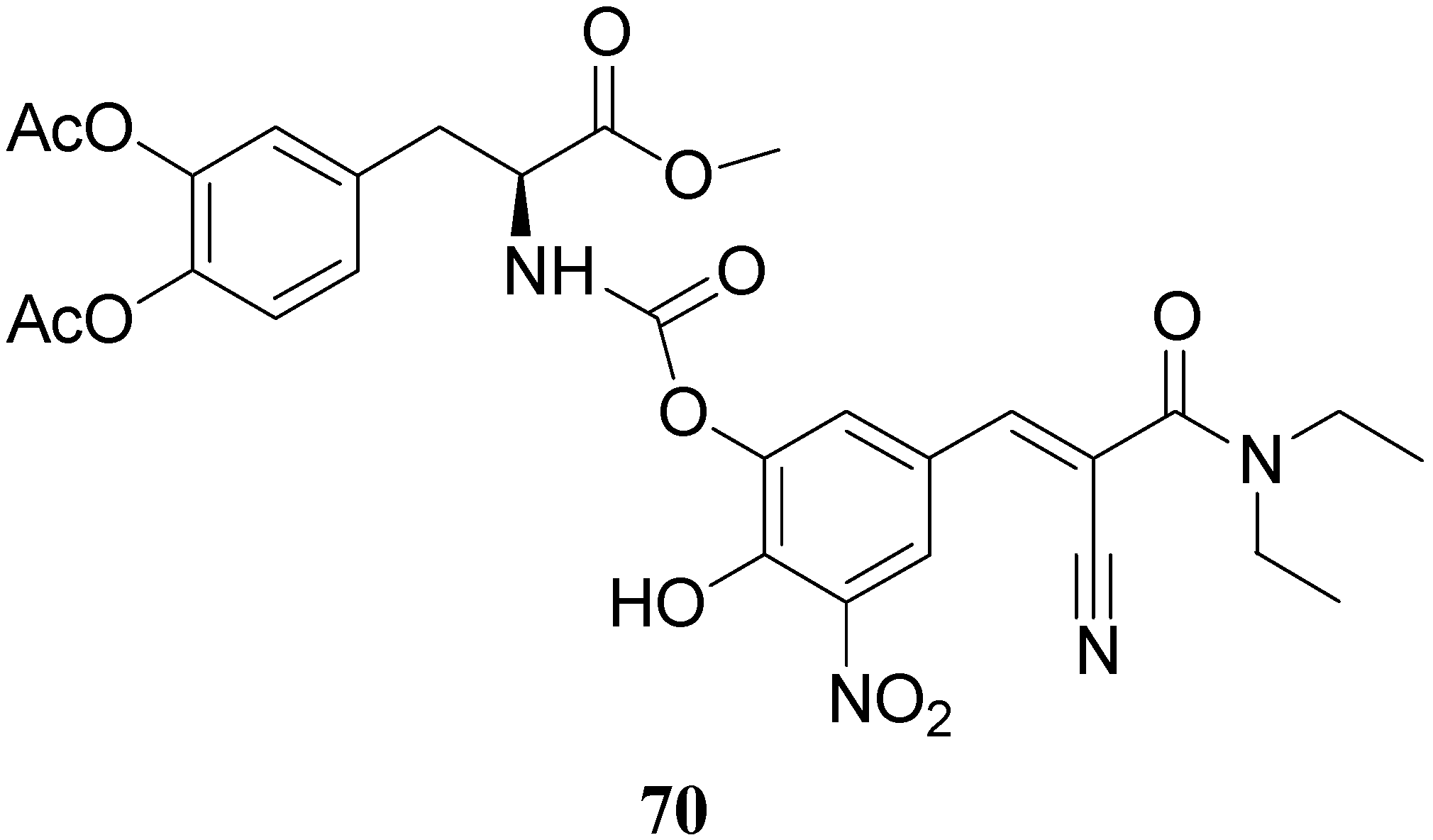
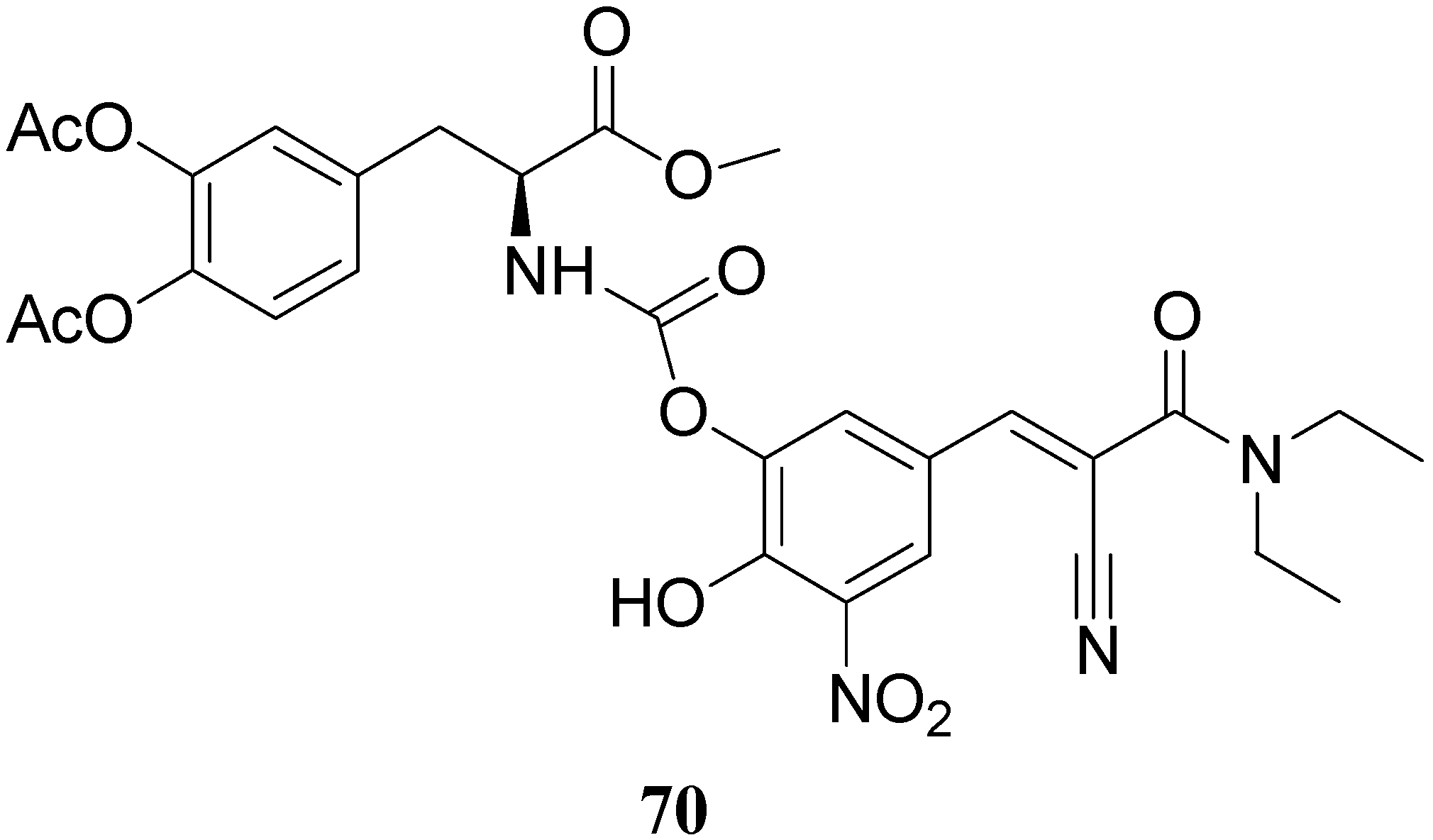
All the new prodrugs showed chemical stability in aqueous buffer solutions (pH 1.3 and 7.4) and a slow release of LD in human plasma was observed. Microdialysis studies in rat striatum after oral administration of prodrugs 62-69 (compared with LD) indicate that the dialisate concentration of DA decreased more slowly using these prodrugs, in particular the compounds 66 and 68 led to a significant increase that could not achieved with LD. Furthermore, neostriatum DA concentration after administration of dimeric derivatives of LD vs. prodrugs in liposomal formulations was compared. Taken together the results suggest that these prodrugs containing liposomes can improve the release of DA in rat brain and demonstrate the potential of these formulations as a method for the controlled delivery of antiparkinson agents [52,53]. During the last few years a codrug approach has been used for delivery LD in CNS. Many diseases are treated by a combination of therapeutic agents that are coadministered in separate dosage forms. When two synergistic drugs are administered individually but simultaneously, they will be transported to the site of action with different efficiencies [54]. However, there are potential advantages in giving codrugs in which two different synergistic drugs are linked together. When it is desirable to have that two drugs reach a site simultaneously, the mutual prodrug strategies may be used; in this case we can have the following advantages: the codrugs could be well absorbed and the dual acting drugs can release the parent drugs at the desired site of action with improved deliver properties [55]. LD is usually administered in combination with peripheral decarboxilase inhibitor (benserazide or carbidopa) or with a reversible peripheral catechol-O-methyltransferase (COMT) (entacapone or tolcapone). In a previous publication Leppanen et al. described a novel codrug 70 (Figure 11), in which LD and entacapone are linked via a biodegradable carbamate spacer to form a single chemical entity: this carbamate codrug provides adequate stability against chemical hydrolysis but rapidly hydrolyzes to LD and entacapone in liver homogenate at 37 °C [56,57]. The results suggest that 70 may release LD and entacapone in the body after absorption. Furthermore, biodegradation of 70 and the simultaneous release of both LD and entacapone results in the inhibition of COMT, which is expected to facilitate a decrease in metabolism of LD more efficiently than if either therapeutic agents were administered in separate dosage forms. Thus, the novel LD-entacapone codrug approach warrants further studies.
Figure 11.
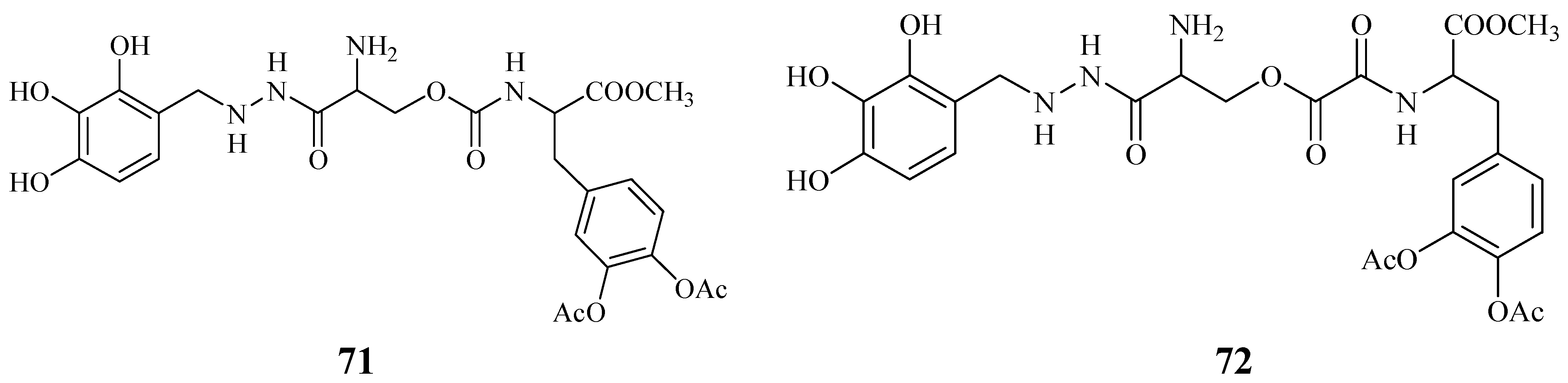
Starting from these data, were proposed the synthesis of two potential dual acting codrugs (71 and 72, Figure 12) in which LD and benserazide are covalently coupled together by different oxalyl and carbonyl spacers [58]. The compounds were evaluated as potential prodrugs with improved physicochemical properties.
