Results and Discussion
In order to get an impression about the hydrophobization effect of the incorporation of one or more O-2’,3’-cyclic ketal derivative of inosine , I*, (with three CH
2 groups in the ketal side chain) into an oligonucleotide, we first calculated the log
P values [
6] of a series of inosine trimers with varying substitutions of inosine by inosine ketals (I*) with a chain length of three methylene groups.
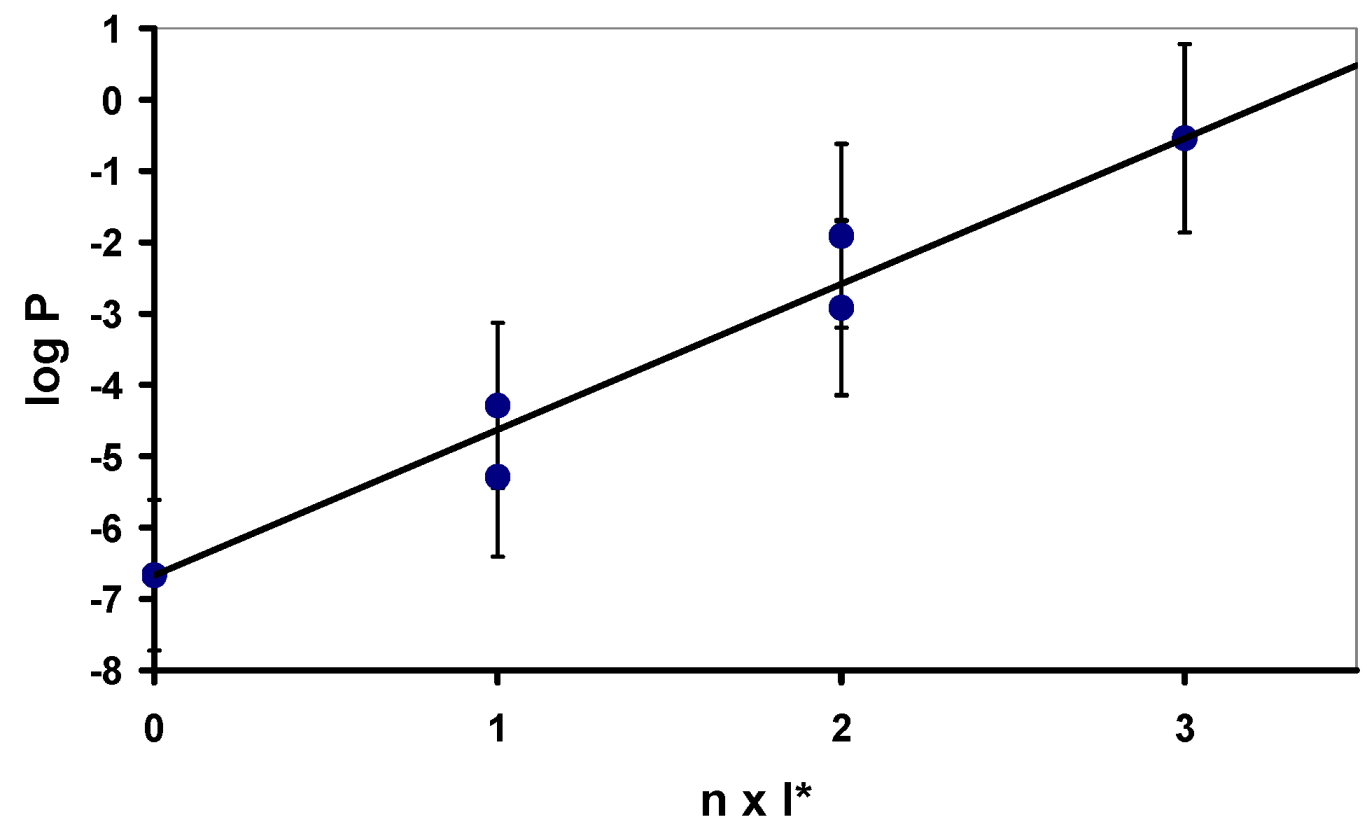
Figure 3 and
Table 1 display the increase of lipophilicity with the increasing number of modifications as well as the influence of the position of incorporation.
