Radiation- and Photo-induced Activation of 5-Fluorouracil Prodrugs as a Strategy for the Selective Treatment of Solid Tumors

Abstract
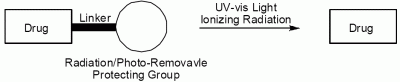
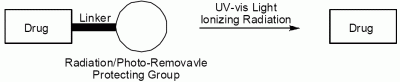
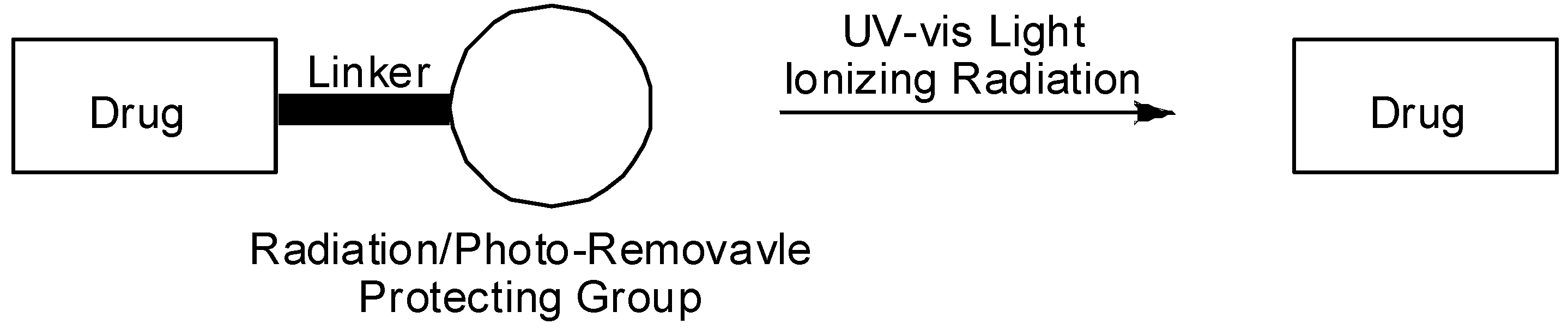
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Introduction

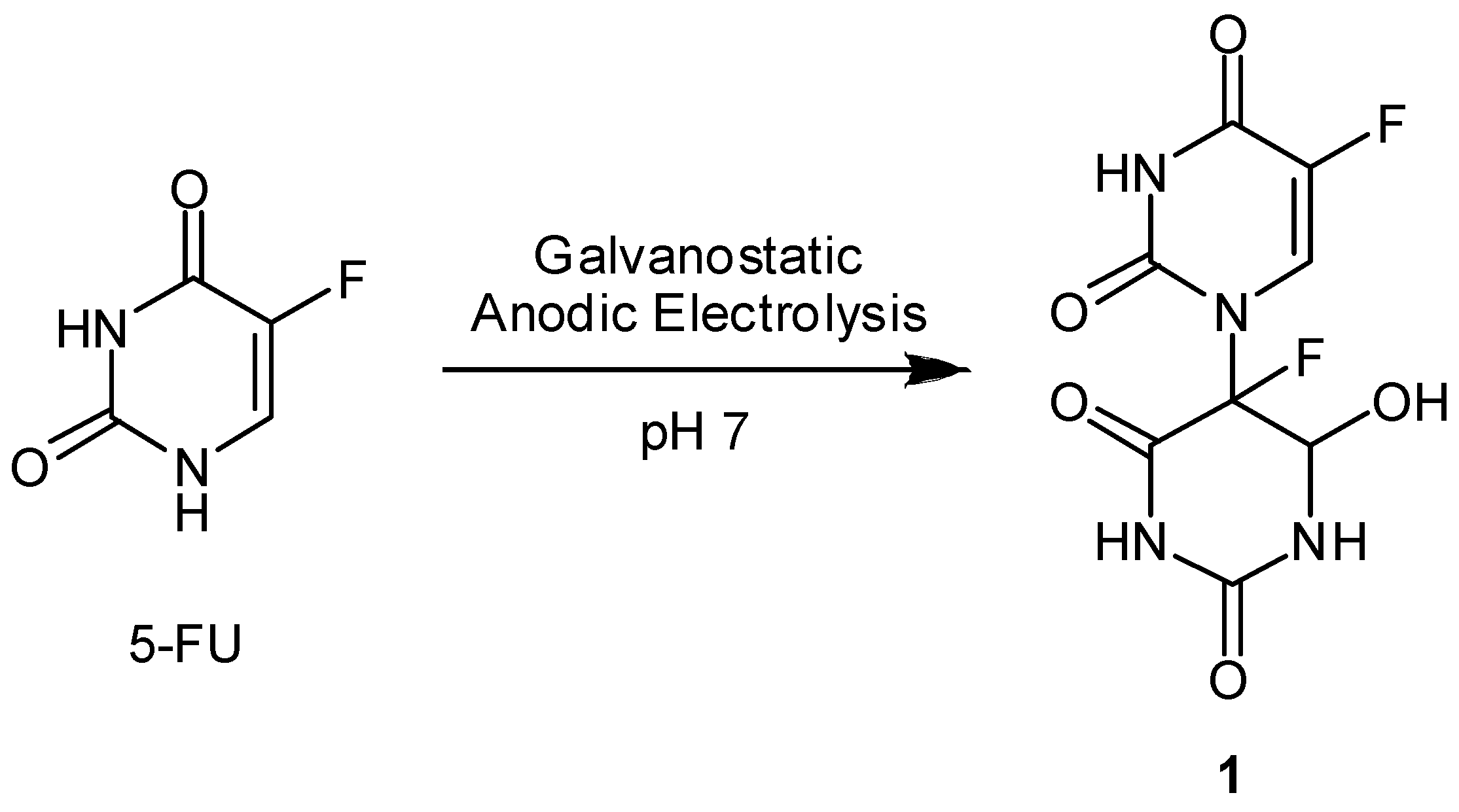
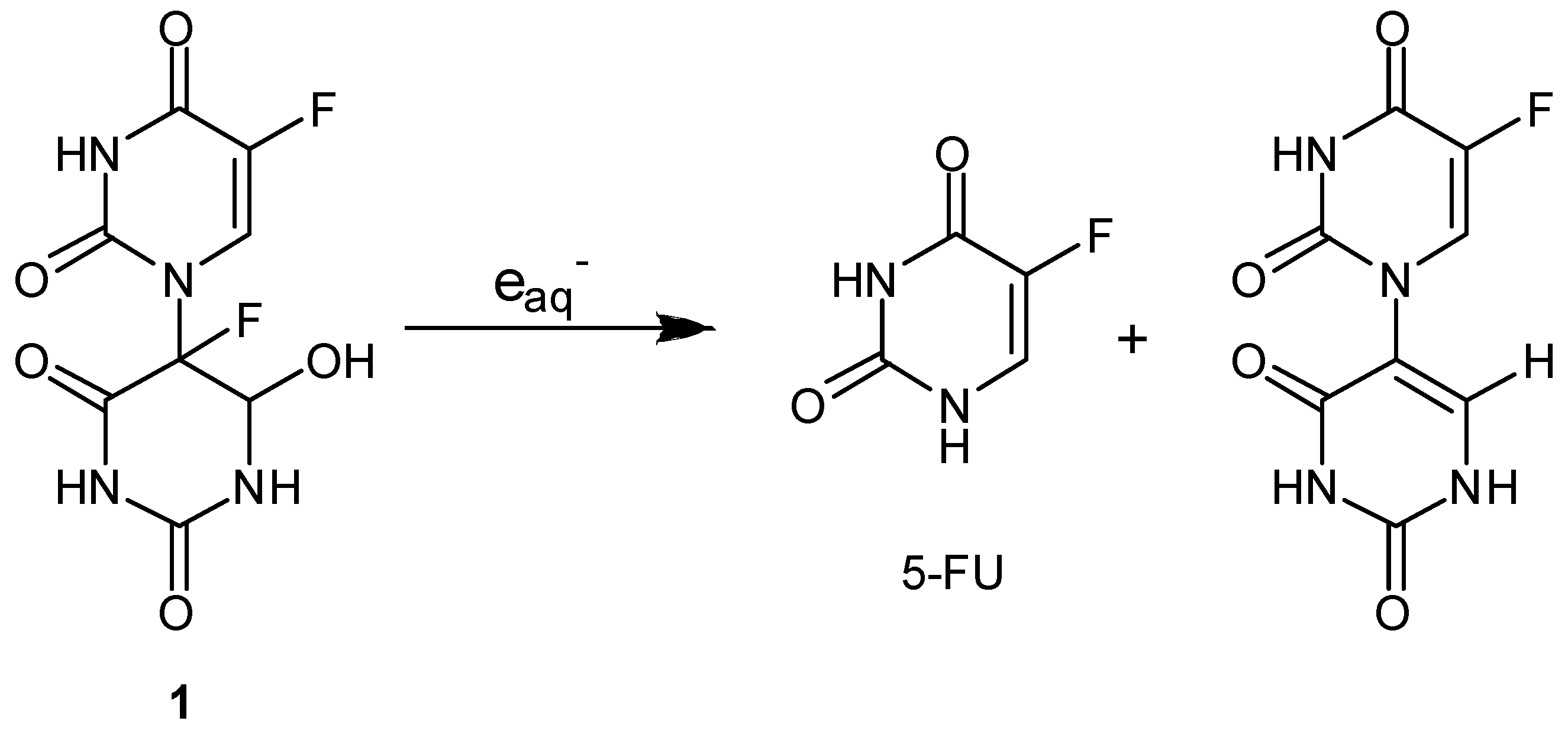
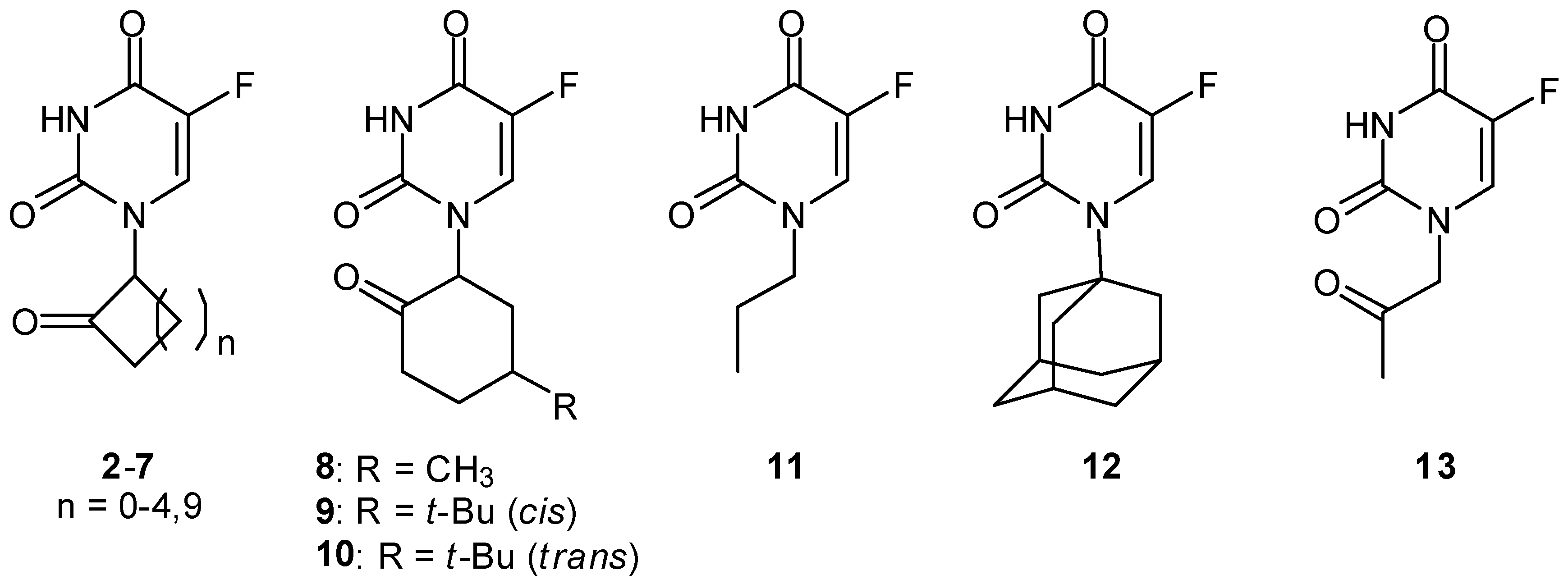
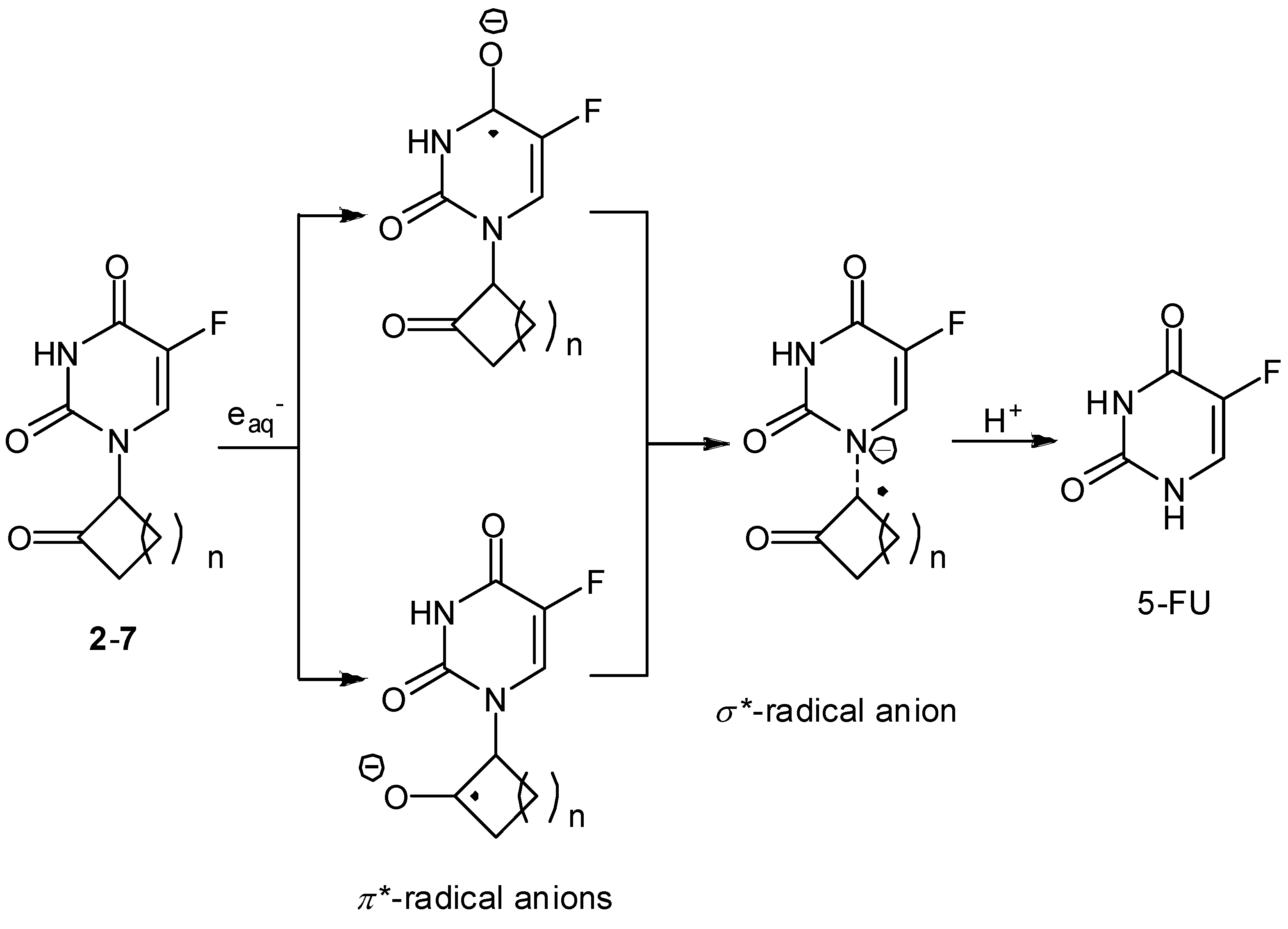
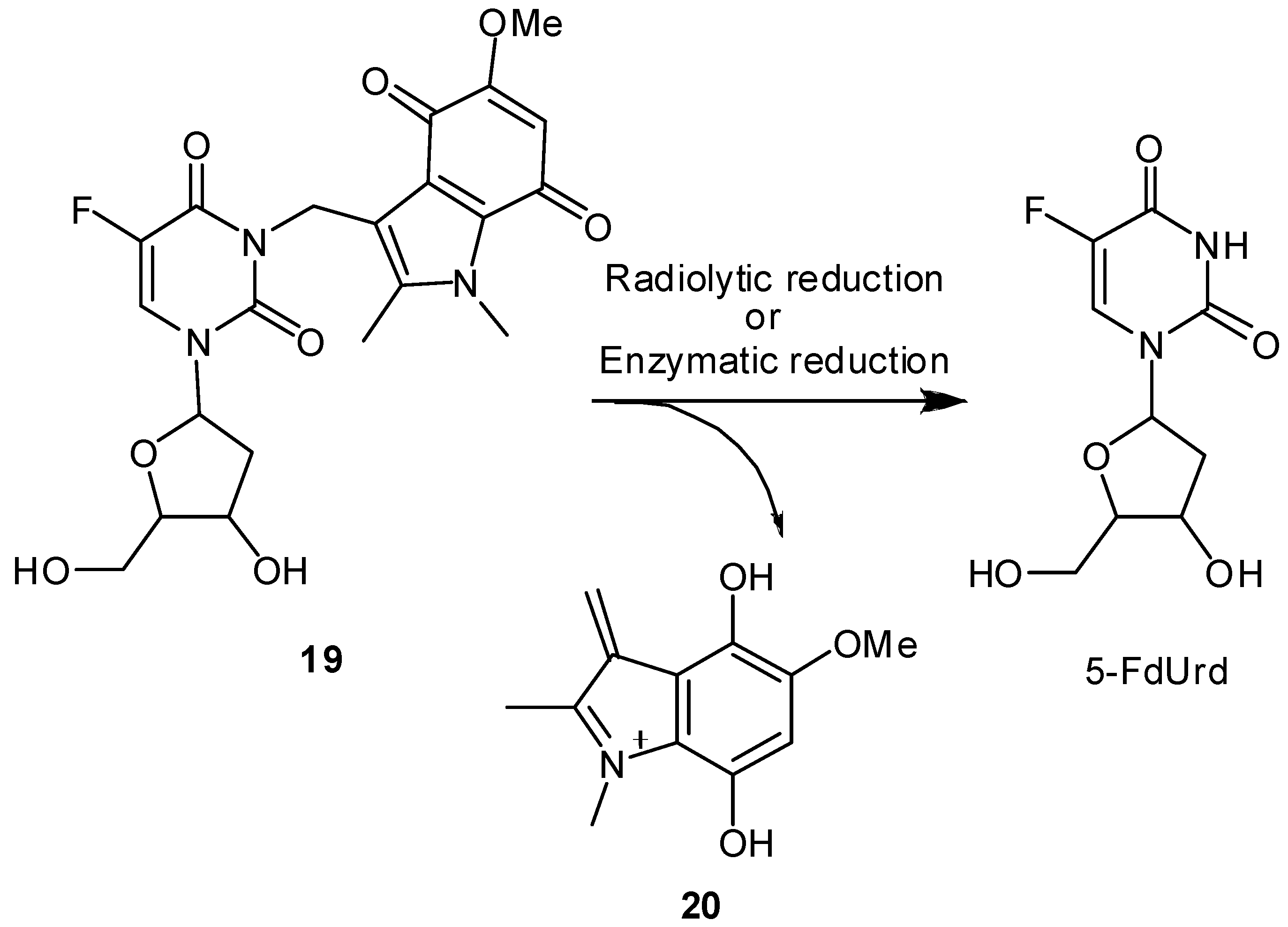
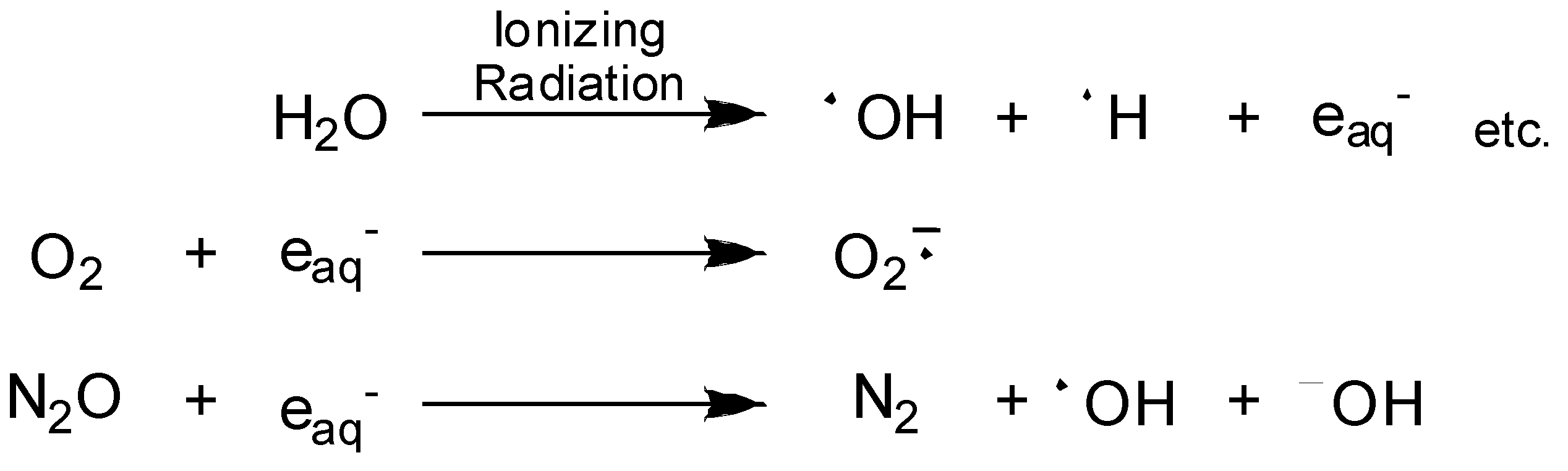
Radiation-activatable 5-FU Prodrugs







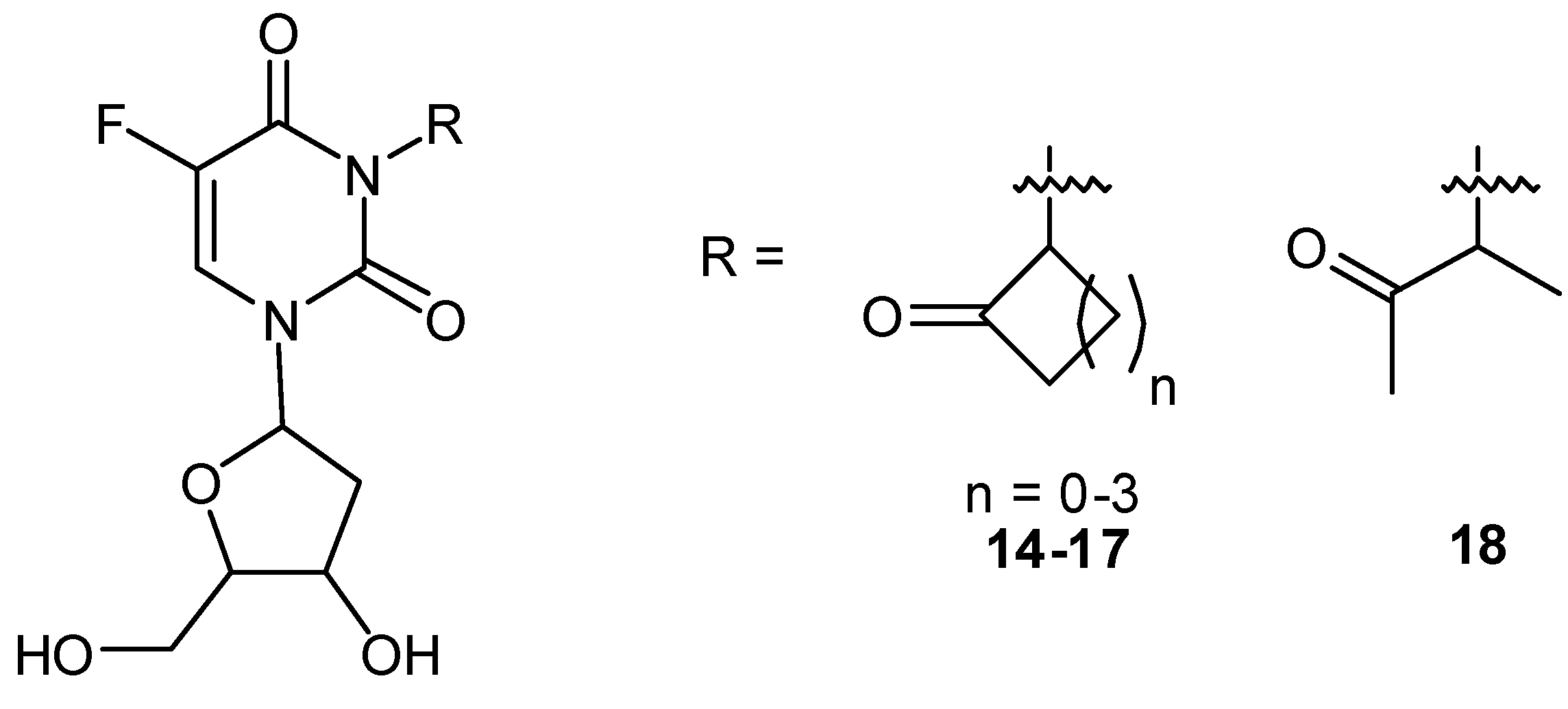
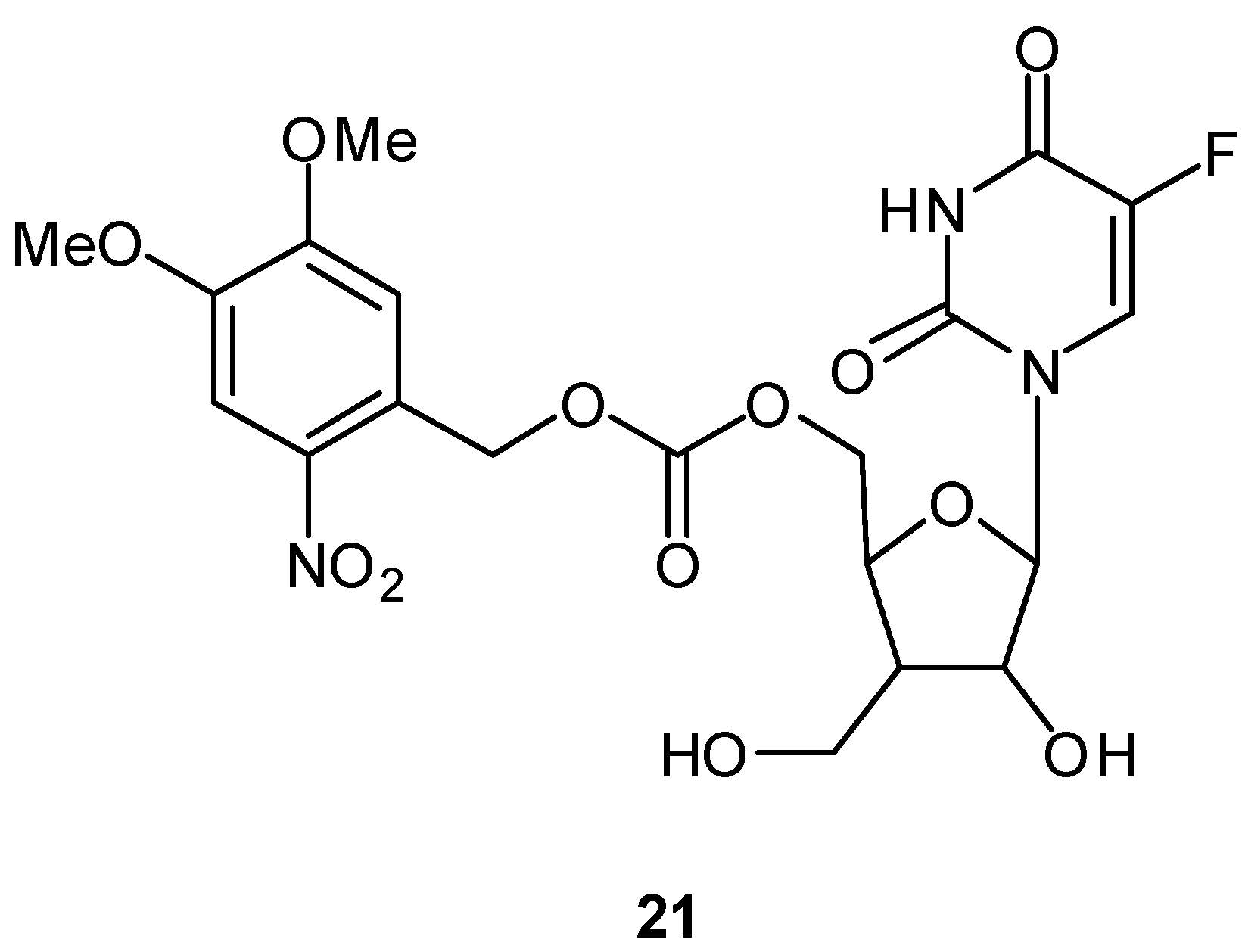
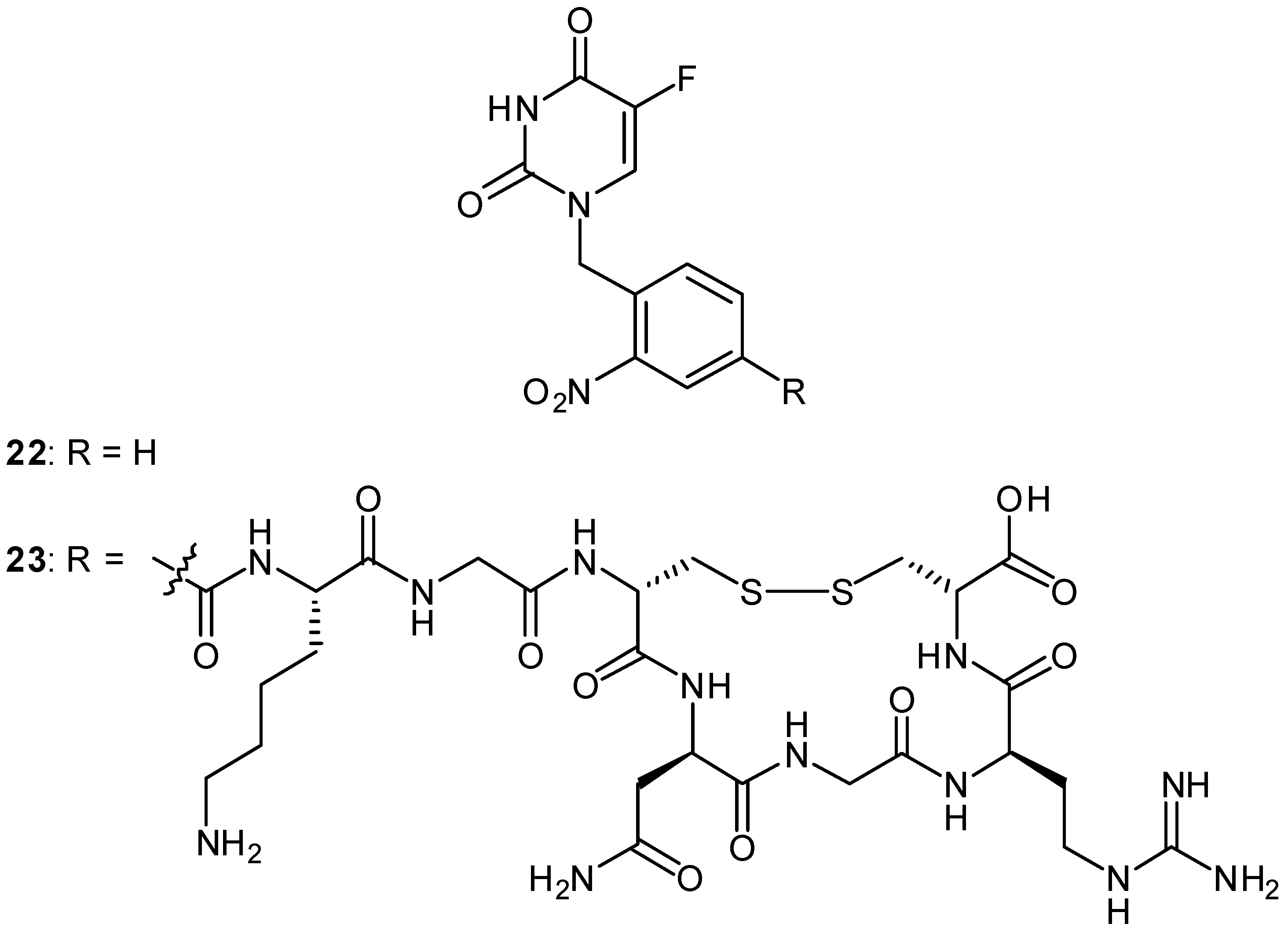
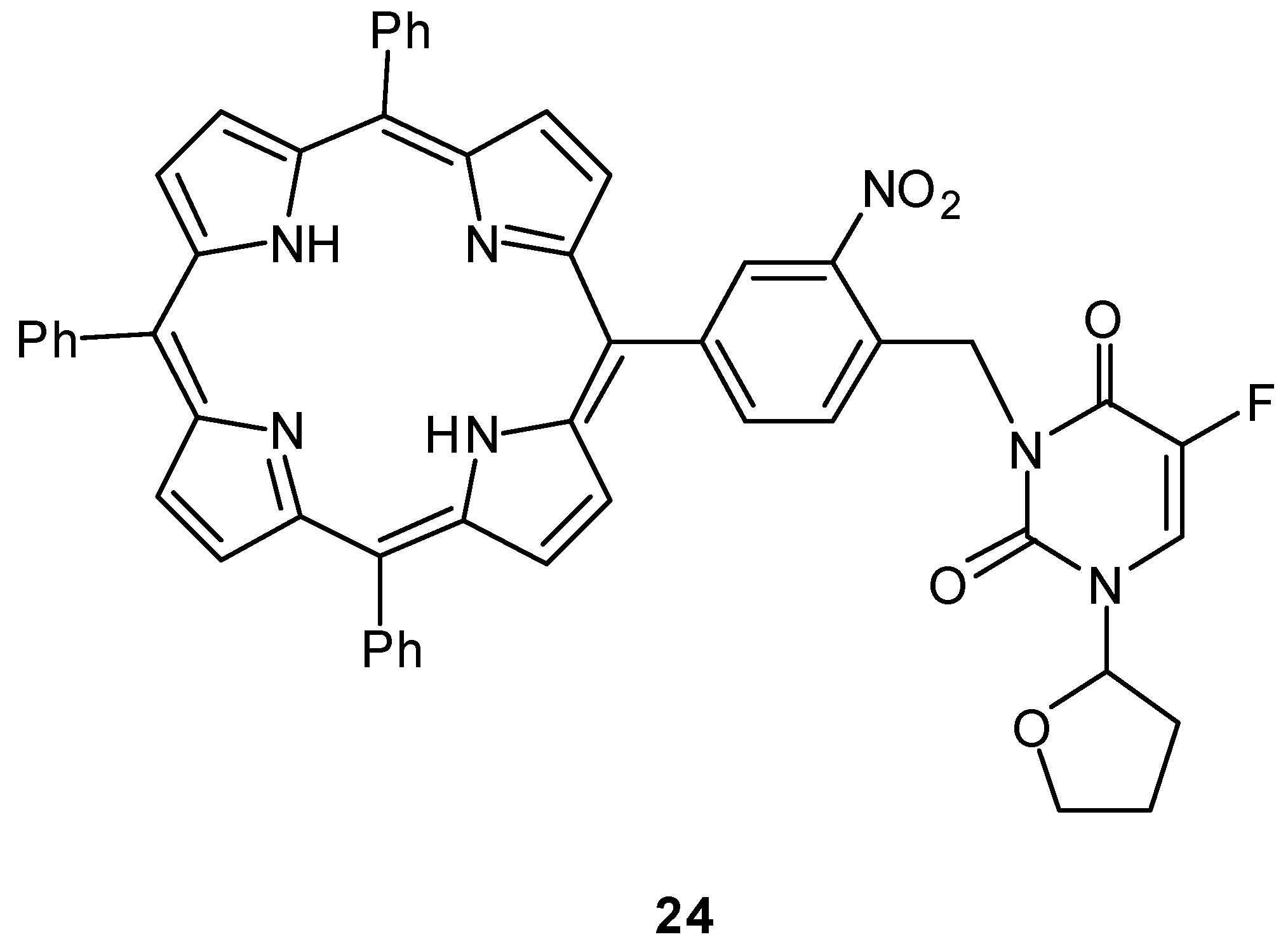
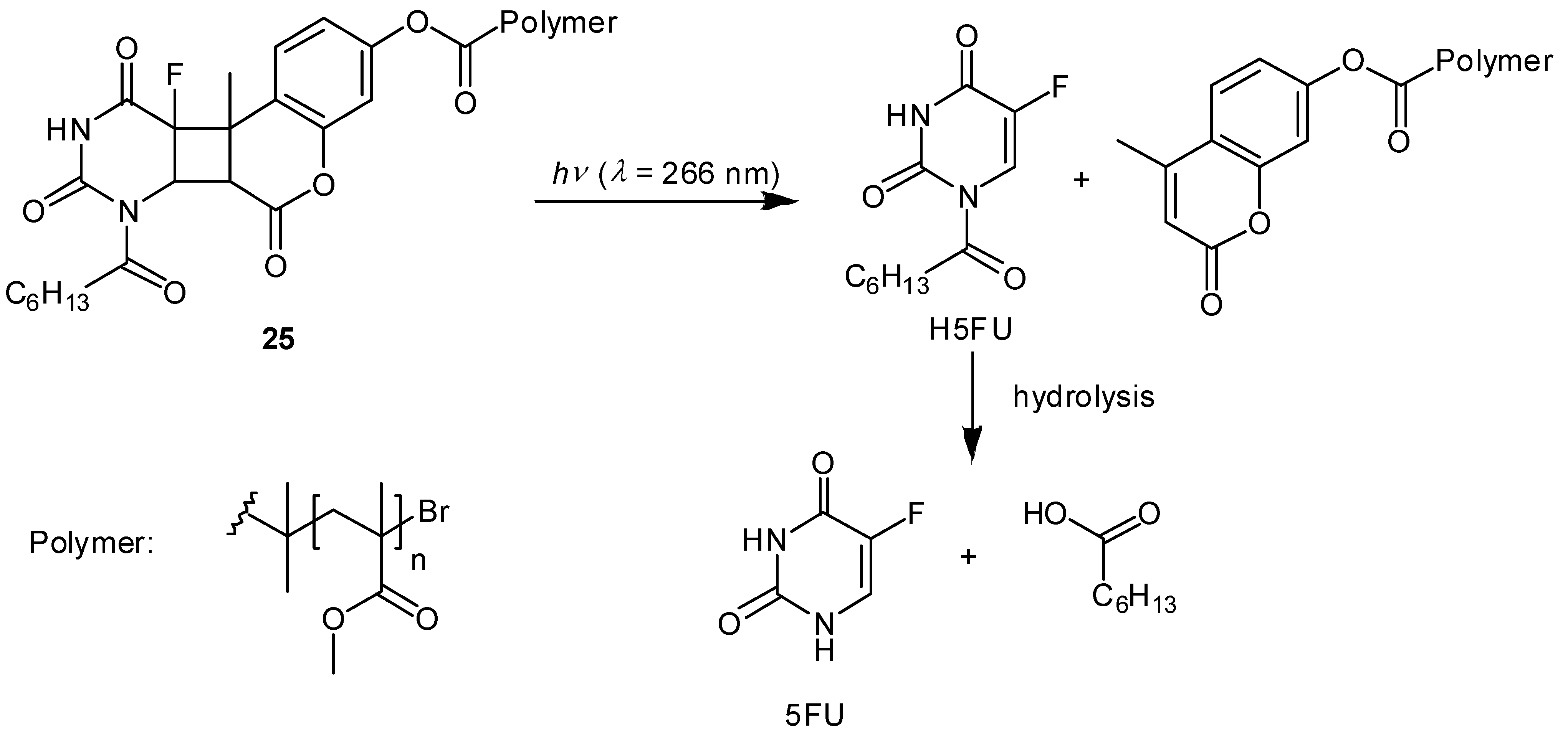
Photoactivatable 5-FU Prodrugs




Conclusions
References
- Rominger, C.J.; Gelber, R.D.; Gunderson, L.L.; Conner, N. Radiation-therapy alone or in combination with chemotherapy in the treatment of residual or inoperable carcinoma of the rectum and rectosigmoid or pelvic recurrence following colorectal surgery - Radiation-therapy oncology group-study. Am. J. Clin. Oncol. 1985, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Wolmark, N.; Fisher, B.; Rockette, H.; Redmond, C.; Wickerham, D.L.; Fisher, E.R.; Jones, J.; Glass, A.; Lerner, H.; Lawrence, W.; Prager, D.; Wexler, M.; Evans, J.; Cruz, A.; Dimitrov, N.; Jochimsen, P. Postoperative adjuvant chemotherapy or BCG for colon cancer - Results from NSABP protocol-C-01. J. Natl. Cancer Inst. 1988, 80, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.L.; Baptiste, N.; Wadler, S.; Makower, D. Thymidine phosphorylase mediates the sensitivity of human colon-carcinoma cells to 5-fluorouracil. J. Biol. Chem. 1995, 270, 19073–19077. [Google Scholar] [PubMed]
- Evrard, A.; Cuq, P.; Robert, B.; Vian, L.; Pelegrin, A.; Cano, J.P. Enhancement of 5-fluorouracil cytotoxicity by human thymidine-phosphorylase expression in cancer cells: In vitro and in vivo study. Int. J. Cancer 1999, 80, 465–470. [Google Scholar] [CrossRef]
