Synthesis and Characterization of Some Chiral Metal-Salen Complexes Bearing a Ferrocenophane Substituent

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
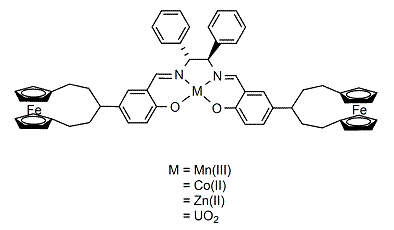
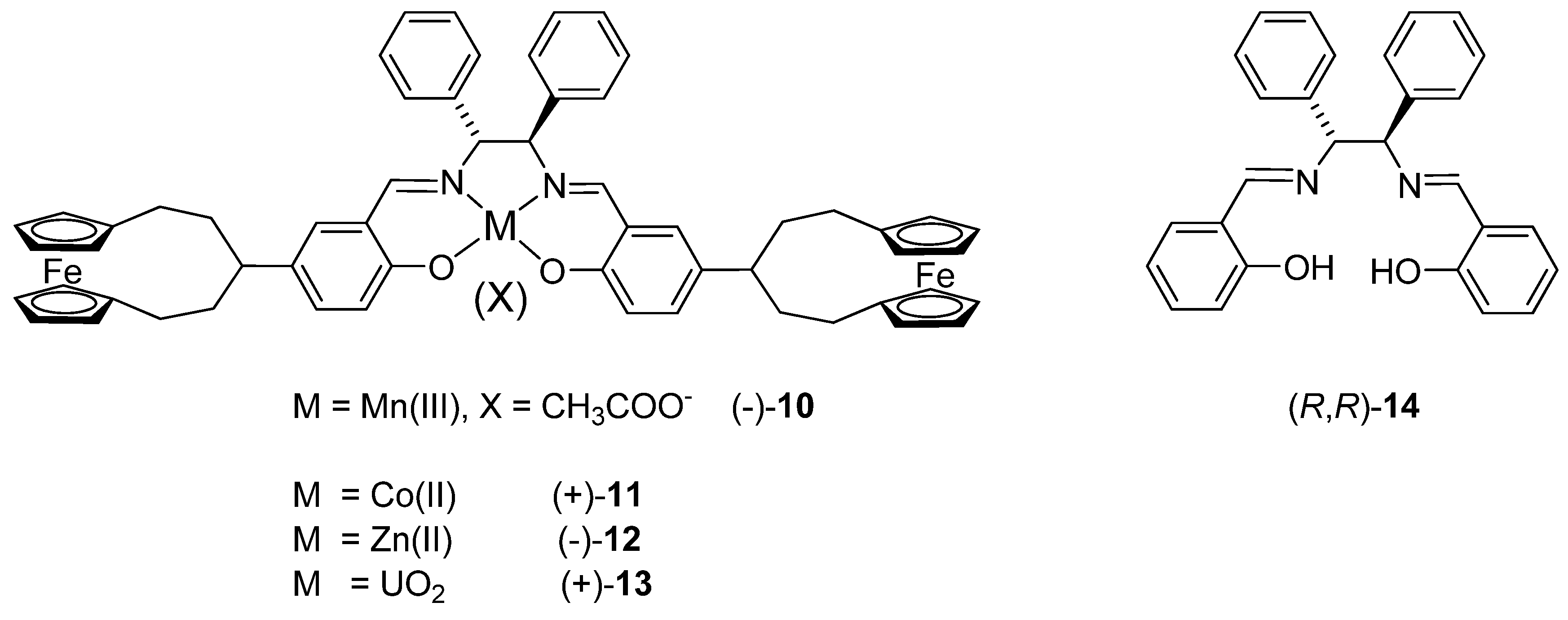
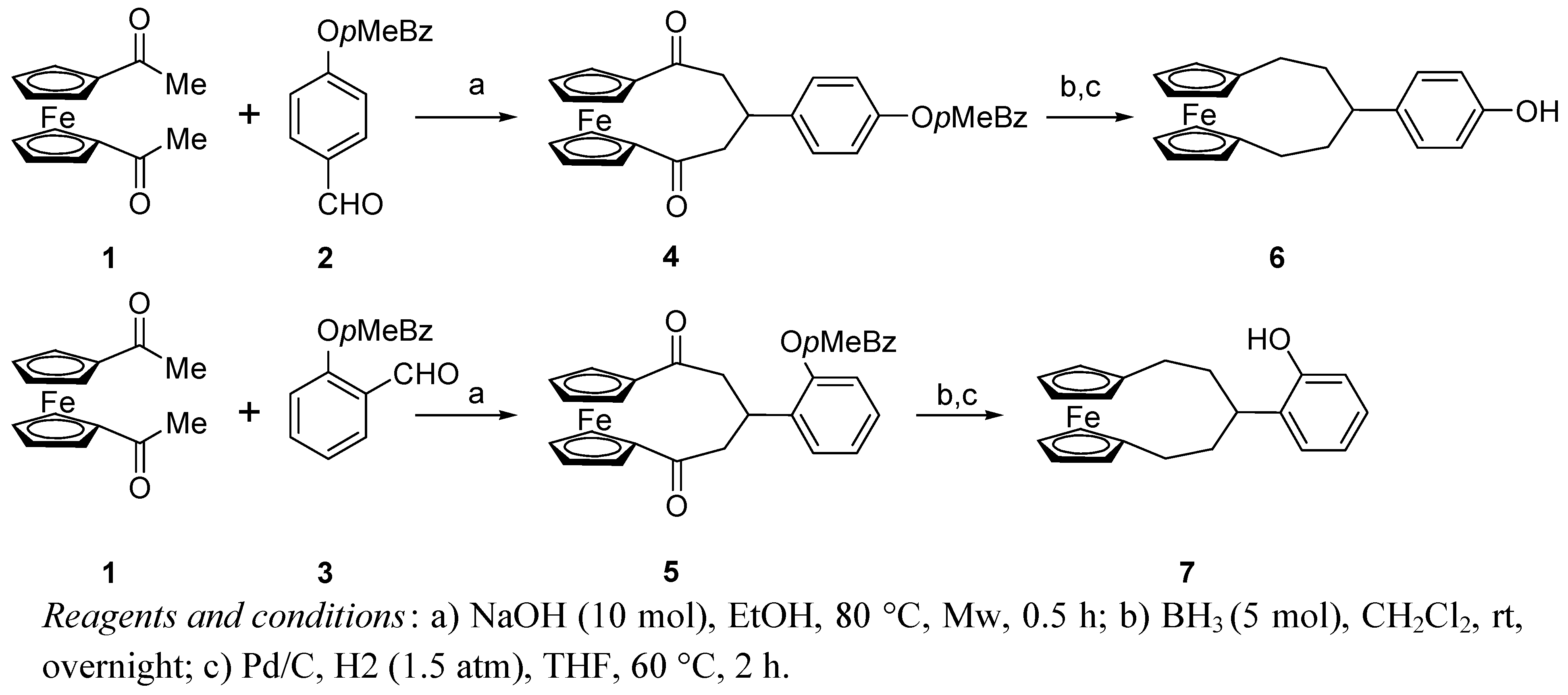
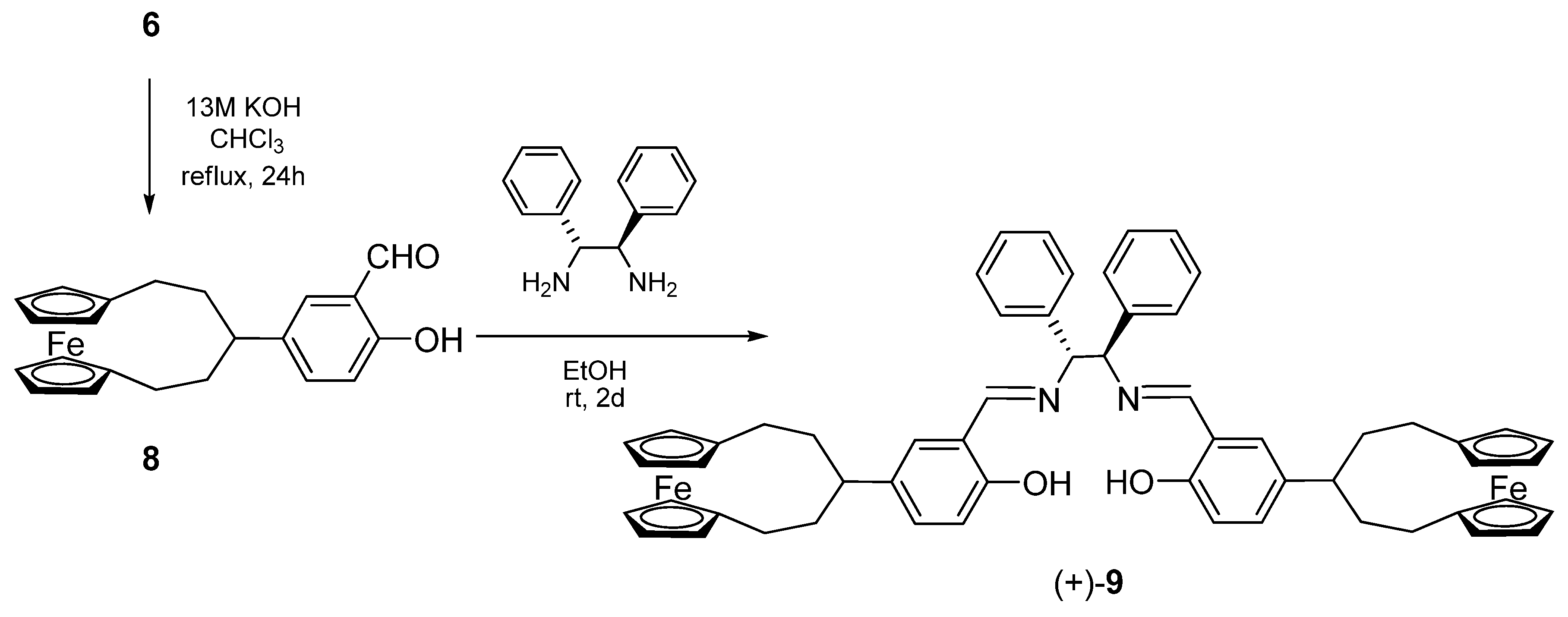
2.1. Synthesis of the ligand and its metal complexes

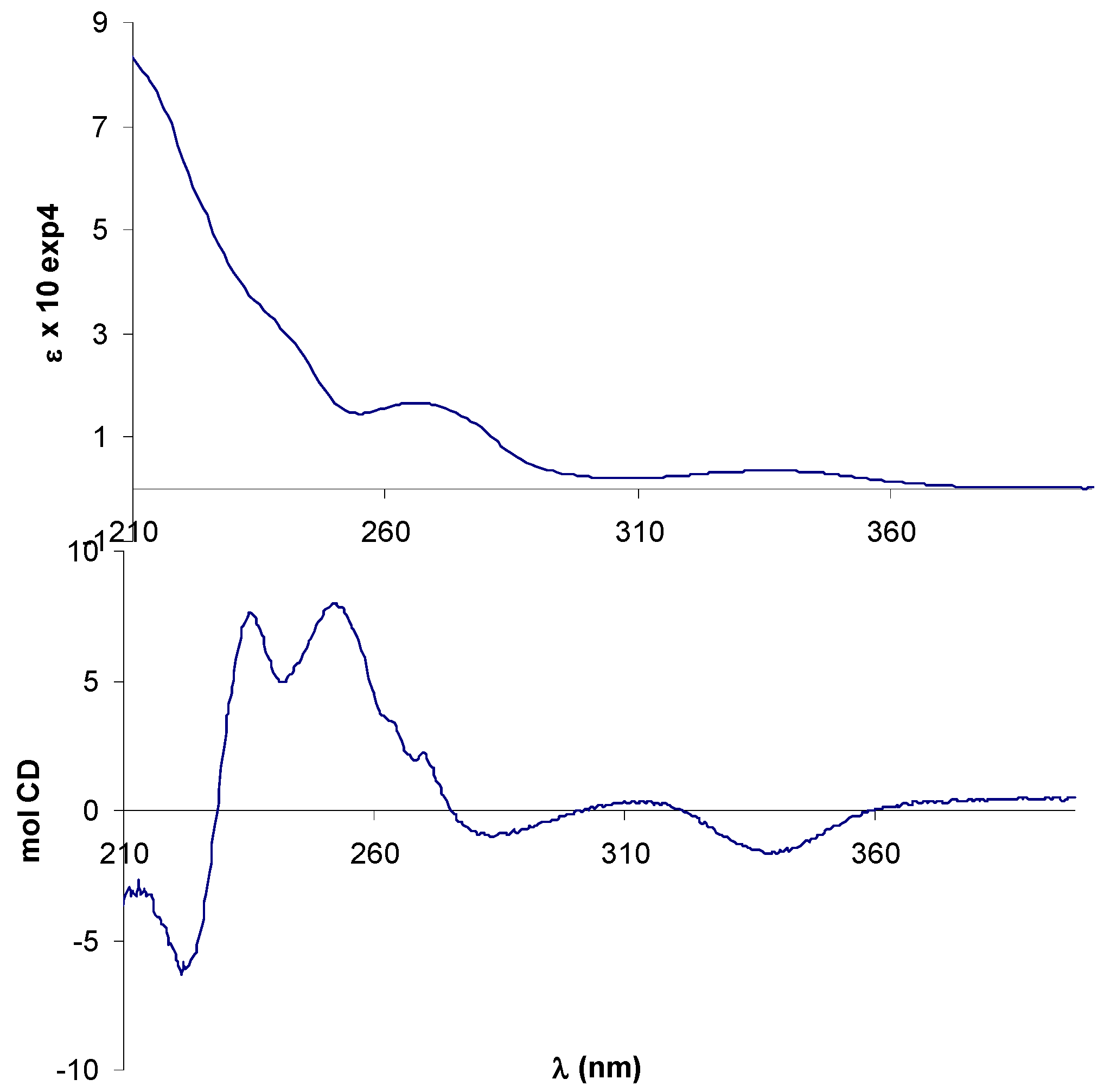
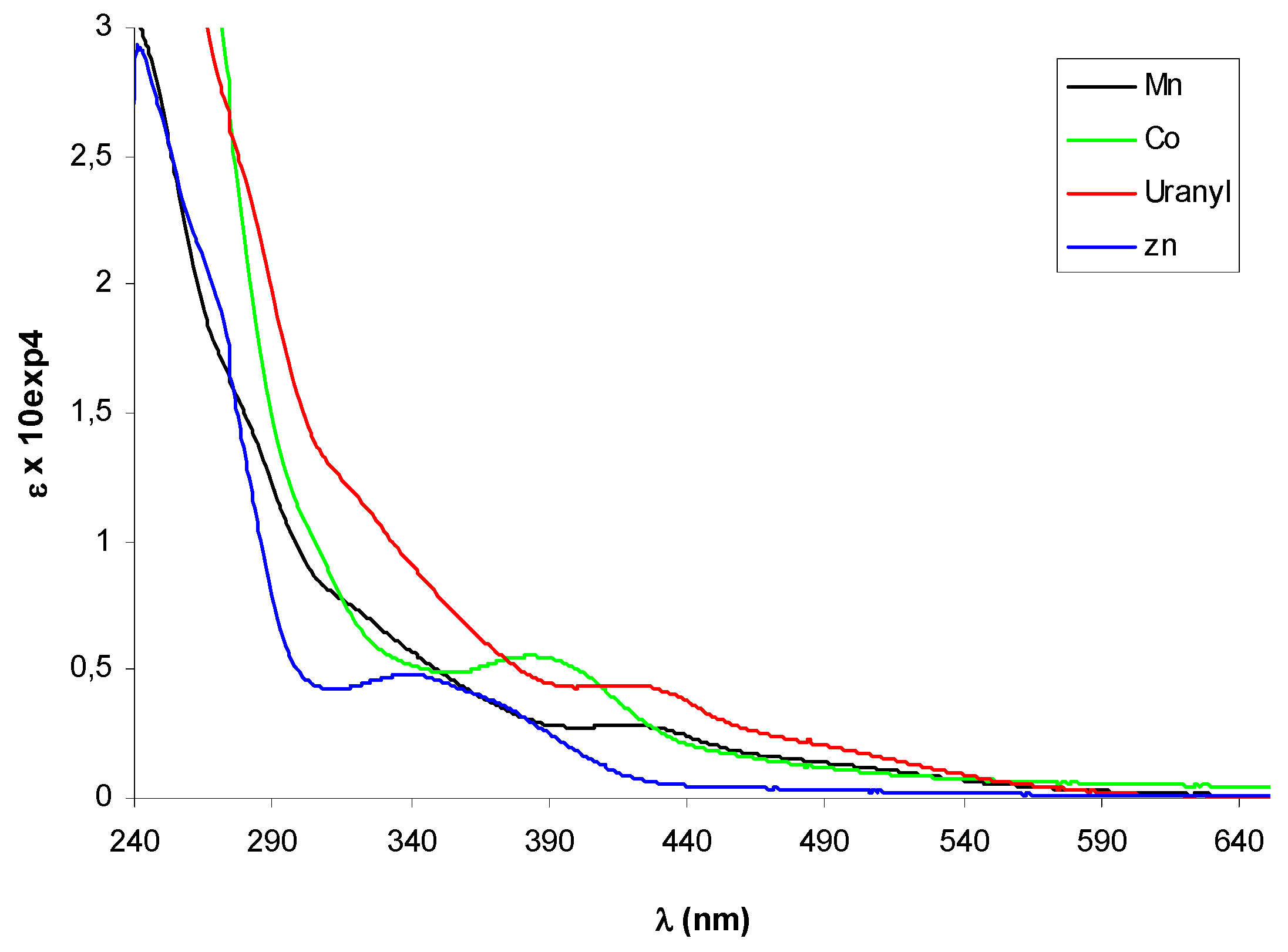
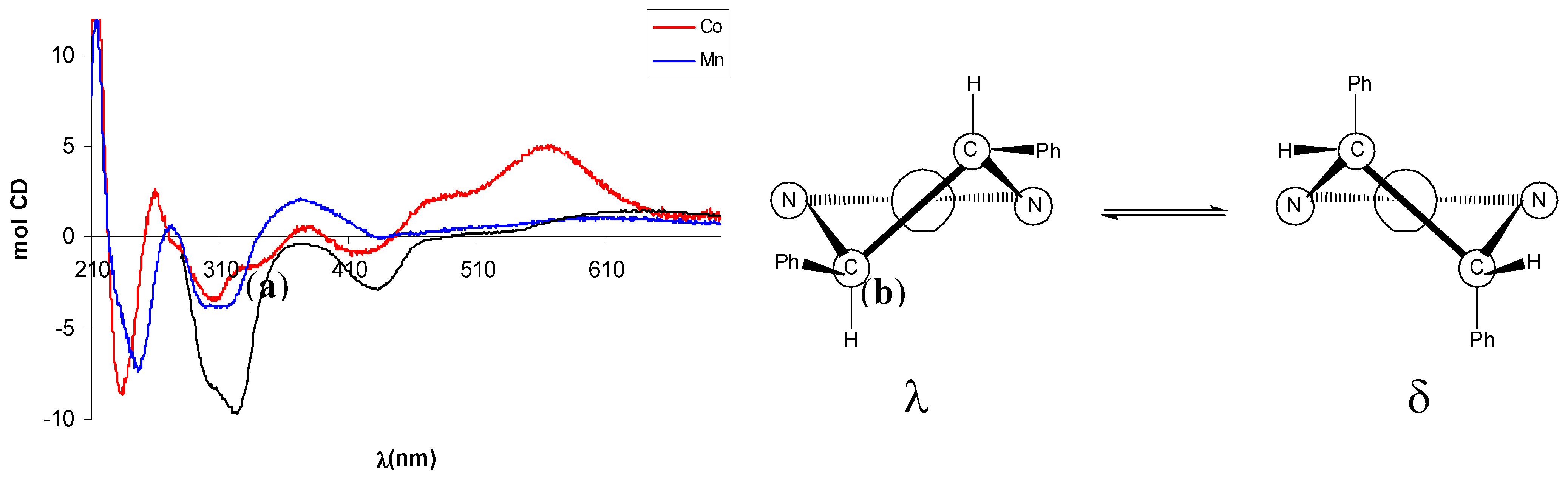
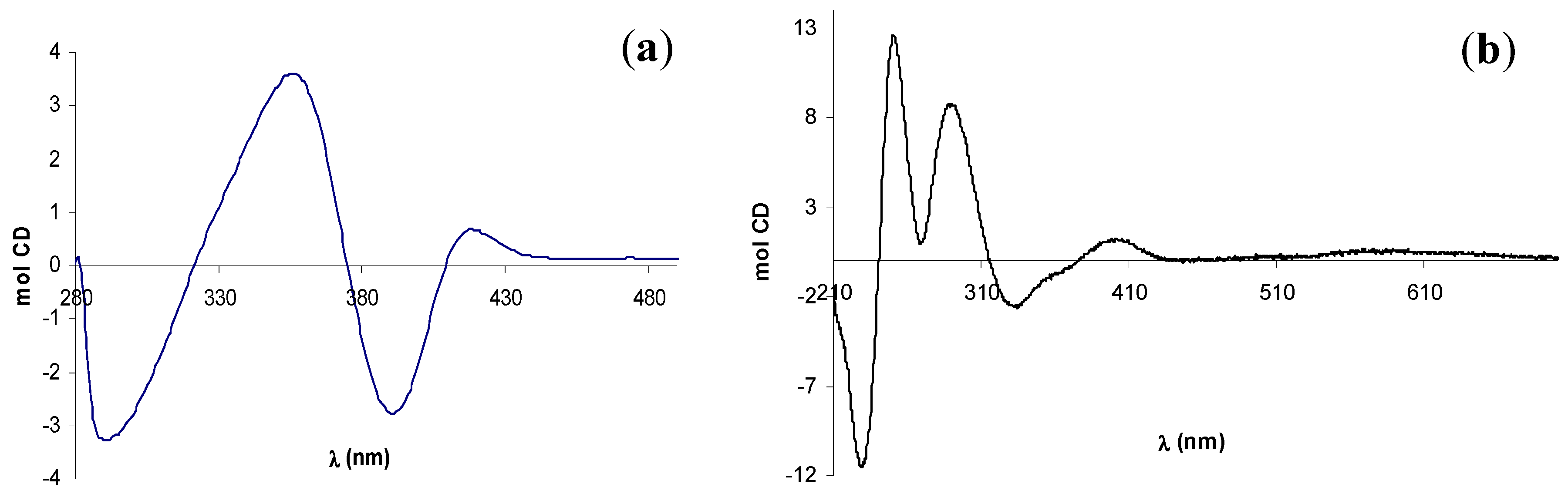
2.2. Circular dichroism spectra




3. Experimental
3.1. General methods
3.2. Synthesis of 1,5-dioxo-3-(p-methylbenzyloxyphenyl)[5]ferrocenophane (4)
3.3. Synthesis of 4-([5]ferrocenophane-3-yl)-phenol (6)
3.4. Synthesis of 2-([5]ferrocenophane-3-yl)-phenol (7)
3.5. Synthesis of 4-([5]ferrocenophane-3-yl)-salicylaldehyde (8)
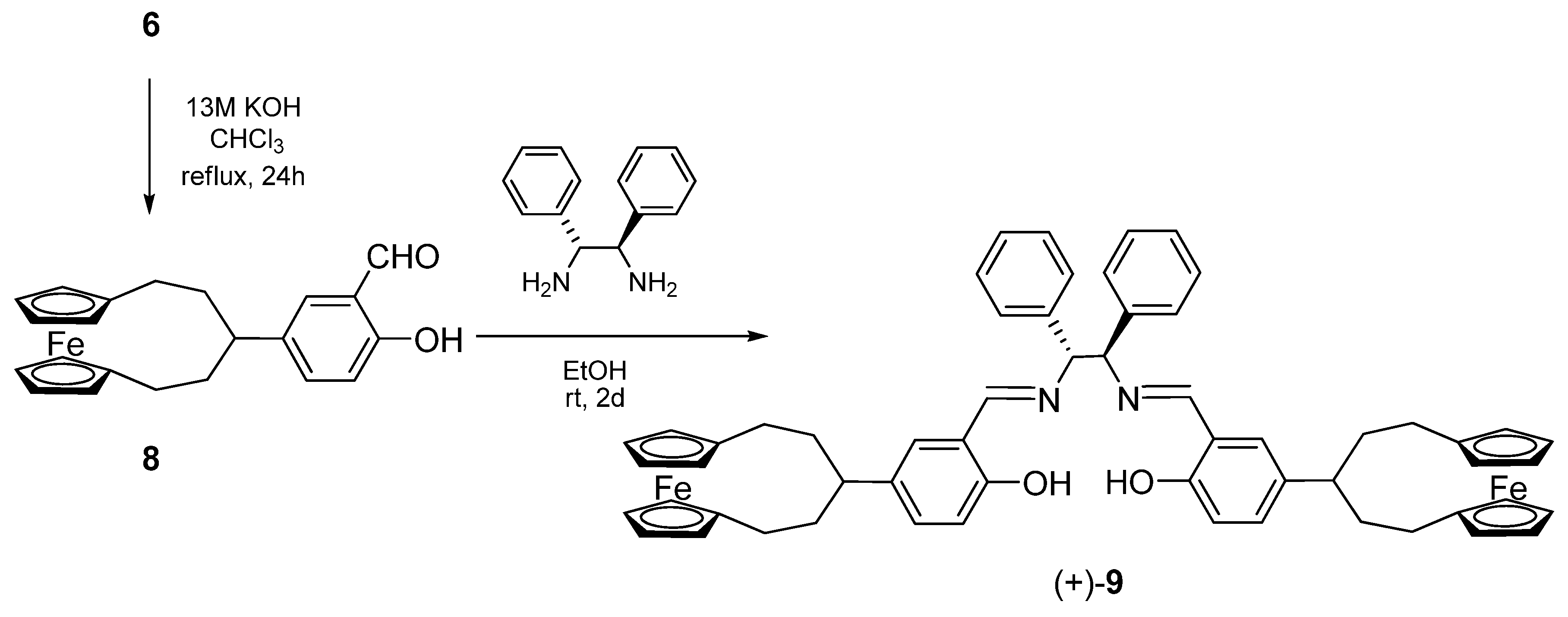
3.6. Synthesis of (+)-(R,R)-N,N’-bis[4-([5]ferrocenophane-3-yl)-salicylidene]-1,2-diphenylethyle-nediamine [(+)-9]
3.7. General procedure for the synthesis of metal complexes
3.8. Synthesis of Zn(II) complex (-)-12
4. Conclusion
Acknowledgements
- Sample Availability: Samples of compounds 4, 5 and 9 are available from the authors.
References
- Cozzi, P.G. Metal-salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef]