It can be seen that in case of incorporation of either one or two modified units into an inosine trimer the resulting log
P value depends on the position of incorporation: Substitution of one inosine by an inosine ketal at the 3’-end results in an oligomer (entry 2,
Table 1) with a free primary hydroxyl group at the 5’-terminus; this oligomer shows a significantly lower lipophilicity than the corresponding trimers carrying the modified inosine derivative either in the middle position (entry 3) or at the 5’-end (entry 4). An analogous result can be seen for trimers carrying two modifications (entries 5-7,
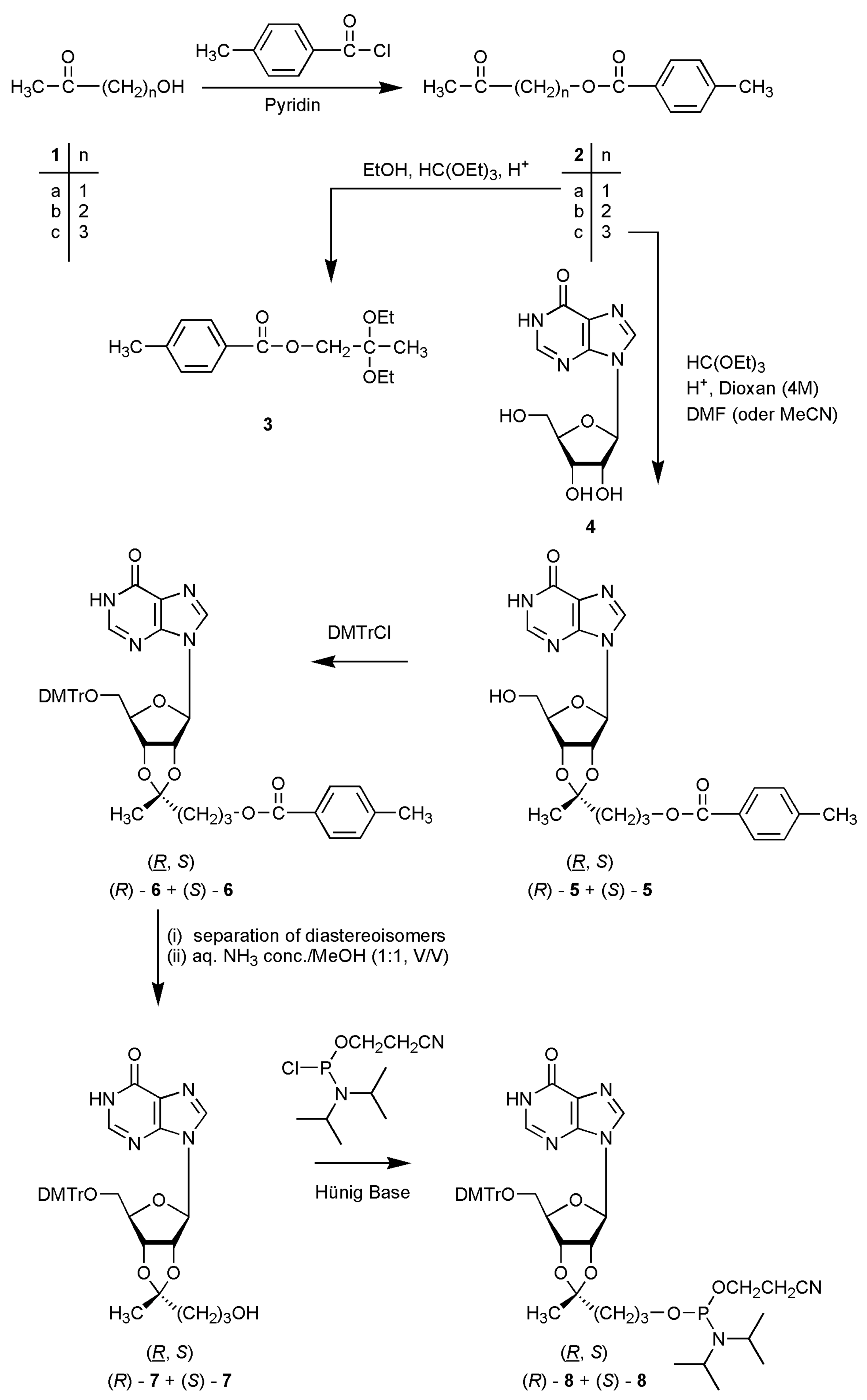
Table 1). The synthesis of 2’-cyanoethyl phosphoramidite building blocks of inosine O-2’,3’-cyclic ketals is shown in
Scheme 1. First, we converted the (ω-1) ketoalcohols
1a-
c to the tolyl-protected compounds
2a-
c. Exemplary,
Figure 4 displays the three-dimensional structure of
2b obtained from an X-ray analysis [
7].