- Hartmann, K.U.; Heidelberger, C. Studies on fluorinated pyrimidines. 13. Inhibition of thymidylate synthetase. J. Biol. Chem. 1961, 236, 3006–3013. [Google Scholar] [PubMed]
- Peters, G.J.; Vanderwilt, C.L.; Vantriest, B.; Codaccipisanelli, G.; Johnston, P.G.; Vangroeningen, C.J.; Pinedo, H.M. Thymidylate synthase and drug-resistance. Eur. J. Cancer 1995, 31A, 1299–1305. [Google Scholar] [CrossRef]
- Yeh, K.H.; Shun, C.T.; Chen, C.L.; Lin, J.T.; Lee, W.J.; Lee, P.H.; Chen, Y.C.; Cheng, A.L. High expression of thymidylate synthase is associated with the drug resistance of gastric carcinoma to high dose 5-fluorouracil-based systemic chemotherapy. Cancer 1998, 82, 1626–1631. [Google Scholar] [CrossRef]
- Albert, A. Chemical aspects of selective toxicity. Nature 1958, 182, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Himmelstein, K.J. Prodrugs and site-specific drug delivery. J. Med. Chem. 1980, 23, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Kratz, F.; Muller, I.A.; Ryppa, C.; Warnecke, A. Prodrug strategies in anticancer chemotherapy. ChemMedChem 2008, 3, 20–53. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Rooseboom, M.; Commandeur, J.N.M.; Vermeulen, N.P.E. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol. Rev. 2004, 56, 53–102. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, M.; Yamawaki, I.; Marunaka, T.; Hashimoto, S. Studies on anti-tumor agents.2. Syntheses and anti-tumor activities of 1-(tetrahydro-2-furanyl)-5-fluorouracil and 1,3- bis(tetrahydro-2-furanyl)-5-fluorouracil. J. Med. Chem. 1978, 21, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.B.; Levenson, C.H. Structure of two hydroxylated metabolites of ftorafur. Biochem. Pharmacol. 1980, 29, 665–668. [Google Scholar] [CrossRef]
- Au, J.L.; Sadee, W. The pharmacology of ftorafur (R, S-1-(tetrahydro-2-furanyl)-5-fluorouracil). Recent Results Cancer Res. 1981, 76, 100–114. [Google Scholar]
- Iigo, M.; Hoshi, A.; Nakamura, A.; Kuretani, K. Anti-tumor activity of 1-alkylcarbamoyl derivatives of 5-fluorouracil in a variety of mouse tumors. Cancer Chemother. Pharmacol. 1978, 1, 203–208. [Google Scholar] [PubMed]
