4-Azatricyclo[5.2.2.02,6]undecane-3,5,8-triones as Potential Pharmacological Agents

Abstract
:Introduction
Results and Discussion





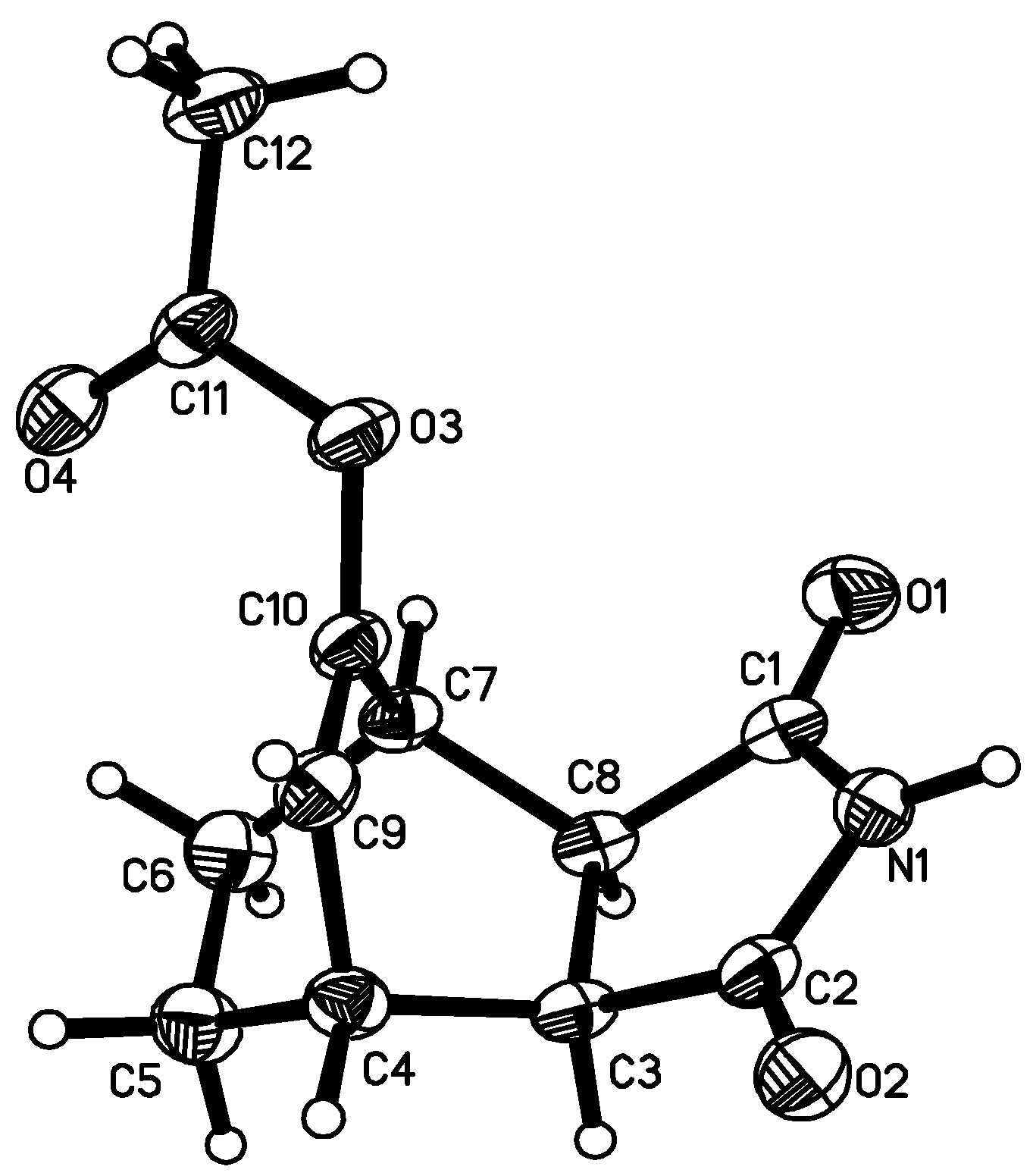{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | aCC50 | bEC50 |
|---|---|---|
| MT-4 | HIV-1 | |
| 2, 3, 3a, 3b, 3c, 3d, 3f, 3h, 4a, 4b, 4c, 4d, 4e, 4f, 4g, 4h, 5a, 5b, 5c, 5d, 5e, 5f, 5g, 5h, 5i, 5j | >100 | >100 |
Experimental
General
Synthesis of 3,5-dioxo-4-azatricyclo[5.2.2.02,6]undec-8-en-8-yl acetate (1)
Synthesis of 4-azatricyclo[5.2.2.02,6]undecane-3,5,8-trione (2)
General method for preparation of 4-(3-bromopropyl)- and 4-(4-bromobutyl)-4-aza-tricyclo[5.2.2.02,6]undecane-3,5,8-trione (3 and 4)
General method for preparation of 4-substituted arylpiperazines with derivatives 3 and 4 (3a–4h)
Synthesis of 4-(oxiran-2-ylmethyl)-4-azatricyclo[5.2.2.02,6]undecane-3,5,8-trione (5)
General method for preparation of 4-(amino)-2-hydroxypropyl derivatives of 4-aza-tricyclo[5.2.2.02,6]undecane-3,5,8-trione 5a–5j
Biological Assays: Compounds
Cells and Viruses
Cytotoxicity Assays
Acknowledgments
References
- Carpenter, C.C.; Fischl, M.A.; Hammer, S.M.; Hirsch, M.S.; Jacobsen, D.M.; Katzenstein, D.A.; Montaner, J.S.; Richman, D.D.; Saag, M.S.; Schooley, R.T.; Thompson, M.A.; Vella, S.; Yeni, P.G.; Volberding, P.A. Antiretroviral therapy for HIV infection in 1998: updated recommendations of the International AIDS Society-USA Panel. J. Am. Med. Assoc. 1998, 280, 78–86. [Google Scholar] [CrossRef]
- Romero, D.L.; Morge, R.A.; Genin, M.J.; Biles, C.; Busso, M.; Resnick, L.; Althaus, I.W.; Reusser, F.; Thomas, R.C.; Tarpley, W.G. Bis(heteroaryl)piperazine (BHAP) reverse transcriptase inhibitors: structure-activity relationships of novel substituted indole analogues and the identification of 1-[(5-methanesulfonamido-1H-indol-2-yl)-carbonyl]-4-[3-[(1-methylethyl)-amino]-pyridinyl]piperazine monomethanesulfonate (U-90152S), a second-generation clinical candidate. J. Med. Chem. 1993, 36, 1505–1508. [Google Scholar] [CrossRef]