Figure 3.
Calculatedlog
P values of inosine oligonucleotide trimers as a function of the number of inosine O-2’,3’-cyclic ketal derivatives (see
Figure 2).
Figure 3.
Calculatedlog
P values of inosine oligonucleotide trimers as a function of the number of inosine O-2’,3’-cyclic ketal derivatives (see
Figure 2).
Table 1.
Calculatedlog
P values of inosine oligonucleotide trimers as a function of the number and position of inosine O-2’,3’-cyclic ketal derivatives (see
Figure 2).
Table 1.
Calculatedlog P values of inosine oligonucleotide trimers as a function of the number and position of inosine O-2’,3’-cyclic ketal derivatives (see Figure 2).
| Entry | Compound | Number of modified units | Calculated logP Values |
|---|
| 1 | 5’-d(IpIpI) | 0 | -6.67 ± 1.06 |
| 2 | 5’-d(IpIpI*) | 1 | -5.29 ± 1.12 |
| 3 | 5’-d(IpI*pI) | 1 | -4.29 ± 1.16 |
| 4 | 5’-d(I*pIpI) | 1 | -4.29 ± 1.16 |
| 5 | 5’-d(IpI*pI*) | 2 | -2.92 ± 1.22 |
| 6 | 5’-d(I*pIpI*) | 2 | -2.92 ± 1.22 |
| 7 | 5’-d(I*pI*pI) | 2 | -1.91 ± 1.29 |
| 8 | 5’-(I*pI*pI*) | 3 | -0.54 ± 1.32 |
Earlier, it had been shown that reaction of inosine (
4) with (ω-1) ketoesters such as ethyl levulinate or unsymmetrical ketones such as pentan-2-one leads to O-2’,3’-ketals with predominant or even exclusive formation of the (
R) configuration at the newly formed stereogenic center [
8,
9,
10,
11,
12] [the (
R)- and (
S)-notation within this manuscript refers always to the configuration at the stereogenic center of the ketal moiety]. We now prepared the keto esters
2a-
c, of which only compound
2c had been described earlier [
13]. All attempts to react inosine with compound
2a in the presence of triethyl orthoformate and 4M HCl in 1,4-dioxane in various solvents failed. Next, we, therefore, converted compound
2a into the open acetal
3. A subsequent transacetalisation with inosine (
4) which usually occurs under mild reaction conditions also failed. Also ketalisation of inosine with the elongated (ω-1) ketoester
2b did not show the desired result. The reason for this result might be an electrostatic repulsion of the ester oxygen atom and the ribose oxygen.
Figure 4.
Ball and stick model of compound 2b (with the exception of the hydrogen atoms, which are represented by use of spheres with a common isotropic radius, all other atoms are represented as thermal displacement ellipsoids showing 50% of the probability of the corresponding atom.
Figure 4.
Ball and stick model of compound 2b (with the exception of the hydrogen atoms, which are represented by use of spheres with a common isotropic radius, all other atoms are represented as thermal displacement ellipsoids showing 50% of the probability of the corresponding atom.
Scheme 1.
Synthesis of the target structures. In case of the ketals (5–8) the R diastereoisomers always are shown.