- Sasaki, H.; Takahashi, T.; Mori, Y.; Nakamura, J.; Shibasaki, J. Transdermal delivery of 5- fluorouracil and its alkylcarbamoyl derivatives. Int. J. Pharm. 1990, 60, 1–9. [Google Scholar] [CrossRef]
- Grem, J.L. 5-Fluorouracil: forty-plus and still ticking. A review of its preclinical and clinical development. Invest. New Drugs 2000, 18, 299–313. [Google Scholar] [CrossRef] [PubMed]
- van Laar, J.A.M.; Rustum, Y.M.; Ackland, S.P.; van Groeningen, C.J.; Peters, G.J. Comparison of 5-fluoro-2'-deoxyuridine with 5-fluorouracil and their role in the treatment of colorectal cancer. Eur. J. Cancer 1998, 34, 296–306. [Google Scholar] [CrossRef]
- Denizli, A.; Kiremitci, M.; Piskin, E. Subcutaneous polymeric matrix system p(hema-bga) for controlled release of an anticancer drug (5-fluorouracil). 2. Release kinetics. Biomaterials 1988, 9, 363–366. [Google Scholar] [CrossRef]
- Endoh, H.; Kawaguchi, T.; Seki, T.; Hasegawa, T.; Juni, K. Controlled release of 5-fluoro-2'-deoxyuridine by the combination of prodrug and polymer matrix. Chem. Pharm. Bull. 1991, 39, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Kim, T.K.; Graham, N.B. Controlled release behavior of prodrugs based on the biodegradable poly(L-glutamic acid) microspheres. Polym. J. 1999, 31, 813–816. [Google Scholar] [CrossRef]
- Michael, N.P.; Chester, K.A.; Melton, R.G.; Robson, L.; Nicholas, W.; Boden, J.A.; Pedley, R.B.; Begent, R.H.; Sherwood, R.F.; Minton, N.P. In vitro and in vivo characterisation of a recombinant carboxypeptidase G(2)::anti-CEA scFv fusion protein. Immunotechnology 1996, 2, 47–57. [Google Scholar] [CrossRef]
- Coelho, V.; Dernedde, J.; Petrausch, U.; Panjideh, H.; Fuchs, H.; Menzel, C.; Dubel, S.; Keilholz, U.; Thiel, E.; Deckert, P.M. Design, construction, and in vitro analysis of A33scFv::CDy, a recombinant fusion protein for antibody-directed enzyme prodrug therapy in colon cancer. Int. J. Oncol. 2007, 31, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Madec-Lougerstay, R.; Florent, J.C.; Monneret, C. Synthesis of self-immolative glucuronide spacers based on aminomethylcarbamate. Application to 5-fluorouracil prodrugs for antibody-directed enzyme prodrug therapy. J. Chem. Soc. Perkin Trans. 1 1999, 1369–1375. [Google Scholar] [CrossRef]