Figure 12.
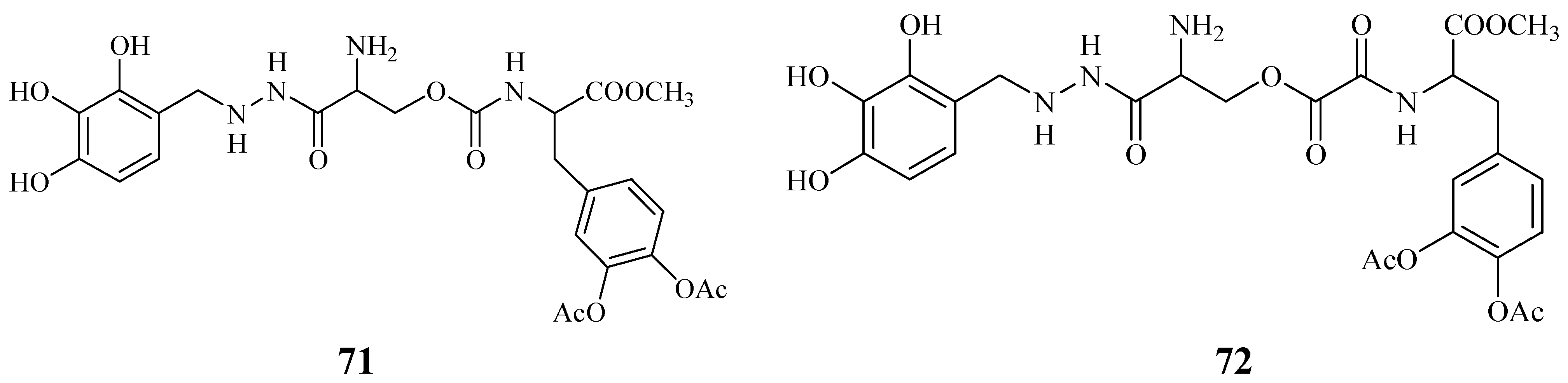
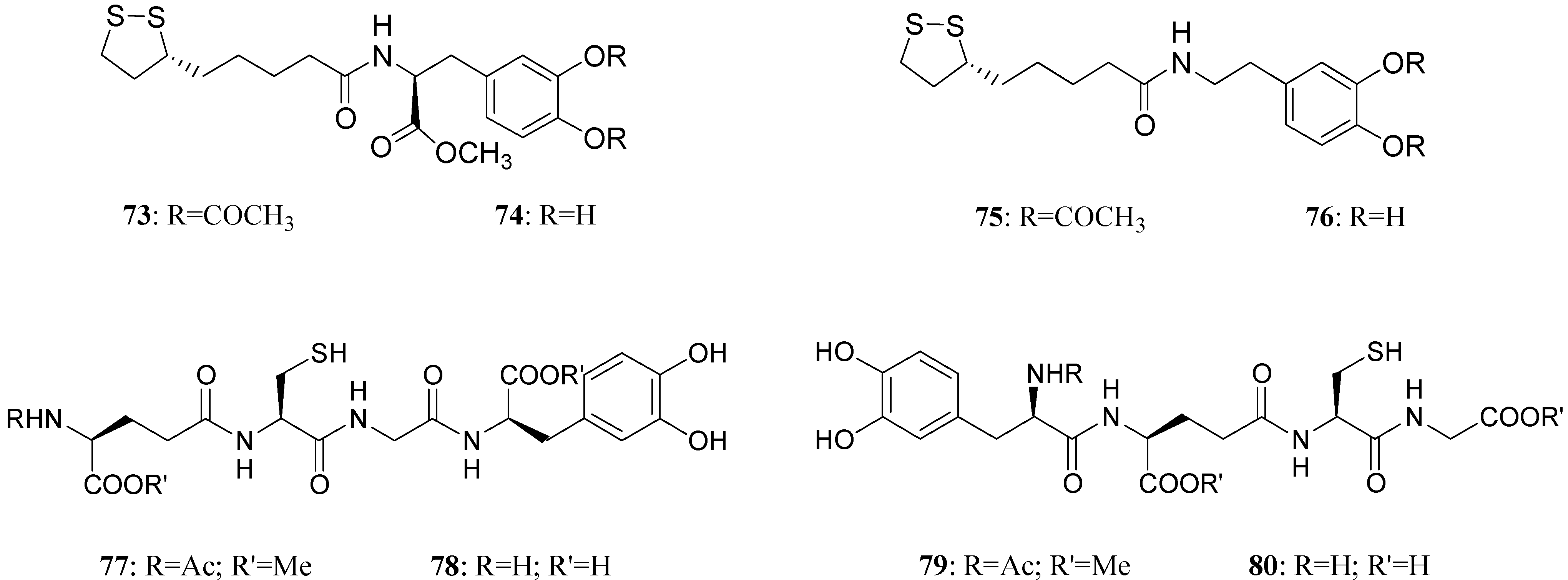
LD conjugates 71 and 72 were adequately stable against chemical hydrolysis and released the parent drugs by enzymatic hydrolysis in rat plasma. Further studies were focused on providing molecular combinations obtained joining an antioxidant molecule with a therapeutic compound also able to generate a targeted antioxidant. These compounds could permit a targeted delivery of the antioxidant moiety directly to specific groups of cells, including neurons, where cellular stress is associated with pathology [59,60]. The synthesis of novel molecular combinations 73-80 in which LD and DA are linked to antioxidant and iron-chelating agents such as (R)-α-lipoic acid (LA) and glutathione (GSH) were proposed (Figure 13).
Figure 13.
Multifunctional LD and DA codrugs, containing antioxidant molecules.
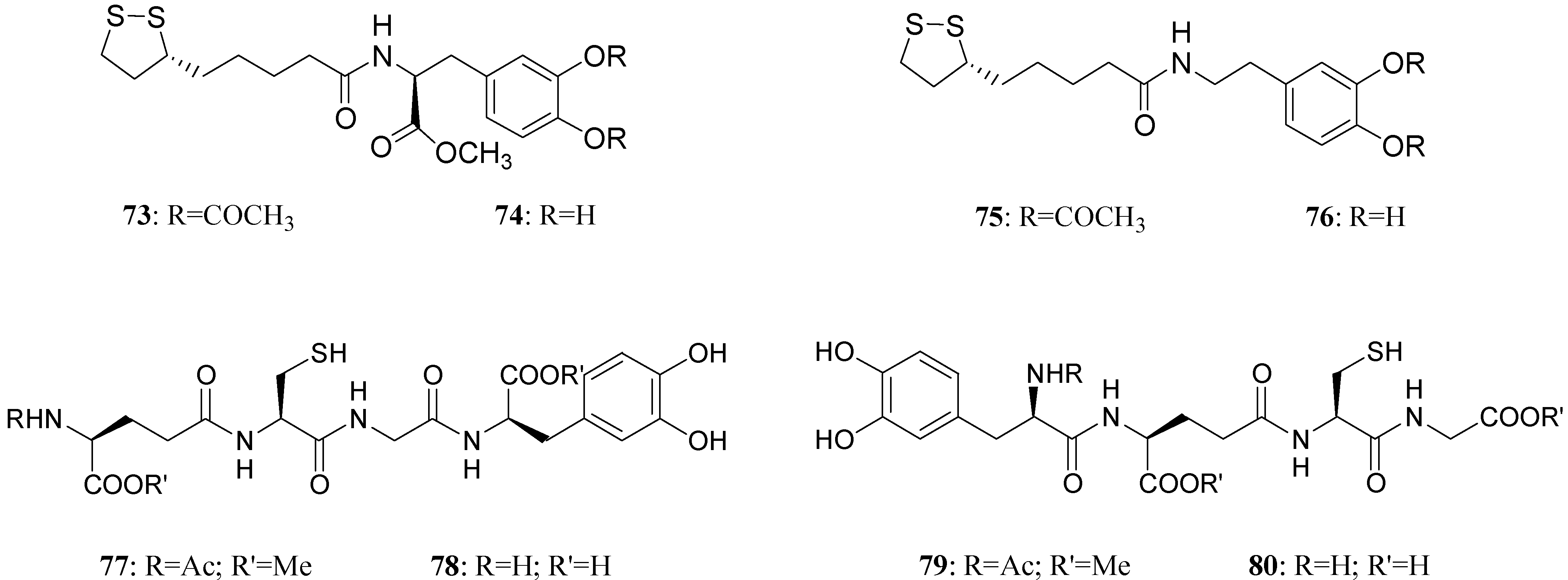
The series of prodrugs with LA was synthesized and evaluated as potential LD and DA codrugs with antioxidant and iron-chelating properties. These multifunctional molecules were synthesized to overcome the pro-oxidant effect associated with LD therapy. The radical scavenging activities of new compounds were evaluated using a test involving the Fe (II)-H2O2-induced degradation of deoxyribose. Furthermore the evaluation of peripheral markers of oxidative stress such as plasmatic activities of superoxide dismutase (SOD) and glutathione peroxidase (GPx) in rat plasma was performed. Codrugs 73-76 possess good lipophilicity (log P > 2 for all tested compounds). Compounds 73 and 74 seem to protect partially against the oxidative stress deriving from auto-oxidation and MAO-mediated metabolism of DA. This evidence, together with the “in vivo” dopaminergic activity and a sustained release of the parent drug in human plasma, allowed pointing out the potential advantages of using 73 and 74 rather than LD in treating Parkinson’s disease [61]. The series of molecular combinations, in which LD is linked covalently via an amide bond with GSH (77-80), were also evaluated as potential anti-Parkinson agents with antioxidant properties. Tested compounds prolonged the plasma LD levels and were able to induce sustained delivery of DA in rat striatum with respect to an equimolar dose of LD. The results suggest that compounds 77 and 79 could represent useful new anti-Parkinson agents devoid of the pro-oxidant effects associated with LD therapy and potentially able to restore the GSH depletion evidenced in the substantia nigra pars compacta (SNpc) of PD patients [62].