- Gulick, R.M.; Su, Z.; Flexner, C.; Hughes, M.D.; Skolnik, P.R.; Wilkin, T.J.; Gross, R.; Krambrink, A.; Coakley, E.; Greaves, W.L.; Zolopa, A.; Reichman, R.; Godfrey, C.; Hirsch, M.; Kuritzkes, D.R. Phase 2 study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-Infected, treatment-experienced patients: AIDS clinical trials group 5211. J. Infect. Dis. 2007, 196, 304–312. [Google Scholar] [CrossRef]
- Pinna, G.A.; Loriga, G.; Murineddu, G.; Grella, G.; Mura, M.; Vargiu, L.; Murgioni, C.; La Colla, P. Synthesis and anti-HIV-1 activity of new delavirdine analogues carrying arylpyrrole moieties. Chem. Pharm. Bull. 2001, 49, 1406–1411. [Google Scholar] [CrossRef]
- Tagat, J.R.; McCombie, S.W.; Nazareno, D.; Labroli, M.A.; Xiao, Y.; Steensma, R.W.; Strizki, J.M.; Baroudy, B.M.; Cox, K.; Lachowicz, J.; Varty, G.; Watkins, R. Piperazine-based CCR5 antagonists as HIV-1 inhibitors. IV. Discovery of 1-[(4,6-dimethyl-5-pyrimidinyl)carbonyl]- 4-[4-[2-methoxy-1(R)-4-(trifluoromethyl)phenyl]ethyl-3(S)-methyl-1-piperazinyl]- 4-methylpiperidine (Sch-417690/Sch-D), a potent, highly selective, and orally bioavailable CCR5 antagonist. J. Med. Chem. 2004, 47, 2405–2408. [Google Scholar] [CrossRef]
- Richter, S.; Parolin, C.; Palumbo, M.; Palu, G. Antiviral properties of quinolone-based drugs. Curr. Drug. Targets Infect. Disord. 2004, 4, 111–116. [Google Scholar] [CrossRef]
- Chan, T.M.; Cox, K.; Feng, W.; Miller, M.W.; Weston, D.; McCombie, S.W. Piperidinyl piperazine derivatives useful as inhibitors of chemokine receptors. U.S. Pat. 2006223821, 2006. [Google Scholar]
- Filosa, R.; Peduto, A.; de Caprariis, P.; Saturnino, C.; Festa, M.; Petrella, A.; Pau, A.; Pinna, G.A.; La Colla, P.; Busonera, B.; Loddo, R. Synthesis and antiproliferative properties of N3/8-disubstituted 3,8-diazabicyclo[3.2.1]octane analogues of 3,8-bis[2-(3,4,5-trimethoxyphenyl)-pyridin-4-yl]methyl-piperazine. Eur. J. Med. Chem. 2007, 42, 293–306. [Google Scholar]
- Shaw, Y.J.; Yang, Y.T.; Garrison, J.B.; Kyprianou, N.; Chen, C.S. Pharmacological exploitation of the alpha1-adrenoreceptor antagonist doxazosin to develop a novel class of antitumor agents that block intracellular protein kinase B/Akt activation. J. Med. Chem. 2004, 47, 4453–4462. [Google Scholar] [CrossRef]