- Bailezao, C.; Garcia, H. Chiral salen complexes: An overview to recoverable and reusable homogeneous and heterogeneous catalysts. Chem. Rev. 2006, 106, 3987–4043. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by (salen) manganese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Zhang, W.; Muci, A.R.; Ecker, J.R.; Deng, L. Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane. J. Am. Chem. Soc. 1991, 113, 7063–7064. [Google Scholar] [CrossRef]
- Srinivasan, K.; Michaud, P.; Kochi, J.K. Epoxidation of olefins with cationic (salen) manganese(III) complexes. The modulation of catalytic activity by substituents. J. Am. Chem. Soc. 1986, 108, 2309–2320. [Google Scholar] [CrossRef]
- Cavallo, L.; Jacobsen, H. Electronic effects in (salen)Mn-based epoxidation catalysts. J. Org. Chem. 2003, 68, 6202–6207. [Google Scholar] [CrossRef]
- Schaus, S.E.; Brandes, B.D.; Larrow, J.F.; Togunaga, M.; Hansen, K.B.; Gould, A.E.; Furrow, M.E.; Jacobsen, E.N. Highly selective hydrolytic kinetic resolution of terminal epoxides catalyzed by chiral (salen) CoIII complexes. Practical synthesis of enantioenriched terminal epoxides and 1,2-Diols. J. Am. Chem. Soc. 2002, 124, 1307–1315. [Google Scholar]
- Oh, C.R.; Choo, D.J.; Shim, W.H.; Lee, D.H.; Roh, E.J.; Lee, S.; Song, C.E. Chiral Co(III) (salen)-catalysed hydrolytic kinetic resolution of racemic epoxides in ionic liquids. Chem. Commun. 2003, 1100–1101. [Google Scholar]