Dopamine receptor agonist prodrugs
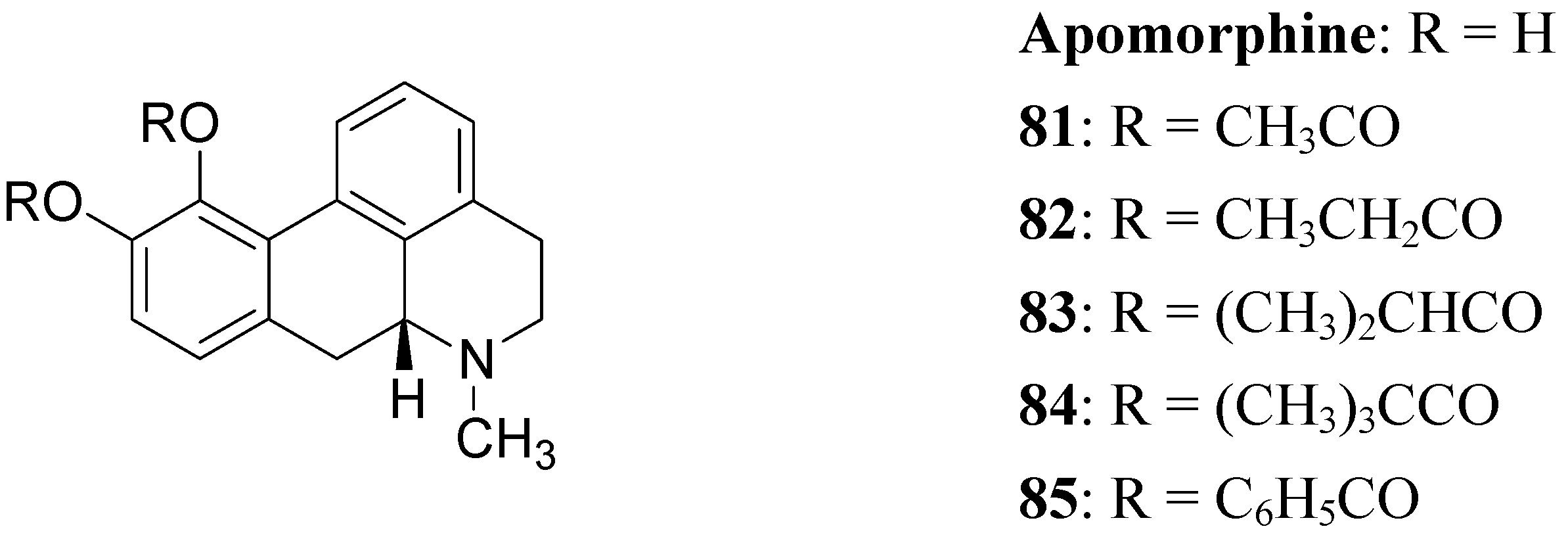
The actions of DA are mediated by five different receptor subtypes classified into two families: D1-like (D1 and D5) and D2-like (D2, D3, and D4). Several structurally different compounds are available as potent and selective ligands for D1 and D2 receptors. Pathological conditions such as PD, schizophrenia, and hyperprolactinemia have been linked to a dysfunction of dopaminergic transmission [63]. DA receptor agonists have been developed to alleviate the symptoms of PD, whereas DA receptor antagonists are effective in treatment of schizophrenia and other neurological and psychiatric disorders [64]. In the treatment of PD, an important property of DA agonists has been suggested to be a long duration of action, which could counteract the development of dyskinesias [65,66]. Prodrugs of dopaminergic agonists represent a new type of prodrug in the research area of antiparkinson agents. About 30 years ago Baldessarini et al. proposed a series of the O,O’-diesters of apomorphine 81-85 (Figure 14) as DA receptors agonists [67,68,69]. O,O′-Diesters of apomorphine exert behavioral effects identical to those of apomorphine, but prolonged in proportion to the bulk of the esters. The increase in duration of action was small for compounds 81 and 82, but prodrugs 83-85 produced dose dependent and prolonged stereotyped behaviour. Furthermore studied compounds were hydrolyzed “in vivo” to yield free catechols capable of stimulating central DA receptors.
Figure 14.
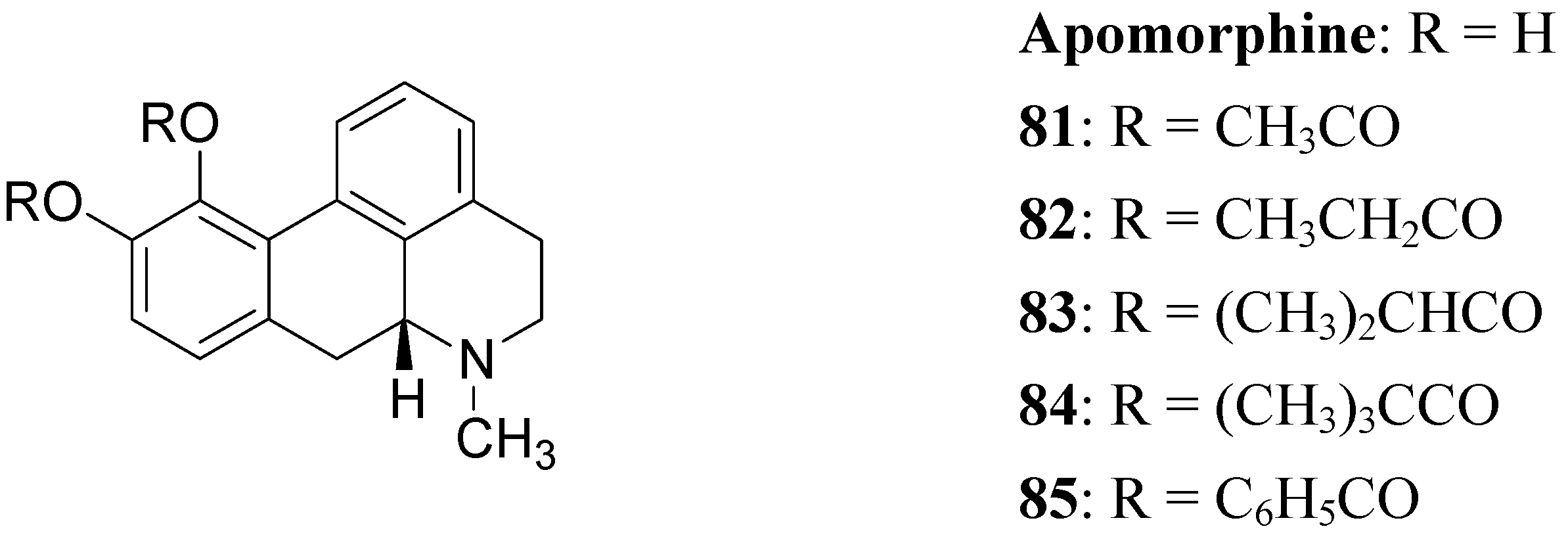
This prolonged activity may reflect depot properties of the esters, and/or decreasing rates of hydrolysis to the presumably active metabolite, apomorphine. The obtained results supported the suggestion that the rate of hydrolysis may be related to desirable sustained action of apomorphine ester prodrugs. During the 1970’s much interest was given to 2-amino-6,7-dihydroxytetrahydronaphthalene (ADTN, 86, Figure 15), which is known from various “in vitro” and “in vivo” studies to be a potent, selective and long-acting DA agonist [70,71]. One of the drawbacks in using this molecule is that, like DA, it does not readily pass the BBB, so that it must be administered to animals intraventricularly. Although “in vivo” ADTN is not the most potent member of 2-aminotetralin derivatives, it has the advantage of being structurally very similar to DA, but metabolically more stable [72]. For these reasons a dibenzoyl-ester derivative of 6,7-ADTN (87) as dopaminergic prodrug was proposed [73]. The regional levels of the DA agonists in rat brain were studied: ADTN levels in brain after administration of the dibenzoyl prodrug were about 5 times higher than when non-esterified ADTN was applied. An accumulation of ADTN in DA-rich areas of the brain was observed after administration of the dibenzoyl derivative.
Furthermore a series of ether derivatives of the purported DA agonist 3,4-dihydroxyphenylimino-2-imidazolidine (DPI, 88, Figure 15) has been prepared as potential prodrugs of the parent compound due to its relatively poor penetration into the brain. Their effects on both DA and noradrenaline utilization in the rat brain have been investigate. Apart from the parent compound, DPI, the diphenylmethane ether analogue (89) showed some dopaminergic activity [74].
Figure 15.

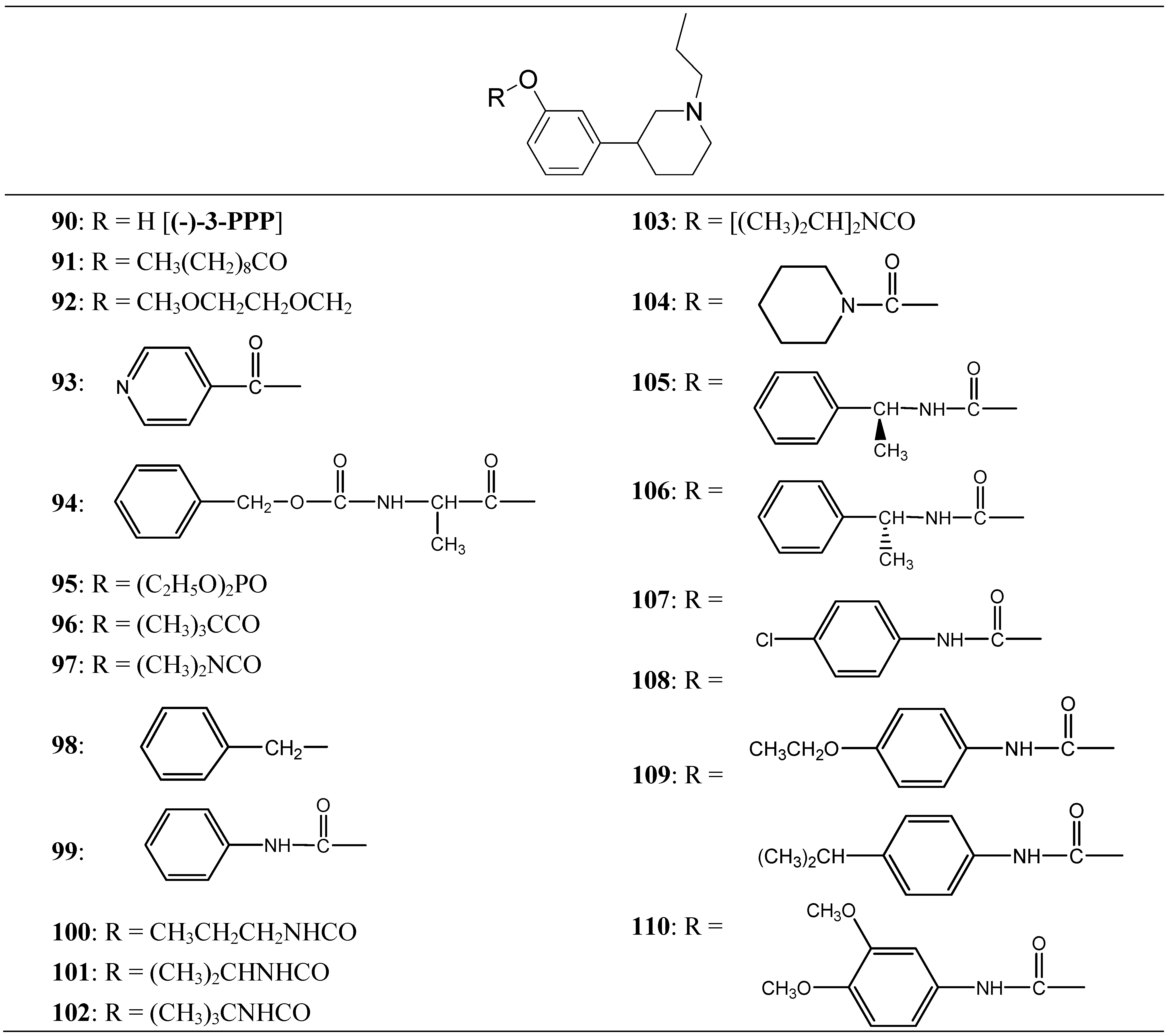
Thorberg et al. proposed the synthesis of twenty derivatives bearing substituents on the phenolic function of (-)-3-(3-hydroxyphenyl)-N-propylpiperidine [(-)-3-PPP (90)], the new compounds 91-110 (Figure 16) were tested as dopaminergic prodrugs [75].