- Kimura, M.; Masuda, T.; Yamada, K.; Kawakatsu, N.; Kubota, N.; Mitani, M.; Kishii, K.; Inazu, M.; Kiuchi, Y.; Oguchi, K.; Namiki, T. Antioxidative activities of novel diphenylalkyl piperazine derivatives with high affinities for the dopamine transporter. Bioorg. Med. Chem. Lett. 2004, 14, 4287–4290. [Google Scholar] [CrossRef]
- Foroumadi, A.; Emami, S.; Hassanzadeh, A.; Rajaee, M.; Sokhanvar, K.; Moshafi, M.H.; Shafiee, A. Synthesis and antibacterial activity of N-(5-benzylthio-1,3,4-thiadiazol-2-yl) and N-(5-benzylsulfonyl-1,3,4-thiadiazol-2-yl)piperazinyl quinolone derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 4488–4492. [Google Scholar] [CrossRef]
- Bartok, M.; Felfoldi, K.; Karpati, E.; Molnar, A.; Szporny, L. 3-[4-(2'-Pyridyl)-piperazin-1-yl]-1-(3,4,5-trimethoxybenzoyloxy)-propane or a pharmaceutically acceptable acid-addition salt thereof in a composition with anti-arrhythmic activity. U.S. Pat. US4196206, 1980. [Google Scholar]
- Mlynárová, R.; Tazká, D.; Racanská, E.; Kyselovic, J.; Svec, P. Effects of a (fluorophenyl) piperazine derivative (substance IIIv) on cardiovascular function. Ceska Slov. Farm. 2000, 49, 177–180. [Google Scholar]
- Cecchetti, V.; Schiaffella, F.; Tabarrini, O.; Fravolini, A. (1,4-Benzothiazinyloxy) alkylpiperazine derivatives as potential antihypertensive agents. Bioorg. Med. Chem. Lett. 2000, 10, 465–468. [Google Scholar] [CrossRef]
- Dutta, A.K.; Venkataraman, S.K.; Fei, X.S.; Kolhatkar, R.; Zhang, S.; Reith, M.E. Synthesis and biological characterization of novel hybrid 7-[[2-(4-phenyl-piperazin-1-yl)-ethyl]-propyl-amino]-5,6,7,8-tetrahydro-naphthalen-2-ol and their heterocyclic bioisosteric analogues for dopamine D2 and D3 receptors. Bioorg. Med. Chem. 2004, 12, 4361–4373. [Google Scholar] [CrossRef]
- Betti, L.; Zanelli, M.; Giannaccini, G.; Manetti, F.; Schenone, S.; Strappaghetti, G. Synthesis of new piperazine-pyridazinone derivatives and their binding affinity toward alpha1-, alpha2-adrenergic and 5-HT1A serotoninergic receptors. Bioorg. Med. Chem. 2006, 14, 2828–2836. [Google Scholar] [CrossRef]