- Dioos, B.M.L.; Jacobs, P.A. Microwave-assisted Cr(salen)-catalysed asymmetric ring opening of epoxides. J. Catal. 2005, 235, 428–430. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, W.; Lough, A.J.; Kim, B.M.; Chin, J. A cobalt(III)-salen complex with an axial substituent in the diamine backbone: Stereoselective recognition of amino alcohols. J. Am. Chem. Soc. 2005, 127, 16776–16777. [Google Scholar] [CrossRef]
- Dzygiel, P.; Reeve, T.B.; Piarulli, U.; Krupicka, M.; Tvaroska, I.; Gennari, C. Resolution of racemic N-benzyl α-amino acids by liquid-liquid extraction. Eur. J. Org. Chem. 2008, 1253–1264. [Google Scholar]
- Belokon, Y.N.; Fuentes, J.; North, M.; Steed, J.W. Influence of the metal and chiral diamine on metal(II)salen catalysed, asymmetric synthesis of α-methyl α-amino acids. Tetrahedron 2004, 60, 3191–3204. [Google Scholar] [CrossRef]
- Cozzi, P.G. Enantioselective alkynylation of ketones catalyzed by Zn(salen) complexes. Angew. Chem. Int. Ed. 2003, 42, 2895–2898. [Google Scholar] [CrossRef]
- Pathak, K.; Bhatt, A.P.; Abdi, S.H.R.; Kureshy, R.I.; Khan, N.H.; Ahmad, I.; Jasra, R. Enantioselective phenylacetylene addition to aldehydes and ketones catalyzed by recyclable polymeric Zn (salen) complex. Chirality 2007, 19, 82–88. [Google Scholar] [CrossRef]
- Wezemberg, S.J.; Kleij, A.W. Material application for salen frameworks. Angew. Chem. Int. Ed. 2008, 47, 2354–2364. [Google Scholar] [CrossRef]
- Kleij, A.W. New templating strategies with salen scaffolds (salen = N,N’-Bis (salicylidene) ethylenediamine dianion). Chem. Eur. J. 2008, 14, 10520–10529. [Google Scholar] [CrossRef]