Figure 16.
Chemical structures of (-)-3-PPP prodrugs.
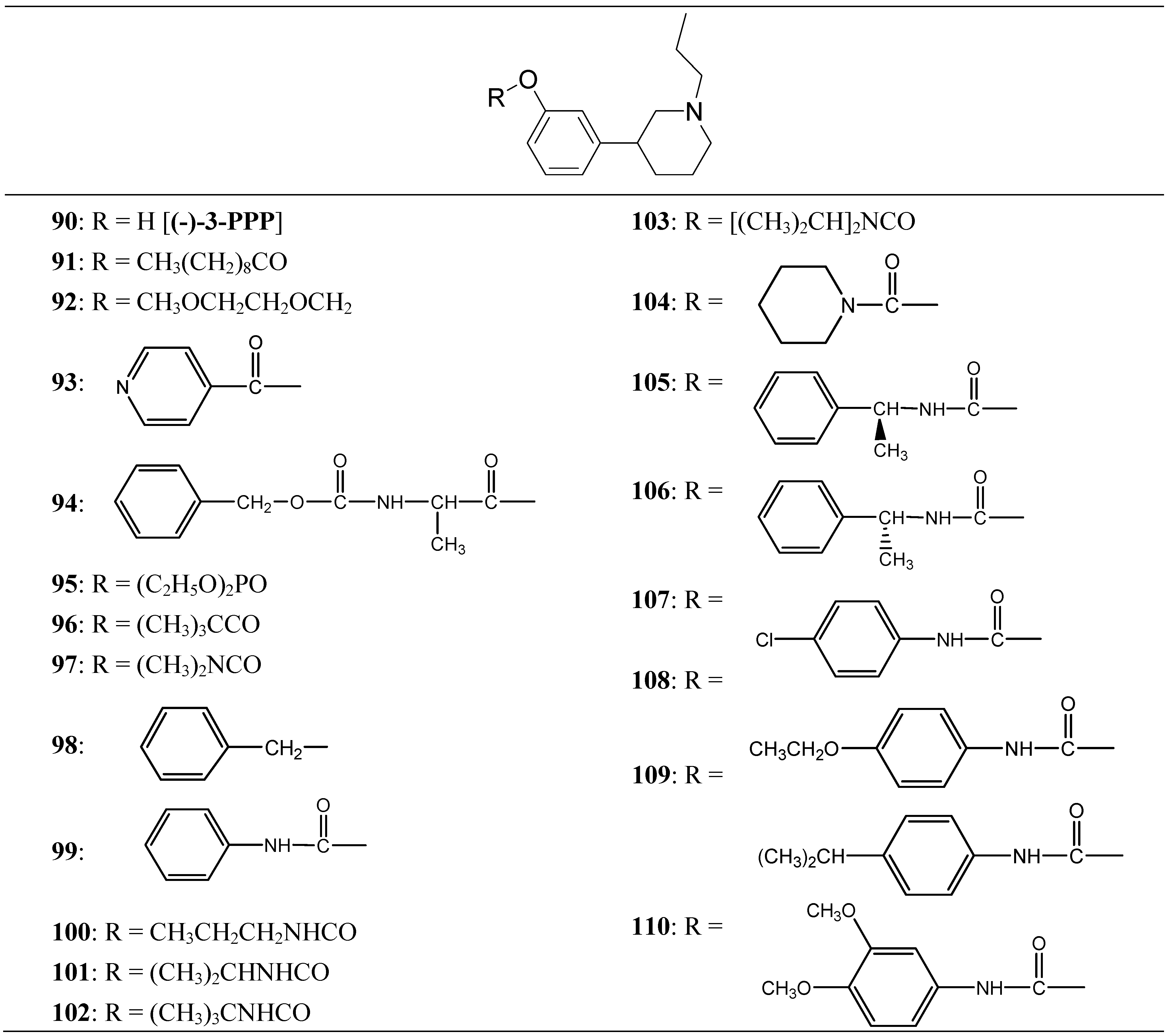
The carbamate ester derivatives were found to be the most suitable prodrugs, and especially the 4-isopropylphenylcarbamate 108 was capable of escaping the first-pass metabolism and still generating high plasma levels of the parent compound. Four hours after an oral dose of 100 µmol/kg to rats, a plasma level of 2400 nmol/L of (-)-3-PPP was found. This was 90 times the level reached after 4 h (27 nmol/L) when (-)-3-PPP itself was given orally at the same dose. The substituted aryl carbamate prodrug seems to offer the solution of a low bioavailability after oral administration of (-)-3-PPP.
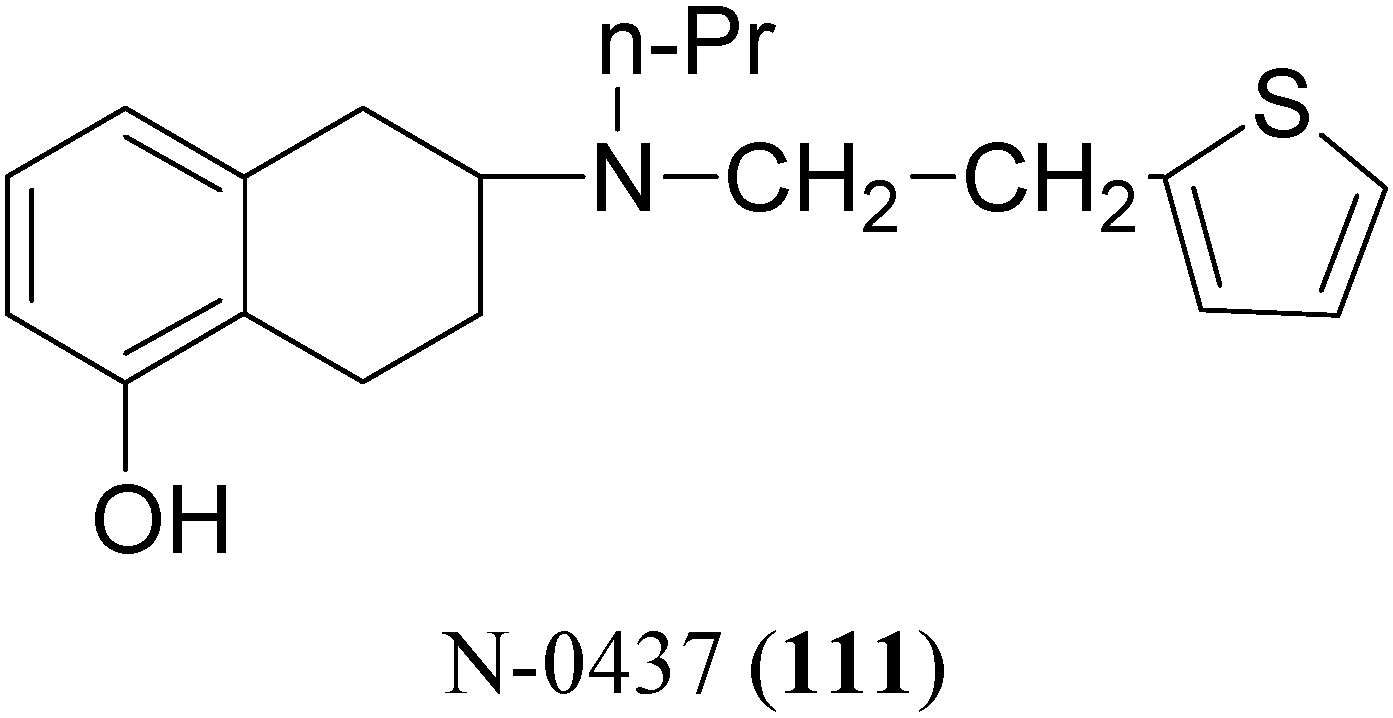
The potent and selective D2-agonist N-0437 [2-(N-propyl-N-2-thienylethylamino)-5-hydroxy-tetralin, (111, Figure 17)] undergoes considerable first-pass metabolism due to glucuronidation of the phenolic group after oral administration. In an attempt to improve the bioavailability, eight ester prodrugs of N-0437 were synthesized by Den Daas et al., i.e. the acetyl, isobutyryl, pivaloyl, benzoyl, 2-methylbenzoyl, 2-methoxybenzoyl, 2,4-dimethylbenzoyl and 2-aminobenzoyl analogues [76].
Figure 17.