Scheme 1.
Synthesis of the target structures. In case of the ketals (5–8) the R diastereoisomers always are shown.
At least, reaction of
4 with 4-oxopentyl-4-methylbenzoate, (
2c) under the reaction conditions mentioned above gave the desired product
5, however,
1H- and
13C-NMR spectroscopy proved the formation of a diastereoisomeric mixture [(
R)-
5 + (
S)-
5]. Integration of the
1H-NMR resonances of the clearly separated ketal Me groups indicated a ratio of 47% of the (
R)- and 53% of the (
S)-diastereoisomer. The assignment of the NMR resonances of the different methyl groups as well as of other signals was made on the basis of a comparison with the
1H- and
13C-NMR spectra of (
R)-2’,3’-O-(3-carboxy-1-methylpropyliden)adenosine from which an X-ray analysis had been performed earlier [
14] as well as by gradient-selected homo- and heteronuclear correlation spectroscopy.
A chromatographic separation of the mixture (R)-5 + (S)-5 proved extremely difficult; only a TLC with a 20-fold development in EtOAc/toluene (97:3, v/v) showed the presence of two products. Because the positioning of the ketal side chain is of decisive influence on the topology of oligonucleotides carrying such modified building blocks, a separation of the diastereoisomers is at least inevitable. Next, we converted the mixture (R)-5 + (S)-5 into the 4,4’-dimethoxytriphenylmethyl derivatives (R)-6 + (S)-6. On this stage the diastereoisomeric mixture could be separated silica gel chromatographically by elution with EtOAc/toluene (97:3, v/v). Both diastereoisomers were characterized by 1H-NMR spectra.
In order to prove if a chromatographic separation is also possible on the stage of the de-toluoylated compounds a mixture of (
R)-
6 + (
S)-
6 was deprotected by treatment with conc. aq. ammonia/MeOH (1:1, v/v). After 72 h a mixture of (
R)-
7 + (
S)-
7 was isolated in moderate 38% yield. As it was found that a chromatographic separation of the diastereoisomers was very difficult, the separation was performed on the stage of compounds
6. De-toluoylation was performed on either the stage of a diastereoisomeric mixture or on the stage of the separated isomers and afforded (
R)-
7 and (
S)-
7. A first subsequent phosphitylation [
15] of a mixture of (
R)-
7 + (
S)-
7 with chloro-(2-cyanoethoxy)-N,N-diisopropylethylaminophosphane (CH
2Cl
2, Hünig’s base, 3 h) gave, after chromatography, a mixture of four diastereoisomers (additional R
Pand S
P diastereoisomers) in 67% total yield. The phosphitylation of the separated (
R)-