- Obniska, J.; Kołaczkowski, M.; Bojarski, A.J.; Duszyńska, B. Synthesis, anticonvulsant activity and 5-HT1A, 5-HT2A receptor affinity of new N-[(4-arylpiperazin-1-yl)-alkyl] derivatives of 2-azaspiro[4.4]nonane and [4.5]decane-1,3-dione. Eur. J. Med. Chem. 2006, 41, 874–881. [Google Scholar] [CrossRef]
- Tandon, M.; O'Donnell, M.M.; Porte, A.; Vensel, D.; Yang, D.; Palma, R.; Beresford, A.; Ashwell, M.A. The design and preparation of metabolically protected new arylpiperazine 5-HT1A ligands. Bioorg. Med. Chem. Lett. 2004, 14, 1709–1712. [Google Scholar] [CrossRef]
- Kossakowski, J.; Raszkiewicz, A.; Bugno, R.; Bojarski, A.J. Introduction of a new complex imide system into the structure of LCAPs. The synthesis and a 5-HT1A, 5-HT2A and D2 receptor binding study. Pol. J. Pharmacol. 2004, 56, 843–848. [Google Scholar]
- Sheldrick, G.M. Shelxs-97. In Program for a crystal structure solution; University of Göttingen: Göttingen , Germany, 1997.
- Sheldrick, G.M. Shelxl-97. In Program for the refinement of a crystal structure from diffraction data; University of Göttingen: Göttingen, Germany, 1997.
- Pawels, R.; Balzarini, J.; Baba, M.; Snoeck, R.; Schols, D.; Herdewijn, P.; Desmyster, J.; De Clercq, E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Meth. 1988, 20, 309–321. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kossakowski, J.; Bielenica, A.; Mirosław, B.; Kozioł, A.E.; Dybała, I.; Struga, M. 4-Azatricyclo[5.2.2.02,6]undecane-3,5,8-triones as Potential Pharmacological Agents. Molecules 2008, 13, 1570-1583. https://doi.org/10.3390/molecules13081570
Kossakowski J, Bielenica A, Mirosław B, Kozioł AE, Dybała I, Struga M. 4-Azatricyclo[5.2.2.02,6]undecane-3,5,8-triones as Potential Pharmacological Agents. Molecules. 2008; 13(8):1570-1583. https://doi.org/10.3390/molecules13081570
Chicago/Turabian StyleKossakowski, Jerzy, Anna Bielenica, Barbara Mirosław, Anna E. Kozioł, Izabela Dybała, and Marta Struga. 2008. "4-Azatricyclo[5.2.2.02,6]undecane-3,5,8-triones as Potential Pharmacological Agents" Molecules 13, no. 8: 1570-1583. https://doi.org/10.3390/molecules13081570