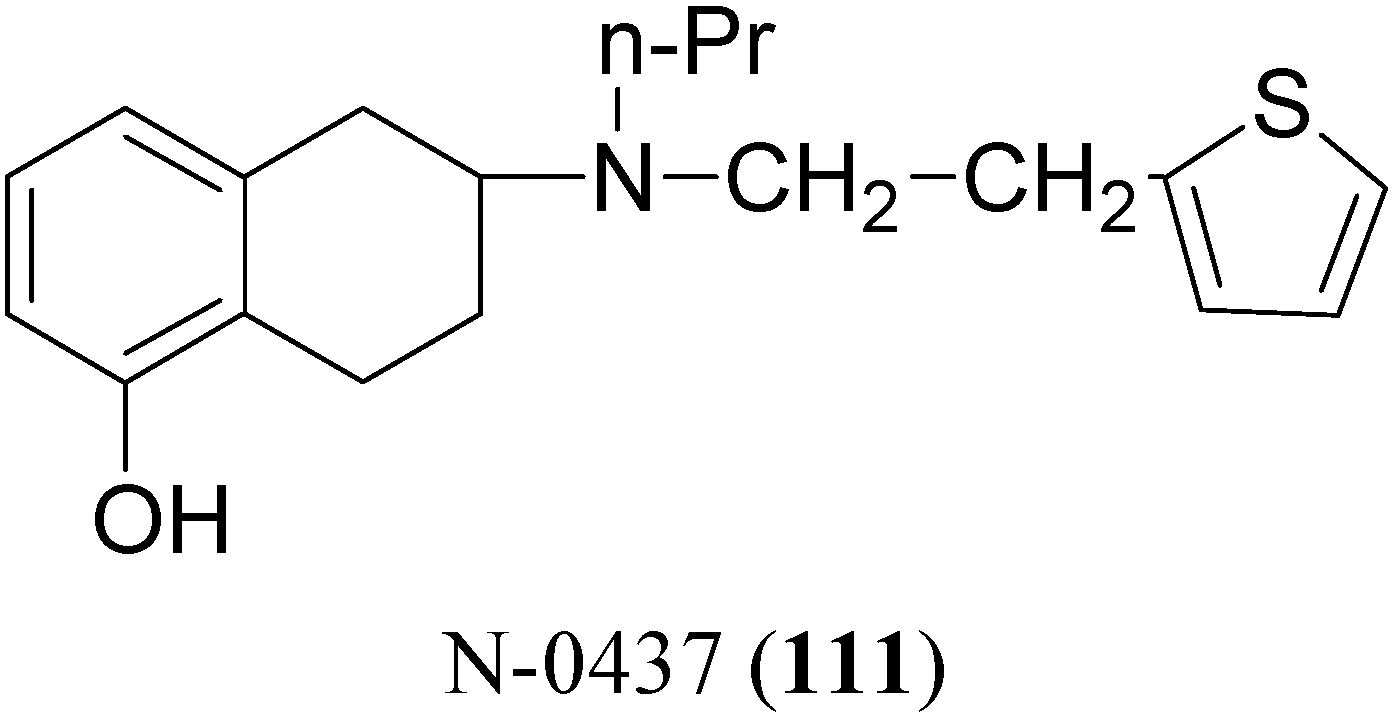
The “in vivo” activities were determined by testing the prodrugs in rats with unilateral 6-OHDA lesions of the striatum. A significantly improved duration of action was found for those prodrugs which have a slow “in vitro” hydrolysis rate. However no significant differences in total activity of these slowly hydrolysing prodrugs compared with N-0437 could be demonstrated, although the 2-amino-benzoyl and the 2,4-dimethylbenzoyl derivatives show interesting behavioural profiles. In contrast the isobutyryl ester, a prodrug with a relatively rapid hydrolysis rate, gave an improvement of turning behaviour over the whole time course in comparison with N-0437.
It was found that slowly hydrolyzing prodrugs, which are known to show an improved duration of action after oral administration, are devoid of activity after transdermal application. The acetyl-, the propionyl- and the isobutyryl analogues, which are prodrugs with a relatively high hydrolysis rate, were found to have interesting and promising profiles following transdermal application [77]. Furthermore the in-vivo activities of carbamate prodrugs of N-0437 were determined by examining the effects of the prodrugs, after their oral administration in rats with unilateral 6-OHDA lesions of the striatum. A comparison of the area under the curve of the time-effect curves of the prodrugs, revealed a significantly improved duration of action compared with N-0437 during the period 11-15 h after administration, for the propylcarbamate and the dimethoxyphenylcarbamate derivatives [78,79]. More recently the (S)-enantiomer of compound N-0437 (rotigotine) has been formulated as a transdermal patch (NEUPRO®) [80]. Transdermal rotigotine significantly improved "off" time in subjects with PD not optimally controlled with LD and was safe and well tolerated, with typical dopaminergic side effects and occasional application site reactions.
The findings with D1 full agonists demonstrate their acute efficacy in the MPTP-lesioned primate models of PD. A D1 agonist may also have advantages over LD and D2 agonists in terms of an improved side-effect profile. The D1 agonist [(-)-(5aR,11bS)-4,5,-5a,6,7,11b-hexahydro-2-propyl-3-thia-5-azacyclopent-fena[c]phenanthrene-9,10-diol], A-869293 (112, Figure 18) has been identified as a D1-selective agonist that maintains efficacy upon repeated administration in the rat rotation model. Its diacetyl prodrug derivative 113 (ABT-431, Figure 18) is rapidly cleaved to the parent compound both and was shown to have improved long-term solid-state stability [81].
Stimulation of the D2 receptor, which is associated with all current DA agonist-based therapies, may contribute to their dose-limiting side effects. An agent such as ABT-431, which selectively stimulates the D1 receptor, may represent a novel mechanism for PD therapy with the potential for an improved side-effect profile and, consequently, improved patient compliance [82].
Figure 18.

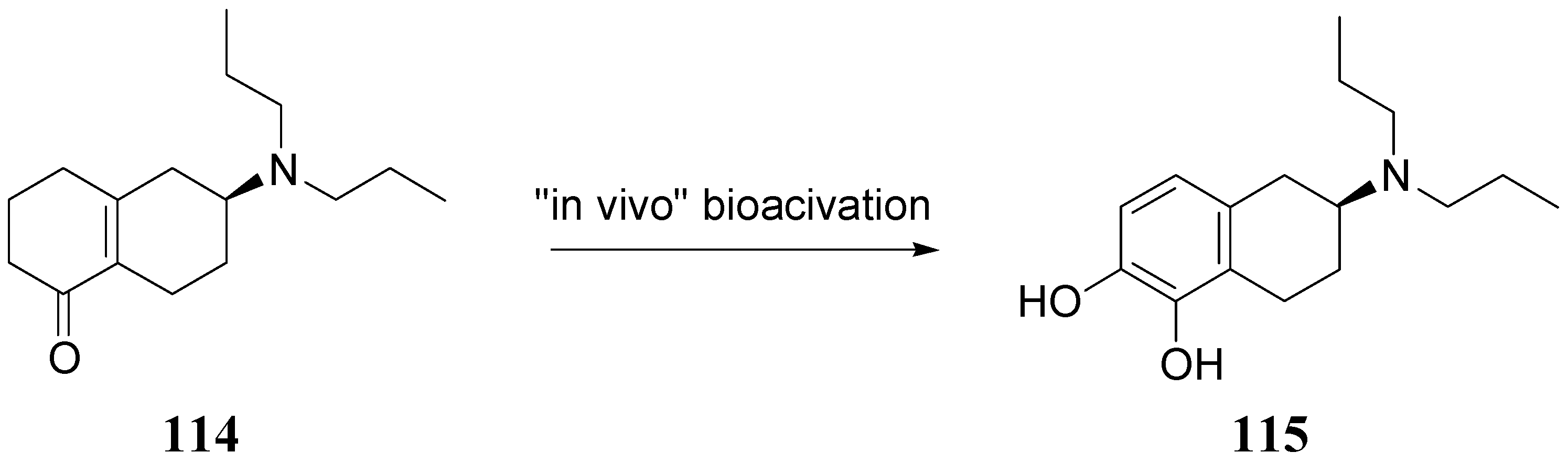
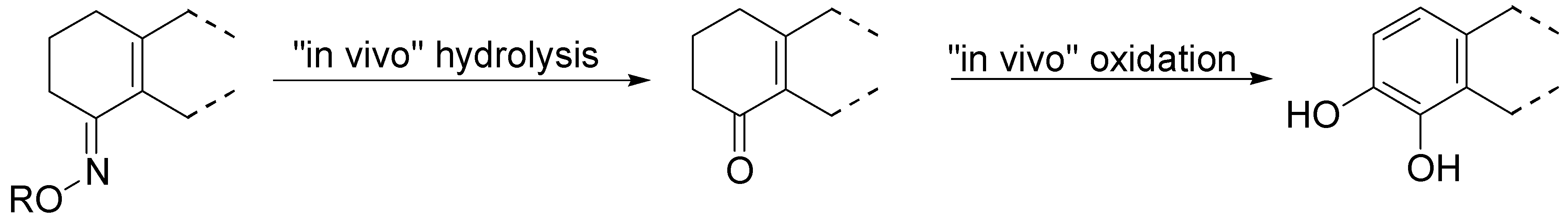
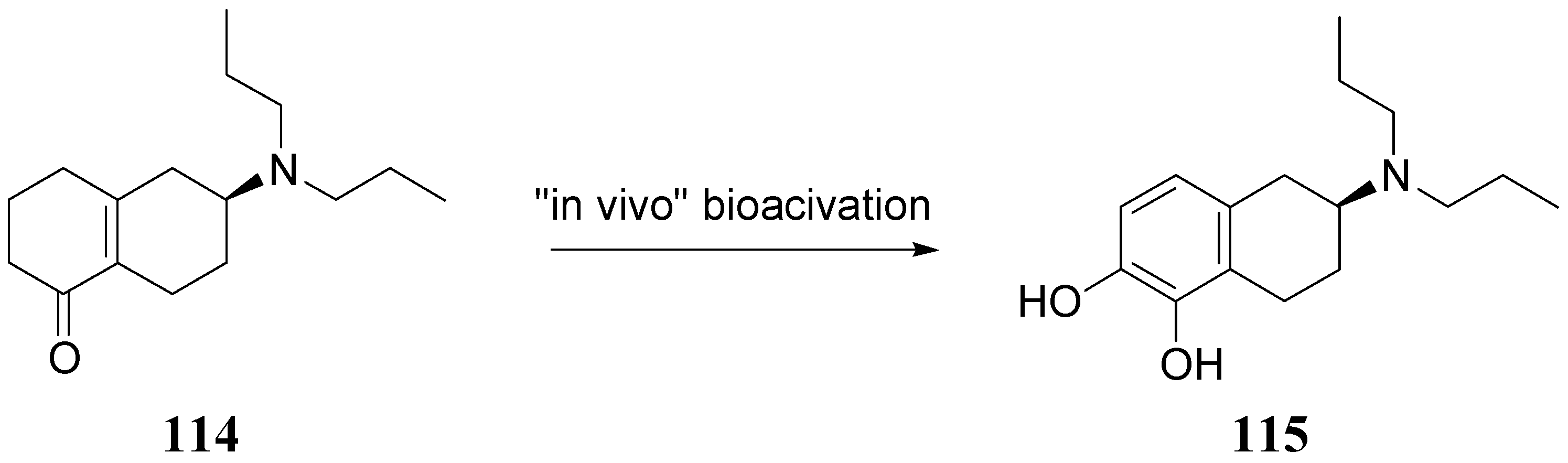
The authors concluded that ABT-431, the prodrug of A-86929, may have good efficacy in PD without the consequence of dyskinesia, one of the main limitations to treatment with LD. A pilot study performed in the Netherlands showed ABT-431 to be well tolerated in patients with PD [83]. ABT-431 can also attenuate the ability of cocaine to induce cocaine-seeking behaviour and does not itself induce cocaine-seeking behaviour in a rodent model of cocaine craving and relapse [84]. During the last years Venhuis et al. proposed the synthesis and pharmacological evaluation of the orally active enone prodrug S-(-)-6-(N,N-di-n-propylamino)-3,4,5,6,7,8-hexahydro-2H-naphthalen-1-one [(-)-114 (S-PD148903)]. 1-4 Bioactivation of enones by metabolic conversion to their corresponding catecholamines, as was demonstrated for (-)-114, is thought to be the general bioactivation mechanism (Figure 19) [85,86,87]. Compound S-PD148903 represents a new type of prodrug which in the rat is bioactivated to the catecholamine S-5,6-diOH-DPAT (115), known to display mixed DA D1/D2 receptor agonist properties just like apomorphine. This prodrug has an enone structure which by an oxidative bioactivation mechanism is converted to the corresponding catechol and is delivered enantioselectively into the CNS.