7 and (
S)-
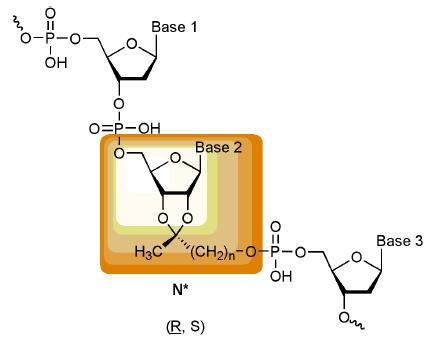
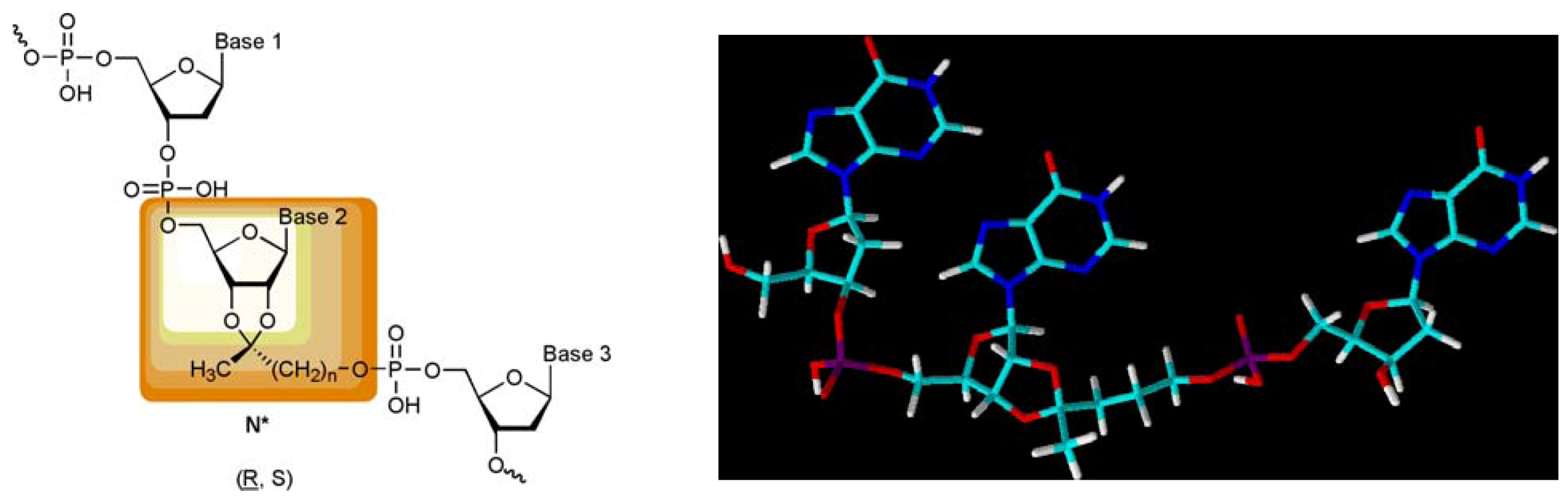
7 isomers, their incorporation into oligonucleotides as well as the base pairing properties of such oligomers will be published in following manuscripts. We anticipate that appending of the novel building blocks to one or both termini of an unmodified oligonucleotide will lead to gap-mers with unaltered binding to their complementary strands but that their incorporation into the innermost part of a nucleic acid will give new and autonomous nucleic acid pairing systems.
Experimental
General
Thin-layer chromatography (TLC): Silica gel 60 F
254 plates (VWR, Darmstadt, Germany). UV-Spectroscopy: U-3200 spectrophotometer (Hitachi, Japan); λ
max in nm; ε in M
-1cm
-1. NMR Spectra were recorded on AC-250 and AMX-500 spectrometers (Bruker BioSpin, Rheinstetten , Germany). Operational frequencies:
1H-NMR: 250.13, 500.14 MHz;
13C-NMR: 62.896, 125.700 MHz. Chemical shifts (δ values) are in parts per million relative to tetramethylsilane as internal standard. Microanalyses were performed by Mikroanalytisches Labor Beller-Matthies (Göttingen, Germany). Melting points were measured on a Büchi SMP 20 apparatus and are not corrected. The 3D-optimized structures were obtained using the program
ChemSketch/3D Viewer, version 12.0, from Advanced Chemistry Development Inc., Toronto, Canada (
http://www.acdlabs.com).
2-Oxopropyl-4-methylbenzoate (2a)
3-Hydroxopropan-2-one (1a, 1.13 g, 15 mmol) was dissolved in anhydr. pyridine (3 mL) and cooled in an ice bath. After drop-wise addition of tolyl chloride (1.55 g, 10 mmol) the mixture was stirred for 30 min, warmed up to ambient temperature and stirred overnight. Then, the reaction mixture was poured into ice-water (100 mL), acidified by addition of conc. hydrochloric acid and warmed up to ambient temperature. After extraction with CHCl3 the organic layer was separated and washed with 5% aqueous NaHCO3. The organic phase was dried (Na2SO4) and evaporated. After drying under high vacuum 1.7 g (88%) of the title compound were obtained as colourless crystals. M.p. 48 °C; TLC (silica gel, EtOAc/n-hexane, 33:67, v/v): Rf, 0.7; Anal. calcd. for C11H12O3 (192.211): C, 68.74; H, 6.29. Found: C, 68.65; H, 6.18; 1H-NMR (d6-DMSO): δ, 2.16 (3H, s, CH3-arom.); 2.40 (3H, s, CH3-C=O); 4.99 (2H, s, CH2); 7.35, 7.89 (2 × 2H, d, J = 7.5 Hz, H-arom.); 13C-NMR (d6-DMSO): δ, 21.6 (CH3-arom.); 26.4 (CH3-C=O); 69.1 (CH2); 126.9, 129.80, 144.5 (4 C-arom.); 165.6 (C=O-ester); 202.3 [C(=O)-CH3].