- Fichera, A.; Michelassi, F.; Arenas, R.B. Selective expression of carcinoembryonic antigen promoter in cancer cell lines- Targeting strategy for gene therapy in colorectal cancer. Dis. Colon Rectum. 1998, 41, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Aquino, A.; Formica, V.; Prete, S.P.; Correale, P.P.; Massara, M.C.; Turriziani, M.; De Vecchis, L.; Bonmassar, E. Drug-induced increase of carcinoembryonic antigen expression in cancer cells. Pharmacol. Res. 2004, 49, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Topf, N.; Worgall, S.; Hackett, N.R. Regional 'pro-drug' gene therapy: intravenous administration of an adenoviral Vector expressing the E-coli cytosine deaminase gene and systemic administration of 5-fluorocytosine suppresses growth of hepatic metastasis of colon carcinoma. Gene Ther. 1998, 5, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A. Prodrugs for gene-directed enzyme-prodrug therapy (suicide gene therapy). J. Biomed. Biotechnol. 2003, 48–70. [Google Scholar] [CrossRef] [PubMed]
- Hatta, H.; Zhou, L.; Mori, M.; Teshima, S.; Nishimoto, S. N(1)-C(5')-linked dimer hydrates of 5-substituted uracils produced by anodic oxidation in aqueous solution. J. Org. Chem. 2001, 66, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Hatta, H.; Ueshima, H.; Kagiya, T. 1-(5'-Fluoro-6'-hydroxy-5',6'-dihydrouracil-5'- yl)-5-fluorouracil, a novel N(1)-C(5’)-linked dimer that releases 5-fluorouracil by radiation activation under hypoxic conditions. J. Med. Chem. 1992, 35, 2711–2712. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood-flow, oxygen and nutrient supply, and metabolic microenvironment of human-tumors- A review. Cancer Res. 1986, 49, 6449–6465. [Google Scholar]
- Vaupel, P.; Schlenger, K.; Knoop, C.; Höckel, M. Oxygenation of human tumors - Evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res. 1991, 51, 3316–3322. [Google Scholar] [PubMed]
- Denny, W.A. The design of drugs that target tumour hypoxia. Aust. J. Chem. 2004, 57, 821–828. [Google Scholar] [CrossRef]