Figure 19.
Bioactivation of compound S-PD148903 [86].
Figure 19.
Bioactivation of compound S-PD148903 [86].
To further increase the usefulness of these enone prodrugs a series of oxime derivatives 116-127 (Figure 20) of the dopaminergic prodrug S-PD148903 were proposed as orally active agents [88].
Figure 20.
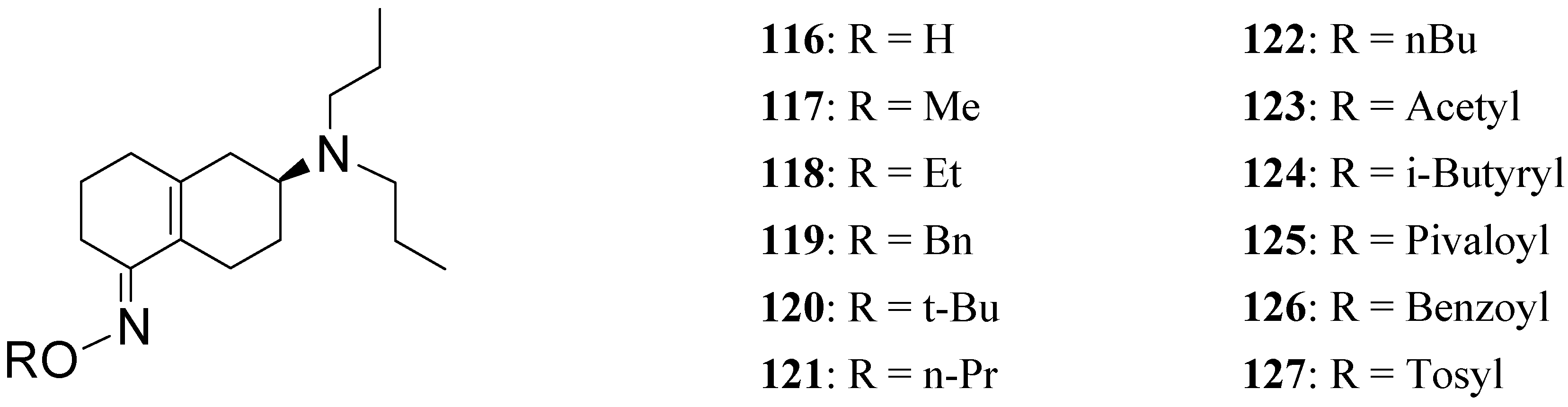
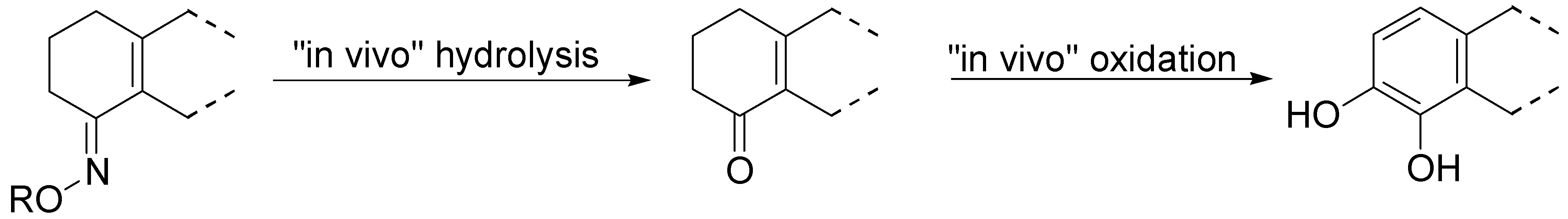
Though less potent than the parent prodrug, the oxime derivatives of S-PD148903 can be orally active, acting as cascade prodrugs (Scheme 3). The only inactive compound in the series was 127, that also was the only oxime stable under aqueous conditions.
Scheme 3.
An oxime acting as a “cascade” prodrug of a catecholamine [88].
Scheme 3.
An oxime acting as a “cascade” prodrug of a catecholamine [88].
Through the proposed bioactivation mechanism prodrug (-)-128 is converted to the corresponding N-(n-propyl)-6,7-di-OH-benzo-[g]quinoline [(-)6,7-di-OH-PBGQ, (-)-129] (Scheme 4). Significant effects of (-)-128 were observed in microdialysis studies after administration of 1 nmol kg-1 sc and 3 nmol kg-1 po. With a potency comparable to that of the most potent apomorphines, (-)-128 could potentially compete with LD and apomorphine in the treatment of PD [89]. Further studies in this field led out to discovery of the first benzo[g]quinoline-derived enone that induces potent DA agonist effects similar to aminotetralin-derived enones.
Scheme 4.
“In vivo” activation of enone prodrug (-)-128 [89].
Scheme 4.
“In vivo” activation of enone prodrug (-)-128 [89].

Conclusions
With the aim to prolong the pharmacological activity of LD enhancing absorption and providing a protection against metabolism and in order to improve the antiparkinson therapeutic strategies, the prodrug approach appeared to be the most promising and many prodrugs have been prepared in an effort to solve the problems related to LD therapy. An ideal prodrug should be soluble in water and in lipids, completely absorbed by the gastrointestinal tract without any chemical degradation or metabolism, and thus deliver the parental drug in the blood stream at reproducible therapeutic levels; furthermore the new entity should improve the brain delivery of the active drug. Despite of the fact that prodrugs strategies has been around for many years, only very few drugs such as LD methyl ester (SIRIO®) have found clinical application in the field on antiparkinson therapy [90]; however, the study of new dopaminergic prodrugs will continue for improving the antiparkinson therapy based on LD administration.
References
- Abou-Sleiman, P.M.; Muqit, M.M.K.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V; Bezard, E.; Brotchie, J.; Calon, F.; Collingridge, G.L.; Ferger, B.; Hengerer, B.; Hirsch, E.; Jenner, P.; Le Novère, N.; Obes, J.A; Schwarzschild, M.A.; Spampinato, U.; Davidai, G. Novel pharmacological targets for the treatment of Parkinson’s disease. Nat. Rev. Drug Discov. 2006, 5, 845–854. [Google Scholar]
- Blandini, F.; Greenamyre, J.T. Protective and Symptomatic Strategies for Therapy of Parkinson's. Drugs Today 1999, 35, 473–483. [Google Scholar]
- Bodor, N; Farag, H.H. Improved delivery through biological membranes. 13. Brain specific delivery of dopamine with a dihydropyridine-pyridinium salt type redox delivery system. J. Med. Chem 1983, 26, 528–534. [Google Scholar]
- Kao, H.D.; Traboulsi, A.; Itoh, S.; Dittert, L.; Hussain, A. Enhancement of the Systemic and CNS Specific Delivery of L-Dopa by the Nasal Administration of Its Water. Soluble Prodrugs. Pharmaceut. Res. 2000, 17, 978–984. [Google Scholar]
- Standaert, D.; Young, A. Treatment of Central Nervous System Degenerative Disorders. In The Pharmacological Basis of Therapeutics; Gilman, A. G., Hardman, J. G., Limbird, L. E., Molinoff, P. B., Ruddon, R.W., Eds.; McGraw Hill: New York, 1996; pp. 503–519. [Google Scholar]
- Hagan, J.J.; Middlemiss, D.N.; Sharpe, P.C; Poste, G.H. Parkinson’s disease: prospects for improved drug therapy. TIPS 1997, 18, 156–163. [Google Scholar]
- Sasahara, K.; T. Nitanai, T.; Habara, T.; Morioka, T.; Nakajimai, E. Dosage form design for improvement of bioavailability of levodopa III: Influence of dose on pharmacokinetic behaviour of levo-dopa administration in dogs and parkinsonian patients. J. Pharm. Sci. 1980, 69, 1374–1378. [Google Scholar]
- Nutt, J.G.; Woodward, W.R.; Anderson, J.L. The Effect of carbidopa on the pharmacokinetics of intravenously administered levodopa: The mechanism of action in the treatment of parkinsonism. Annal. Neurol. 1986, 18, 537–543. [Google Scholar]
- Wingard, L.B.; Brody, T.M.; Larner, J.; Schwartz, A. Human Pharmacology; Mosby Year Book: St Louis, 1991. [Google Scholar]
- Cedarbaum, J.M. Antiparkinsonian drugs. Clin. Pharmacokin. 1987, 13, 141–178. [Google Scholar] [CrossRef]
- Nutt, J.G.; Woodward., W.R. Levodopa pharmacokinetics and pharmacodynamics in fluctuating parkinsonian patients. Neurology 1986, 36, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Hardie, R.; Lees, A.J.; Stern, G.M. On-off fluctuations in parkinson’s disease. Brain 1984, 107, 487–506. [Google Scholar] [CrossRef] [PubMed]
- Quinn, N.; Parkes, J.D.; Marsden, C.D. Control of on/off phenomenon by continuous intravenous infusion of levodopa. Neurology 1984, 34, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Birkmayer, W.; Danielcyk, W.; Neumayer, E.; Riederer, P. L-dopa level in plasma, primary condition for the kinetic effect. J. Neural Trans. 1973, 34, 133–143. [Google Scholar] [CrossRef]
- Leppert, P.S.; Cortese, M.; Fix, J.A. The Effects of Carbidopa Dose and Time and Route of Administration on Systemic L-Dopa Levels in Rats. Pharmaceut. Res. 1988, 5, 587–591. [Google Scholar] [CrossRef]
- Sasahara, K.; Nitanai, T.; Habara, T.; Morioka, T.; Kawahara, Y.; Nakajima, E. Dosage form design for improvement of bioavailability of levodopa IV: Possible causes of low bioavailability of oral levodopa in dogs. J. Pharm. Sci. 1981, 70, 730–733. [Google Scholar]
- Hutton, J.H.; Morris, J.L.; Gustavo, C.R. Treatment of chronic parkinson’s disease with controlled-release carbidopa/ levodopa. Neurology 1988, 45, 861–864. [Google Scholar]
- Bordor, N.; Sloan, K.B.; Higuchi, T.; Sasahara, K. Improved delivery through biological membranes 4: Prodrugs of L-dopa. J. Med. Chem. 1977, 20, 1435–1445. [Google Scholar]
- Goole, J.; Vanderbist, F.; Amighi, K. Development and evaluation of new multiple-unit levodopa sustained-release floating dosage forms. Int. J. Pharm. 2007, 334, 35–41. [Google Scholar]
- Di Stefano, A.; Mosciatti, B.; Cingolani, G.M.; Giorgioni, G.; Ricciutelli, M.; Cacciatore, I.; Sozio, P.; Claudi, F. Dimeric L-Dopa derivatives as potential prodrugs. Bioorg. Med. Chem. Lett. 2001, 11, 1085–1088. [Google Scholar] [CrossRef] [PubMed]
- Chemuturi, N.V.; Donovan, M.D. Role of Organic Cation Transporters in Dopamine Uptake across Olfactory and Nasal Respiratory Tissues. Mol. Pharmaceut. 2007, 4, 936–942. [Google Scholar]
- Casagrande, C.; Ferrari, G. 3,4-0-diacyl derivatives of dopamine. Farmaco 1973, 28, 143–148. [Google Scholar]
- Borgman, R.J; McPhillips, J.J.; Stitzel, R.E. Synthesis and pharmacology of centrally acting dopamine derivatives and analogs in relation to Parkinson’s disease. J. Med. Chem. 1973, 16, 630–633. [Google Scholar]
- Bodor, N; Farag, H.H.; Brewster, M.E. Site-specific, sustained release of drugs to the brain. Science 1981, 214, 1370–1372. [Google Scholar]
- Fernández, C.; Nieto, O.; Rivas, E.; Montenegro, G.; Fontenla, J. A.; Fernández-Mayoralas, A. Carbohydr. Res. 2000, 327, 353–365.