2,2-Diethoxypropyl-4-methylbenzoate (3)
To compound 2a (0.96 g, 5 mmol) was added triethyl orthoformate (0.82 g, 5.5 mmol), anhydrous ethanol (0.23 g, 5 mmol) and one drop of conc. sulphuric acid. After stirring at room temperature overnight the mixture was evaporated and chromatographed on silica gel 60 (6 × 10 cm, EtOAc/n-hexane, 33:67, v/v). From the main fraction the title compound was obtained as a colourless foam upon evaporation. Yield: 0.43 g (32%). TLC (silica gel, EtOAc/n-hexane, 33:67, v/v): Rf, 0.95; 1H-NMR (d6-DMSO): δ, 1.10 (3H, t, J = 7.5 Hz, CH3-ethyl); 1.36 [CH3-C(OEt)2]; 2.50 (3H, s, CH3-arom.); 3.47 (2H, q, J = 7.5 Hz, CH2-ethyl); 4.22 (2H, s, CH2); 7.34, 7.88 (2 × 2H, d, J = 7.5 Hz, H-arom.); 13C-NMR (d6-DMSO): δ, 15.8 (2 CH3CH2O); 21.6 (2 CH3); 56.1 (2 CH3CH2O); 65.3 (CH2); 127.2, 129.8, 144.3 (4 C-arom.); 99.4 (C-ketal); 165.7 (C=O-ester).
3-Oxobutyl-4-methylbenzoate (2b)
4-Hydroxybutan-2-one (1b, 1.32 g, 15 mmol) was reacted with tolyl chloride (1.55 g, 10 mmol) and worked up as described for 2a. After crystallization from hot propan-2-ol colourless crystals of 2b (1.55 g, 75%) were obtained. M.p. 37 °C; TLC (silica gel, EtOAc/n-hexane, 33:67, v/v): Rf, 0.7; Anal. calcd. for C12H14O3 (206.238): C, 69.88; H, 6.84. Found: C, 69.74; H, 6.65; 1H-NMR (d6-DMSO): δ, 2.10 (3H, s, CH3-arom.); 2.32 (3H, s, CH3-C=O); 2.87 (2H, t, J = 5.5 Hz, CH2-C=O); 4.36 (2H, t, J = 7.5 Hz, CH2-O); 7.25, 7.74 (2 2H, d, J = 7.3 Hz, H-arom.); 13C-NMR (d6-DMSO): δ, 21.6 (CH3-arom.); 30.4 (CH3-C=O); 42.1 (CH2-C=O); 60.2 (CH2-O); 127.4, 129.7, 144.1 (4 C-arom.); 166.1 (C=O-ester); 208.2 [C(=O)-CH3].
4-Oxopentyl 4-methylbenzoate (2c) [13]
5-Hydroxypentan-2-one (1c, 1.53 g, 15 mmol) was reacted with tolyl chloride (1.55 g, 10 mmol) and worked up as described for 2a. After column chromatography (silica gel 60H, 6 × 10 cm, EtOAc/n-hexane, 33:67, v/v) the main fraction was pooled and evaporated to dryness to give a colourless oil of 2c (1.72 g, 78%). TLC (silica gel, EtOAc/n-hexane, 33:67, v/v): Rf, 0.68; Anal. calcd. for C13H16O3 (220.264): C, 70.89, H, 7.32. Found: C, 70.78; H, 7.21; 1H-NMR (d6-DMSO): δ, 1.89 (2H, pq, J = 7.5 Hz, CH2); 2.10 (3H, s, CH3-C=O); 2.38 (3H, s, CH3-arom.); 2.60 (2H, t, J = 7.5 Hz, CH2-C=O); 4.21 (2H, t, J = 7.5 Hz, CH2-O); 7.32, 7.84 (2 × 2H, d, J = 7.5 Hz, H-arom.); 13C-NMR (d6-DMSO): δ, 21.6 (CH2); 23.0 (CH3-arom.); 30.2 [CH3-C(=O)]; 40.3 [CH2-C(=O)]; 64.4 (CH2-O); 127.5, 129.7, 144.0 (4 C-arom.); 166.2 (C=O-ester); 208.2 [C(=O)-CH3].