- Bruno, R.D.; Njar, V.C.O. Targeting cytochrome P450 enzymes: A new approach in anti-cancer drug development. Bioorg. Med. Chem. 2007, 15, 5047–5060. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A. Hypoxia-activated anticancer drugs. Expert Opin. Ther. Pat. 2005, 15, 635–646. [Google Scholar] [CrossRef]
- Tanabe, K.; Zhang, Z.; Ito, T.; Hatta, H.; Nishimoto, S. Current molecular design of intelligent drugs and imaging probes targeting tumor-specic microenvironments. Org. Biomol. Chem. 2007, 5, 3745–3757. [Google Scholar] [CrossRef] [PubMed]
- von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair. A Chemical Perspective; Springer-Verlag: Berlin, Heidelberg, Germany, 2006. [Google Scholar]
- Mori, M.; Hatta, H.; Nishimoto, S. Stereoelectronic effect on one-electron reductive release of 5- fluorouracil from 5-fluoro-1-(2'-oxocycloalkyl)uracils as a new class of radiation-activated antitumor prodrugs. J. Org. Chem. 2000, 65, 4641–4647. [Google Scholar] [CrossRef] [PubMed]
- Shibamoto, Y.; Zhou, L.; Hatta, H.; Mori, M.; Nishimoto, S. A novel class of antitumor prodrug, 1-(2 '-oxopropyl)-5-fluorouracil (OFU001), which releases 5-fluorouracil upon hypoxic irradiation. Jpn. J. Cancer Res. 2000, 91, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Shibamoto, Y.; Zhou, L.; Hatta, H.; Mori, M.; Nishimoto, S. In vivo evaluation of a novel antitumor prodrug, 1-(2 '-oxopropyl)-5-fluorouracil (OFU001), which releases 5-fluorouracil upon hypoxic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 407–413. [Google Scholar] [CrossRef]
- Tanabe, K.; Mimasu, Y.; Eto, A.; Tachi, Y.; Sakakibara, S.; Mori, M.; Hatta, H.; Nishimoto, S. One-electron reduction characteristics of N(3)-substituted 5-fluorodeoxyuridines synthesized as radiation-activated prodrugs. Bioorg. Med. Chem. 2003, 11, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Shibamoto, Y.; Mimasu, Y.; Tachi, Y.; Hatta, H.; Nishimoto, S. Comparison of 5-fluorouracil and 5-fluoro-2'-deoxyuridine as an effector in radiation-activated prodrugs. J. Chemother. 