- Di Mauro, E.F.; Kozlowski, M.C. Salen-derived catalysts containing secondary basic groups in the addition of diethylzinc to aldehydes. Org. Lett. 2001, 3, 3053–3056. [Google Scholar] [CrossRef]
- Jiang, C.J.; Chen, Z.R. Chiral cobalt salen complexes containing Lewis acid: A highly reactive and enantioselective catalyst for the hydrolytic kinetic resolution of terminal epoxides. Kinet. Catal. 2008, 49, 474–478. [Google Scholar] [CrossRef]
- Ferrocenes; Hayashi, T.; Togni, A. (Eds.) Wiley-VCH Verlag: Weinheim, Germany, 2008.
- Arrayas, R.G.; Adrio, J.; Carretero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. In. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef]
- Patti, A.; Pedotti, S. Synthesis of chiral 1-ferrocenylaldols and 1-ferrocenyl-1,3-diols via asymmetric reduction. Tetrahedron Asymmetry 2006, 17, 1824–1830. [Google Scholar] [CrossRef]
- Patti, A.; Pedotti, S. Synthesis of chiral alcohols containing the 1,3-diferrocenylpropane structural motif. Tetrahedron Asymmetry 2008, 19, 1891–1897. [Google Scholar] [CrossRef]
- Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M.; Williams, D.J. Novel chiral (salen)MnIII complexes containing a calix[4]arene unit as catalysts for enantioselective epoxidation reactions of (Z)-aryl alkenes. Eur. J. Org. Chem. 2005, 3562–3570. [Google Scholar]
- Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Sciotto, D.; Tomaselli, G.A.; Toscano, R.M. Synthesis and conformational aspects of 20- and 40-membered macrocyclic mono and dinuclear uranyl complexes incorporating salen and (R)-Binol units. Tetrahedron 2007, 63, 9751–9757. [Google Scholar] [CrossRef]
- Gibson, V.C.; Long, N.J.; Marshall, E.L.; Oxford, P.J.; White, A.J.P.; Williams, D.J. The synthesis and metal coordination chemistry of new 1,1’-N-substituted ferrocenediyl ligands derived from 1,1’-diaminoferrocene. J. Chem. Soc. Dalton Trans. 2001, 1162–1164. [Google Scholar]
- Shafir, A.; Fiedler, D.; Arnold, J. Formation of 1:1 complexes of ferrocene-containing salen ligands with Mg, Ti and Zr. J. Chem. Soc. Dalton Trans. 2002, 555–560. [Google Scholar]
- Gregson, C.K.A.; Gibson, V.C.; Long, N.J.; Marshall, E.L.; Oxford, P.J.; White, A.J.P. Redox control within single-site polymerization catalysts. J. Am. Chem. Soc. 2006, 128, 7410–7411. [Google Scholar]
- Wölfle, H.; Kopacka, H.; Wurst, K.; Ongania, K.H.; Görtz, H.H.; Preishuber-Pflügl, P.; Bildstein, B. Planar chiral ferrocene salen-type ligands featuring additional central and axial chirality. J. Organomet. Chem. 2006, 691, 1197–1215. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Patti, A.; Pedotti, S.; Tomaselli, G.A.; Toscano, R.M. Synthesis of novel chiral ‘salen-type’ ferrocenyl ligands. Tetrahedron Asymmetry 2007, 18, 2377–2380. [Google Scholar] [CrossRef]