3-((3aS,4R,6R,6aS)-4-(hydroxymethyl)-2-methyl-6-(6-oxo-1,6-dihydropurin-9-yl)-tetrahydrofuro[3,4-d][1,3]dioxol-2-yl)propyl-4-methylbenzoate, diastereoisomeric mixture [(R)-5 + (S)-5]
Anhydrous inosine (4, 0.54 g, 2 mmol) was suspended in anhydrous MeCN (7.5 mL), and compound 2c (2.2 g, 10 mmol) and triethyl orthoformate (0.5 ml, 3 mmol) were added. Upon drop-wise addition of 4M HCl in 1,4-dioxane (1.75 mL) the inosine went into solution. The reaction mixture was stirred at ambient temperature for 24 h and then evaporated to dryness. Column chromatography (silica gel 60, 6 × 15 cm, stepwise elution: (i) CHCl3/MeOH, 97:3, 250 ml; (ii) CHCl3/MeOH, 9:1, 250 ml, v/v, each) afforded one main fraction from which a diastereoisomeric mixture of compound 5 (0.56 g, 60%) was obtained after evaporation of the solvent as a colourless solid. M.p. 229 °C (decomp.); TLC (silica gel, CHCl3/MeOH, 9:1, v/v): Rf, 0.62; UV (MeOH): λmax, 248 nm (ε = 28,200 M-1cm-1; ε260 = 18,200 M-1cm-1); Anal. calcd. for C23H26N4O7 (470.514): C, 58.72%; H, 5.57%; N, 11.91%. Found: C, 58.80%, H, 5.49%; N, 11.81%; 1H-NMR (d6-DMSO): δ, 12.5 (1H, br., s, NH); 8.3002 and 8.2970 (2 × 1H, 2 s, H-2); 8.0731 and 8.0668 (2 × 1H, 2 s, H-8); 7.86 and 7.83 (2 × 2H, 2 d, J = 5.0 Hz, H-arom.), 7.34 and 7.30 (2 × 2H, 2 d, J = 5.0 Hz, H-arom.); 6.14 (2 × 1H, 2 d, J(1’,2’) = 2.5 Hz, H-1’); 5.341-5.359 and 5.304-5.286 (2 × 1H, 2 × dd, J(2’,1’) = 2.5 Hz, J(2’,3’) = 6.5 Hz, H-2’); 5.10 (1H, m, 5’-OH); 4.994-4.956 (2 × 1H, 2 × dd, J(3’,2’) = 6.5 Hz), J(3’,4’) = 2.5 Hz, H-3’); 4.33 (1H, m, H-4’); 4.255-4.235 (2H, m, CH2-O-C(O)); 3.52 (2H, m, H2-5’); 2.39 and 2.38 (2 × 3H, 2 × s, CH3-arom.); 1.93 (2H, m, CH2); 1.75 (2H, m, CH2); 1.538 (3H, s, CH3-ketal (R)); 1.333 (3H, s, CH3-ketal (S)); 13C-NMR (d6-DMSO): δ, 146.5 (C-2); 148.2 (C-4); 115.2 (C-5); 157.0 (C-6); 144.0 (C-8); 87.4 (C-1’); 84.3 and 84.7 (C-2’); 81.7 and 82.1 (C-3’); 90.0 (C-4’); 61.9 (C-5’); 23.2 and 23.9 (CH3-ketal); 114.8 (C-quart. ketal); 35.0 and 35.6 (CH2); 21.6 (CH2); 64.8 (CH2-O); 166.2 (C(=O)-ester); 124.9, 129.6, 127.6 and 139.2 (C-arom.); 25.5 (CH3-arom.).