2002, 14, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Shibamoto, Y.; Tachi, Y.; Tanabe, K.; Hatta, H.; Nishimoto, S. In vitro and in vivo evaluation of novel antitumor prodrugs of 5-fluoro-2'-deoxyuridine activated by hypoxic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Makimura, Y.; Tachi, Y.; Imagawa-Sato, A.; Nishimoto, S. Hypoxia-selective activation of 5-fluorodeoxyuridine prodrug possessing indolequinone structure: Radiolytic reduction and cytotoxicity characteristics. Bioorg. Med. Chem. Lett. 2005, 15, 2321–2324. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.A.; Swann, E.; Everett, S.A.; Jaffar, M.; Nolan, J.; Robertson, N.; Lockyer, S.D.; Patel, K.B.; Dennis, M.F.; Stratford, M.R.L.; Wardman, P.; Adams, G.E.; Moody, C.J.; Stratford, I.J. Indolequinone antitumor agents: reductive activation and elimination from (5- methoxy-1-methyl-4,7-dioxoindol-3-yl)methyl derivatives and hypoxia-selective cytotoxicity in vitro. J. Med. Chem. 1998, 41, 2720–2731. [Google Scholar] [CrossRef] [PubMed]
- Swann, E.; Barraja, P.; Oberlander, A.M.; Gardipee, W.T.; Hudnott, A.R.; Beall, H.D.; Moody, C.J. Indolequinone antitumor agents: correlation between quinine structure and rate of metabolism by recombinant human NAD(P)H: quinine oxidoreductase. Part 2. J. Med. Chem. 2001, 44, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Hernick, M.; Flader, C.; Borch, R.F. Design, synthesis, and biological evaluation of indolequinone phosphoramidate prodrugs targeted to DT-diaphorase. J. Med. Chem. 2002, 45, 3540–3548. [Google Scholar] [CrossRef] [PubMed]
- Everett, S.A.; Swann, E.; Naylor, M.A.; Stratford, M.R.L.; Patel, K.B.; Tian, A.; Newman, R.G.; Vojnovic, B.; Moody, C.J.; Wardman, P. Modifying rates of reductive elimination of leaving groups from indolequinone prodrugs: A key factor in controlling hypoxia-selective drug release. Biochem. Pharmacol. 2002, 63, 1629–1639. [Google Scholar] [CrossRef]
- Skibo, E.B.; Xing, C.; Groy, T. Recognition and cleavage at the DNA major groove. Bioorg. Med. Chem. 2001, 9, 2445–2459. [Google Scholar] [CrossRef]