- Achard, T.; Belokon, Y.N.; Fuentes, J.A.; North, M.; Parsons, T. Influence of aromatic substituents on metal(II)salen catalysed, asymmetric synthesis of α-methyl α-amino acids. Tetrahedron 2004, 60, 5919–5930. [Google Scholar] [CrossRef]
- Malinowska, M.; Kwiatkowski, P.; Jurezac, J. The enantioselective high-pressure Diels-Alder reaction of 1-methoxybuta-1,3-diene with tert-butyldimethylsilyloxy-acetaldehyde catalyzed by (salen)Co(II) and (salen)Cr(III)Cl complexes. Tetrahedron Lett. 2004, 45, 7693–7696. [Google Scholar] [CrossRef]
- Pedotti, S.; Patti, A. Microwave-assisted synthesis of 1,5-dioxo-3-substituted [5]ferrocenophanes. J. Organomet. Chem. 2008, 693, 1375–1381. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Pasquini, C.; Schiaffino, L. Nonsymmetrically substituted uranyl-salophen receptors: New opportunities for molecular recognition and catalysis. Supramol. Chem. 2007, 19, 79–87. [Google Scholar] [CrossRef]
- Cametti, M.; Dalla Cort, A.; Mandolini, L.; Nissinen, M.; Rissanen, K. Specific recognition of fluoride anion using a metallamacrocycle incorporatine a uranyl-salen unit. New J. Chem. 2008, 32, 1113–1116. [Google Scholar] [CrossRef]
- Pasini, A.; Gullotti, M.; Cesarotti, E. Uranyl complexes with tetradentate optically active schiff bases of salicylaldehyde. J. Inorg. Nucl. Chem. 1972, 32, 3821–3833. [Google Scholar]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. BBA-Proteins Proteomics 2005, 1751, 119–139. [Google Scholar] [CrossRef]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef]