- Fernández, C.; Nieto, O.; Fontenla, J. A.; Rivas, E.; de Ceballos, M.L.; Fernández-Mayoralas, A. Org. Biomol. Chem. 2003, 1, 767–771. [CrossRef] [PubMed]
- Bonina, F.; Puglia, C.; Rimoli, M.G.; Melisi, D.; Boatto, G.; Nieddu, M.; Malignano, A.; La Rana, G.; De Caprariis, P. J. Drug Target 2003, 11, 25–36.
- Oldendorf, W.H. Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am. J. Physiol. 1971, 221, 1629–1639. [Google Scholar]
- Mueckler, M. Facilitative glucose transporters. Eur. J. Biochem. 1994, 219, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Prokai, L. Peptide drug delivery into the central nervous system. Prog. Drug Res. 1998, 51, 95–131. [Google Scholar] [PubMed]
- Carlsson, A; Lindqvist, M; Magnusson, T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 1957, 180, 1200. [Google Scholar] [PubMed]
- Birkmayer, W.; Hornykiewicz, O. The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wien Klin. Wochenschr. 1961, 10, 787–788. [Google Scholar]
- Felix, A.M.; Winter, D.P.; Wang, S.S.; Kulesha, I.D.; Pool, W.R.; Hane, D.L.; Sheppard, H. Synthesis and antireserpine activity of peptides of L. Dopa. J. Med. Chem. 1974, 17, 422–426. [Google Scholar] [CrossRef]
- Wang, H.; Lee, J.; Tsai, M.; Lu, H.; Hsu, W. Synthesis and Pharmacological activities of a novel tripeptide mimetic dopamine prodrug. Bioorg. Med. Chem. Lett. 1995, 5, 2195–2198. [Google Scholar] [CrossRef]
- Marrel, C.; Boss, G.; Van De Waterbeemd, H.; Testa, B.; Cooper, D.; Jenner, P.; Marsden, C.D. L-Dopa esters as potential prodrugs. Eur. J. Med. Chem.-Chim. Ther 1985, 20, 459–465. [Google Scholar]
- Cooper, D.R.; Marrel, C.; Van De Waterbeemd, H.; Quinn, N.; Testa, B.; Jenner, P.; Marsden, C.D. L-Dopa methyl ester-a candidate for chronic systemic delivery of L-Dopa in Parkinson's disease. Clin. Neuropharmacol. 1984, 89–98. [Google Scholar] [CrossRef]
- Garzon-Aburbeh, A; Poupaert, JH; Claesen, M; Dumont, P. A lymphotropic prodrug of L-dopa: synthesis, pharmacological properties, and pharmacokinetic behavior of 1,3-dihexadecanoyl-2-[(S)-2-amino-3-(3,4-dihydroxyphenyl)prop anoyl] propane-1,2,3-triol. J. Med. Chem. 1986, 29, 687–691. [Google Scholar]
- Cooper, D.R.; Marrel, C.; Van De Waterbeemd, H.; Testa, B.; Jenner, P.; Marsden, C.D. L-dopa esters as potential prodrugs: effect on brain concentration of dopamine metabolites in reserpinized mice. J. Pharm. Pharmacol. 1987, 39, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.R.; Marrel, C.; Van De Waterbeemd, H.; Testa, B.; Jenner, P.; Marsden, C.D. L-dopa esters as potential prodrugs: behavioural activity in experimental models of Parkinson's disease. J. Pharm. Pharmacol. 1987, 39, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Tsuchiya, Y.; Sawasaki, Y.; Hisaka, A.; Takehana, H.; Tomimoto, K.; Yano, M.A. New potential prodrug to improve the duration of L-dopa: L-3-(3-hydroxy-4-pivaloyloxyphenyl)-alanine. J. Pharm. Sci. 1989, 78, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M; Nakajima, S; Hisaka, A; Tsuchiya, Y; Sakuma, Y; Suzuki, H; Kitani, K; Yano, M. Hydrolysis and acyl migration of a catechol monoester of L-dopa: L-3-(3-hydroxy-4-pivaloyloxyphenyl)alanine. J. Pharm. Sci. 1990, 79, 703–708. [Google Scholar]
- Brime, B.; Ballesteros, M.P.; Frutos, P. Preparation and in vitro characterization of gelatin microspheres containing Levodopa for nasal administration. J. Microencapsul. 2000, 17, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Chavis, C.; Grodenic, F.; Imbach, J.L. Ribosides et dérivés de la L-méthyl dopa et de la L-dopa. Eur. J. Med. Chem.-Chim. Ther. 1981, 16, 219–227. [Google Scholar]
- Cingolani, G.M.; Di Stefano, A.; Mosciatti, B.; Napolitani, F.; Giorgioni, G.; Ricciutelli, M.; Claudi, F. Bioorg. Med. Chem. Lett. 2000, 10, 1385–1388. [CrossRef]
- Langlois, M.; Quintard, D.; Abalain, C. Synthesis of symmetrical pseudopeptides as potential inhibitors of the human immunodeficiency virus-1 protease. Eur. J. Med. Chem. 1994, 29, 639–647. [Google Scholar]
- Christiaans, J. A. M.; Timmerman, H. Cardiovascular hybrid drugs: combination of more than one pharmacological property in one single molecule. Eur. J. Pharm. Sci. 1996, 4, 1–22. [Google Scholar] [CrossRef]
- Mahfouz, N.M.; Aboul-Fadl, T.; Diab, A. K. Metronidazole twin ester prodrugs: synthesis, physicichemical properties, hydrolysis kinetics and antigiardial activity. Eur. J. Med. Chem. 1998, 33, 675–683. [Google Scholar] [CrossRef]
- Giannola, L. I.; Giammona, G.; Alotta, R. Pro-drugs of isoniazid: synthesis and diffusion characteristics of acyl derivatives. Pharmazie 1992, 47, 423–425. [Google Scholar] [PubMed]
- Ducho., C.; Görbig, U.; Jessel, S.; Gisch, N.; Balzarini, J.; Meier, C. Bis-cycloSal-d4T-monophosphates: drugs that deliver two molecules of bioactive nucleotides. J. Med. Chem. 2007, 50, 1335–1346. [Google Scholar] [PubMed]
- Cannazza, G.; Di Stefano, A.; Mosciatti, B.; Braghiroli, D.; Baraldi, M.; Pinnen, F.; Sozio, P.; Benatti, C.; Parenti, C. Detection of levodopa, dopamine and its metabolites in rat striatum dialysates following peripheral administration of l-DOPA prodrugs by mean of HPLC–EC. J. Pharm. Biomed, Anal. 2005, 36, 1079–1084. [Google Scholar]
- Di Stefano, A.; Carafa, M.; Sozio, P.; Pinnen, F.; Braghiroli, D.; Orlando, G.; Cannazza, G.; Ricciutelli, M.; Marianecci, C.; Santucci, E. Evaluation of rat striatal l-dopa and DA concentration after intraperitoneal administration of l-dopa prodrugs in liposomal formulations. J. Control. Release 2004, 99, 293–300. [Google Scholar]
- Di Stefano, A.; Sozio, P.; Iannitelli, A.; Marianecci, C.; Santucci, E.; Carafa, M. Maleic- and fumaric-diamides of (O,O-diacetyl)-L-Dopa-methylester as anti-Parkinson prodrugs in liposomal formulation. J. Drug Target 2006, 14, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Menger, F. M.; Rourk, M.J. Synthesis and Reactivity of 5-Fluorouracil/Cytarabine Mutual Prodrugs. J. Org. Chem. 1997, 62, 9083–9088. [Google Scholar] [CrossRef]
- Singh, G.; Sharma, P.D. Mutual prodrugs : a recent trend in prodrug design. Indian J. Pharm. Sci. 1994, 56, 69–79. [Google Scholar]
- Savolainen, J.; Leppänen, J.; Forsberg, M.; Taipale, H.; Nevalainen, T.; Huuskonen, J.; Gynther, J.; Männistö, P. T.; Järvinen, T. Synthesis and in vitro/in vivo evaluation of novel oral N-alkyl- and N,N-dialkyl-carbamate esters of entacapone. Life Sci. 2000, 67, 205. [Google Scholar]
- Leppanen, J.; Huuskonen, J.; Nevalainen, T.; Gynther, J.; Taipale, H.; Jarvinen, T. Design and Synthesis of a Novel L-Dopa-Entacapone Codrug. J. Med. Chem. 2002, 45, 13791382. [Google Scholar] [CrossRef]
- Di Stefano, A.; Sozio, P.; Iannitelli, A.; Cocco, A.; Orlando, G.; Ricciutelli, M. Synthesis and Preliminary Evaluation of L-Dopa/Benserazide Conjugates as Dual Acting Codrugs. Lett. Drug Des. Discovery 2006, 3, 747–752. [Google Scholar]
- Rosini, M.; Andrisano, V.; Bartolini, M.; Bolognesi, M. L.; Hrelia, P.; Minarini, A.; Tarozzi, A.; Melchiorre, C. Rational Approach to discover multipotent anti-Alzheimer drugs. J. Med. Chem. 2005, 48, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Bharath, S.; Cochran, B. C.; Hsu, M.; Ames, B. N.; Andersen, J. K. Pretreatment with R-Lipoic acid alleviates the effects of GSH depletion in PC12 cells: implications for Parkinson’s disease therapy. Neurotoxicology 2002, 23, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Sozio, P.; Cocco, A.; Iannitelli, A.; Santucci, E.; Costa, M.; Pecci, L.; Nasuti, C.; Cantalamessa, F.; Pinnen, F. L-Dopa and DA/(R)-α-lipoic acid conjugates as multifunctional codrugs with antioxidant properties. J. Med. Chem. 2006, 49, 1486–1493. [Google Scholar]
- Pinnen, F.; Cacciatore, I.; Cornacchia, C.; Sozio, P.; Iannitelli, A.; Costa, M.; Pecci, L.; Nasuti, C.; Cantalamessa, F.; Di Stefano, A. Synthesis and Study of L-Dopa-Glutathione Codrugs as New Anti-Parkinson Agents with Free Radical Scavenging Properties. J. Med. Chem. 2007, 50, 2506–2515. [Google Scholar]