- Dormán, G.; Prestwich, G.D. Using photolabile ligands in drug discovery and development. Trends Biotech. 2000, 18, 64–77. [Google Scholar]
- Reinhard, R.; Schmidt, B.F. Nitrobenzyl-based photosensitive phosphoramide mustards: Synthesis and photochemical properties of potential prodrugs for cancer therapy. J. Org. Chem. 1998, 63, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Sato, M.; Sakamoto, S.; Yamaguchi, K.; Iwamura, M. New dendritic caged compounds: synthesis, mass spectrometric characterization, and photochemical properties of dentrimers with a-carboxy-2-nitrobenzyl caged compounds at their periphery. J. Am. Chem. Soc. 2000, 122, 12588–12589. [Google Scholar] [CrossRef]
- Mizuta, H.; Watanabe, S.; Sakurai, Y.; Nishiyama, K.; Furuta, T.; Kobayashi, Y.; Iwamura, M. Design, synthesis, photochemical properties and cytotoxic activities of water-soluble caged L-leucyl-L-leucine methyl esters that control apoptosis of immune cells. Bioorg. Med. Chem. 2002, 10, 675–683. [Google Scholar] [CrossRef]
- McCoy, C.P.; Rooney, C.; Edwards, C.R.; Jones, D.S.; Gorman, S.P. Light-triggered molecule-scale drug dosing devices. J. Am. Chem. Soc. 2007, 129, 9572–9573. [Google Scholar] [CrossRef] [PubMed]
- McCoy, C.P.; Rooney, C.; Jones, D.S.; Gorman, S.P.; Nieuwenhuyzen, M. Rational design of a dual-mode optical and chemical prodrug. Pharm. Res. 2007, 24, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Tachi, Y.; Dai, W.-M.; Tanabe, K.; Nishimoto, S. Synthesis and DNA cleavage reaction characteristics of enediyne prodrugs activated via an allylic rearrangement by base or UV irradiation. Bioorg. Med. Chem. 2006, 14, 3199–3209. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Noguchi, M.; Hirota, S.; Sohma, Y.; Kimura, T.; Hayashi, Y.; Kiso, Y. Development of first photoresponsive prodrug of paclitaxel. Bioorg. Med. Chem. Lett. 2006, 16, 4492–4496. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Skwarczynski, M.; Prakash, H.; Hirota, S.; Kimura, T.; Hayashi, Y.; Kiso, Y. Development of novel water-soluble photocleavable protecting group and its application for design of photoresponsive paclitaxel prodrugs. Bioorg. Med. Chem. 2008, 16, 5389–5397. [Google Scholar] [CrossRef] [PubMed]