- Kypr, J.; Kejnovská, I.; Renčiuk, D.; Vorlíčková, M. Circular dichroism and conformational polymorphism of DNA. Nucl. Acids Res. 2009, 37, 1713–1725. [Google Scholar] [CrossRef]
- McCann, D.M.; Stephens, P.J. Determination of absolute configuration using density fuctional theory calculations of optical rotation and electronic circular dichroism: Chiral Alkenes. J. Org. Chem. 2006, 71, 6074–6098. [Google Scholar] [CrossRef]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The assignment of absolute stereostructures through quantum chemical circular dichroism calculations. Eur. J. Org. Chem. 2009, 2717–2727. [Google Scholar]
- Pasini, A.; Gullotti, M.; Ugo, R. Optically active complexes of Schiff bases. Part 4. An analysis of the circular dichroism spectra of some complexes of different coordination numbers with quadridentate Schiff bases of optically active diamines. J. Chem. Soc. Dalton Trans. 1977, 346–356. [Google Scholar]
- Hirotsu, M.; Nakajima, K.; Kojima, M.; Yoshikawa, Y. Manganese (III) complexes containing optically active tetradentate Schiff base ligands. Effect of phenyl substituents. Inorg. Chem. 1995, 34, 6173–6178. [Google Scholar] [CrossRef]
- Szlyk, E.; Surdykowski, A.; Barwiolek, M.; Larsen, E. Spectroscopy and stereochemistry of t6he optically active copper(II), cobalt(II) and nickel(II) complexes with Schiff bases N,N’-(1R,2R)-(-)-1,2-cyclohexylenebis(3-methylbenzylideneiminato) and N,N’-(1R,2R)-(-)1,2-cyclohexylene-bis-(5-methylbenzylideneiminato). Polyhedron 2002, 21, 2711–2717. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, H.; Li, L.; Hao, H.Q.; Wang, X.Y.; Chen, J.G. Synthesis and characterization of chiral nickel(II) Schiff base complexes and their CD spectra-absolute configuration correlations. Tetrahedron Asymmetry 2006, 17, 2059–2063. [Google Scholar] [CrossRef]
- Janowska, I.; Zakrzewski, J. Circular dichroism spectra of planar chiral 2-substituted ferrocenecarboxaldehydes and 2-ferrrocenyl-1,1-dicyanoethylenes. Tetrahedron Asymmetry 2003, 14, 3271–3273. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Patti, A.; Pedotti, S.; Ballistreri, F.P.; Sfrazzetto, G.T. Synthesis and Characterization of Some Chiral Metal-Salen Complexes Bearing a Ferrocenophane Substituent. Molecules 2009, 14, 4312-4325. https://doi.org/10.3390/molecules14114312
Patti A, Pedotti S, Ballistreri FP, Sfrazzetto GT. Synthesis and Characterization of Some Chiral Metal-Salen Complexes Bearing a Ferrocenophane Substituent. Molecules. 2009; 14(11):4312-4325. https://doi.org/10.3390/molecules14114312
Chicago/Turabian StylePatti, Angela, Sonia Pedotti, Francesco Paolo Ballistreri, and Giuseppe Trusso Sfrazzetto. 2009. "Synthesis and Characterization of Some Chiral Metal-Salen Complexes Bearing a Ferrocenophane Substituent" Molecules 14, no. 11: 4312-4325. https://doi.org/10.3390/molecules14114312