- Seeman, P.; Bzowej, N.H.; Guan, H.C.; Bergeon, C.; Reynolds, G.P.; Bird, E.D. Human brain D1 and D2 dopamine receptors in schizophrenia, Alzheimer’s, Parkinson’s and Huntington’s diseases. Neuropsycopharmacology 1987, 1, 5–15. [Google Scholar] [CrossRef]
- Missale, C.; Nash, S.R.; Robinson, S.; Jaber, M.; Caron, M.G. Dopamine receptors: from structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [PubMed]
- Uitti, R.Y.; Ahlskog, J.E. Comparative review of dopamine receptor agonists in Parkinson's disease. CNS Drugs 1996, 5, 369–388. [Google Scholar] [CrossRef]
- Maratos, E.C.; Jackson, M.J.; Pearce, R.K.; Jenner, P. Antiparkinsonian activity and dyskinesia risk of ropinirole and L-DOPA combination therapy in drug naive MPTP-lesioned common marmosets (Callithrix jacchus). Mov. Disord. 2001, 16, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Baldessarini, R.J.; Walton, K.G.; Borgman, R.J. Esters of apomorphine and N,N-dimethyldopamine as agonists of dopamine receptors in the rat brain in vivo. Neuropharmacology 1975, 14, 725–731. [Google Scholar]
- Borgman, R.J.; Baldessarini, R.J.; Walton, K.G. Diester derivatives as apomorphine prodrugs. J. Med. Chem. 1976, 19, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Baldessarini, R.J.; Kula, N.S.; Walton, K.G.; Borgman, R.J. Hydrolysis of diester prodrugs of apomorphine. Biochem. Pharmaco. 1977, 26, 1749–1756. [Google Scholar] [CrossRef]
- Woodruff, G.N.; Elkhawad, A.O.; Crossman, A.R.; Walker, R.J. Further evidence for the stimulation of rat brain dopamine. J. Pharm. Pharmac. 1974, 26, 740–741. [Google Scholar] [CrossRef]
- Woodruff, G.N.; Elkhawad, A.O.; Pinder, R.M. Long lasting stimulation of locomotor activity produced by the intraventricular injection of a cyclic analogue of dopamine into conscious mice. Eur. J. Pharmac. 1974, 25, 80–86. [Google Scholar] [CrossRef]
- Costall, B.; Naylor, R. J.; Cannon, J. G.; Lee, T. Differential activation by some 2-aminotetralin derivatives of the receptor mechanisms in the nucleus accumbens of rats which mediate hyperactivity and stereotyped biting. Eur. J. Pharmac. 1977, 41, 307–319. [Google Scholar]
- Westerink, B.H.; Dijkstra, D.; Feenstra, M.G.; Grol, C.J.; Horn, A.S.; Rollema, H; Wirix, E. Dopaminergic prodrugs: brain concentrations and neurochemical effects of 5,6- and 6,7-ADTN after administration as dibenzoyl esters. Eur. J. Pharmac. 1980, 61, 7–15. [Google Scholar] [CrossRef]
- Houwing, H.A.; van Oene, J.C.; Horn, A.S. 3,4-disubstituted phenyliminoimidazolidines as potential prodrugs of the purported dopamine agonist 3,4-dihydroxyphenylimino-2-imidazolidine (DPI). Pharmaceut. Weekblad. Sci. 1983, 5, 177–181. [Google Scholar] [CrossRef]
- Thorberg, S.O.; Berg, S.; Lundstrom, J.; Pettersson, B.; Wijkstrom, A.; Sanchez, D.; Lindberg, P.; Nilsson, J.L. Carbamate ester derivatives as potential prodrugs of the presynaptic dopamine autoreceptor agonist (-)-3-(3-hydroxyphenyl)-N-propylpiperidine. J. Med. Chem. 1987, 30, 2008–2012. [Google Scholar] [CrossRef] [PubMed]
- den Daas, I.; Tepper, P.G.; Horn, A.S. Improvement of the oral bioavailability of the selective dopamine agonist N-0437 in rats: the in vitro and in vivo activity of eight ester prodrugs. N-S Arch. Pharmacol. 1990, 341, 186–191. [Google Scholar]
- den Daas, I.; Tepper, P.G.; Rollema, H.; Horn, A.S. Transdermal administration of the dopamine agonist N-0437 and seven ester prodrugs: comparison with oral administration in the 6-OHDA turning model. N-S Arch Pharmacol. 1990, 342, 655–659. [Google Scholar]
- den Daas, I.; de Boer, P.; Tepper, P.G.; Rollema, H.; Horn, A.S. Orally active carbamate prodrugs of the selective dopamine agonist N-0437: in-vivo activities in the 6-OHDA turning model and in-vitro activities. J. Pharm. Pharmacol. 1991, 43, 11–16. [Google Scholar]
- Hansen, K. T; Faarup, P; Bundgaard, H. Carbamate ester prodrugs of dopaminergic compounds: synthesis, stability, and bioconversion. J. Pharm. Sci. 1991, 80, 793–798. [Google Scholar]
- LeWitt, P.A.; Lyons, K.E.; Pahwa, R. Advanced Parkinson disease treated with rotigotine transdermal system. Neurology 2007, 68, 1262–1267. [Google Scholar]
- Michaelides, M.R.; Hong, Y.; DiDomenico, S.Jr.; Asin, K.E.; Britton, D.R.; Lin, C.W.; Williams, M.; Shiosaki, K. (5aR,11bS)-4,5,5a,6,7,11b-Hexahydro-2-propyl-3-thia-5-azacyclopent-1-ena[c]phenanthrene-9,10-diol (A-86929): a potent and selective dopamine D1 agonist that maintain behavioral efficacy following repeated administration and characterization of its diacetyl prodrug (ABT-431). J. Med. Chem. 1995, 38, 3445–3447. [Google Scholar] [CrossRef] [PubMed]
- Shiosaki, K.; Jenner, P.; Asin, K.E.; Britton, D.R.; Lin, C.W.; Michaelides, M.; Smith, L.; Bianchi, B.; DiDomenico, S.; Hodges, L.; Hong, Y.; Mahan, L.; Mikusa, J.; Miller, T.; Nikkel, A.; Stashko, M.; Witte, D.; Williams, M. ABT-431: the diacetyl prodrug of A-86929, a potent and selective dopamine D1 receptor agonist: in vitro characterization and effects in animal models of Parkinson's disease. J. Pharmacol. Exp. Ther. 1996, 276, 150–160. [Google Scholar]
- Rascol, O.; Blin, O.; Thalamas, C.; Descombes, S.; Soubrouillard, C.; Azulay, P.; Fabre, N.; Viallet, F.; Lafnitzegger, K.; Wright, S.; Carter, J. H.; Nutt, J. G. ABT-431, a D1 receptor agonist prodrug, has efficacy in Parkinson's disease. Ann. Neurol. 1999, 45, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Giardina, W. J.; Williams, M. Adrogolide HCl (ABT-431; DAS-431), a prodrug of the dopamine D1 receptor agonist, A-86929: Preclinical pharmacology and clinical data. Neurological and Urological Diseases Research. CNS Drug Rev. 2001, 7, 305–316. [Google Scholar]
- Dijkstra, D.; Venhuis, B. J.; Wikstro¨m, H. V.; Wise, L. D.; Wustrow, D. J.; Meltzer, L. T. Method for treating Parkinson’s disease by administering of (-)-5-keto-2-N,N-di-propylamino-tetrahydrotetralin. Patent WO 0128977 A1.
- Venhuis, B. J.; Wikstro¨m, H. V.; Rodenhuis, N.; Sundell, S.; Wustrow, D.; Meltzer, L. T.; Wise, L. D.; Johnson, S. J.; Dijkstra, D. A New Type of Prodrug of Catecholamines - An Opportunity to Improve the Treatment of Parkinsons Disease. J. Med. Chem. 2002, 45, 2349–2351. [Google Scholar] [CrossRef] [PubMed]
- Venhuis, B. J.; Dijkstra, D.; Wustrow, D. J.; Meltzer, L. T.; Wise, L. D.; Johnson, S. J.; Heffner, T. G.; Wikstro¨m, H. V. Orally Active Analogues of the Dopaminergic Prodrug 6-(N,N-di-npropylamino)-3,4,5,6,7,8-hexahydro-2H-naphthalen-1-one. Synthesis and Pharmacological Activity. J. Med. Chem. 2003, 46, 584–590. [Google Scholar] [CrossRef]
- Venhuis, B.J.; Dijkstra, D.; Wustrow, D.; Meltzer, L.T.; Wise, Lawrence D.; Johnson, S.J.; Wikstroem, H.V. Orally active oxime derivatives of the dopaminergic prodrug 6-(N,N-Di-n-propylamino)-3,4,5,6,7,8-hexahydro-2H-naphthalen-1-one. Synthesis and pharmacological activity. J. Med. Chem. 2003, 46, 4136–4140. [Google Scholar]
- Liu, D.; Wikstroem, H.V.; Dijkstra, D.; De Vries, J.B.; Venhuis, B. Extremely potent orally active benzo[g]quinoline analogue of the dopaminergic prodrug: 6-(N,N-Di-n-propyl)amino-3,4,5,6,7,8-hexahydro-2H-naphthalen-1-one. J. Med. Chem. 2006, 49, 1494–1498. [Google Scholar] [CrossRef] [PubMed]
- Juncos, J.L.; Mouradian, M.M.; Fabbrini, G.; Serrati, C.; Chase, T.N. Levodopa methyl ester treatment of Parkinson's disease. Neurology 1987, 37, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are not available.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Di Stefano, A.; Sozio, P.; Cerasa, L.S. Antiparkinson Prodrugs. Molecules 2008, 13, 46-68. https://doi.org/10.3390/molecules13010046
AMA Style
Di Stefano A, Sozio P, Cerasa LS. Antiparkinson Prodrugs. Molecules. 2008; 13(1):46-68. https://doi.org/10.3390/molecules13010046
Chicago/Turabian StyleDi Stefano, Antonio, Piera Sozio, and Laura Serafina Cerasa. 2008. "Antiparkinson Prodrugs" Molecules 13, no. 1: 46-68. https://doi.org/10.3390/molecules13010046