- Willner, I.; Willner, B. Biological Applications of Photochemical Switches; Morrison, H., Ed.; John Wiley & Sons, Inc: New York, USA, 1993. [Google Scholar]
- Wei, Y.; Yan, Y.; Pei, D.; Gong, B. A photoactivated prodrug. Bioorg. Med. Chem. Lett. 1998, 8, 2419–2422. [Google Scholar] [CrossRef]
- Zhang, Z.; Hatta, H.; Ito, T.; Nishimoto, S. Synthesis and photochemical properties of photoactivated antitumor prodrugs releasing 5-fluorouracil. Org. Biomol. Chem. 2005, 3, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Peng, D.; Wang, B.; Long, L.; Guo, C.; Yuan, J. A model for light-triggered porphyrin anticancer prodrugs based on an o-nitrobenzyl photolabile group. Eur. J. Org. Chem. 2008, 793, 796. [Google Scholar] [CrossRef]
- Takiuchi, H.; Ajani, J.A. Uracil-tegafur in gastric carcinoma: a comprehensive review. J. Clin. Oncol. 1998, 16, 2877–2885. [Google Scholar] [PubMed]
- Sinkel, C.; Greiner, A.; Agarwal, S. Synthesis, characterization, and properties evaluation of methylcoumarin end-functionalized poly(methyl methacrylate) for photoinduced drug release. Macromolecules 2008, 41, 3460–3467. [Google Scholar] [CrossRef]
- Sample Availability: Samples are not available.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ito, T.; Tanabe, K.; Yamada, H.; Hatta, H.; Nishimoto, S.-i. Radiation- and Photo-induced Activation of 5-Fluorouracil Prodrugs as a Strategy for the Selective Treatment of Solid Tumors. Molecules 2008, 13, 2370-2384. https://doi.org/10.3390/molecules13102370
Ito T, Tanabe K, Yamada H, Hatta H, Nishimoto S-i. Radiation- and Photo-induced Activation of 5-Fluorouracil Prodrugs as a Strategy for the Selective Treatment of Solid Tumors. Molecules. 2008; 13(10):2370-2384. https://doi.org/10.3390/molecules13102370
Chicago/Turabian StyleIto, Takeo, Kazuhito Tanabe, Hisatsugu Yamada, Hiroshi Hatta, and Sei-ichi Nishimoto. 2008. "Radiation- and Photo-induced Activation of 5-Fluorouracil Prodrugs as a Strategy for the Selective Treatment of Solid Tumors" Molecules 13, no. 10: 2370-2384. https://doi.org/10.3390/molecules13102370