3-((3aS,4R,6R,6aS)-4-(bis(4-methoxyphenyl)(phenyl)methoxy)-methyl)-2-methyl-6-(6-oxo-1,6-dihydro-purin-9-yl)-tetrahydrofuro[3,4-d][1,3]dioxol-2-yl)propyl 4-methylbenzoate, diastereoisomeric mixture [(R)-6 + (S)-6]
The diastereoisomeric mixture (R)-5 + (S)-5 (470 mg, 1.0 mmol) was dried by repeated co-evaporation from anhydrous pyridine. The residue was dissolved in anhydrous pyridine (6 mL), 4,4’-dimethoxytriphenylmethyl chloride (643 mg, 1.9 mmol) was added, and the reaction mixture was stirred for 3 h under a N2 atmosphere. Then, 5% aq. NaHCO3 (30 ml) was added, and the mixture was extracted three times with CHCl3 (30 mL, each). The combined organic layers were dried (Na2SO4) and evaporated to dryness (high vacuum). Column chromatography (silica gel 60, 6 × 20 cm, CHCl3/MeOH, 97:3, v/v) afforded one main fraction from which the title compound was obtained as colourless glassy foam (0.41 g, 53%). TLC (silica gel, CHCl3/MeOH, 97:3, v/v): Rf, 0.38; UV (MeOH): λmax, 242 nm (ε = 47,300 M-1cm-1; ε260 = 26,000 M-1cm-1); Anal. calcd. for C44H44N4O9 (772.842): C, 68.38%; H, 5.74%; N, 7.25%. Found: C, 68.45; H, 5.84; N, 7.09. The diastereoisomeric mixture [(R)-6 + (S)-6] was separated by column chromatography (silica gel 60, 6 × 25 cm, EtOAc/toluene, 98:2, v/v).
3-((2R,3aS,4R,6R,6aS)-4-(bis(4-methoxyphenyl)(phenyl)methoxy)-methyl)-2-methyl-6-(6-oxo-1,6-dihydropurin-9-yl)-tetrahydrofuro[3,4-d][1,3]dioxol-2-yl)propyl 4-methylbenzoate [(R)-6]
From the slower migrating zone the (R) diastereoisomer was isolated as colourless foam. TLC (silica gel, EtOAc/toluene, 98:2, v/v): Rf, 0.45; 1H-NMR (d6-DMSO): δ, 13.23 (1H, s, NH); 8.16 (1H, s, H-2); 7.94 (1H, s, H-8); 8.00 (2 × 1H, d, J = 7.5 Hz, DMTr); 7.40-7.19 (13 H, m, DMT); 6.81 (2 × 2H, H-arom.); 6.17 (1H, d, J(1’,2’) = 2.5 Hz, H-1’); 5.46 (1H, dd, J(2’,1’) = 2.5 Hz, J(2’,3’) = 5.0 Hz, H-2’); 5.00 (1H, dd, J(3’,2’) = 5.0 Hz, J(3’,4’) = 3.0 Hz, H-3’); 4.61 (1H, m, H-4’); 4.44 (2H, m, H2-5’); 3.78 (6H, s, 2 × OCH3); 3.33 (2H, m, CH2); 2.44 (3H, s, CH3-arom.); 1.43 (2H, m, CH2); 1.30 (3H, s, CH3-ketal).
3-((2S,3aS,4R,6R,6aS)-4-(bis((4-methoxyphenyl)(phenyl)methoxy)-methyl)-2-methyl-6-(6-oxo-1,6-dihydropurin-9-yl)-tetrahydrofuro[3,4-d][1,3]dioxol-2-yl)propyl 4-methylbenzoate [(S)-6]
From the faster migrating zone the (S) diastereoisomer was isolated as colourless foam. TLC (silica gel, EtOAc/toluene, 98:2, v/v): Rf, 0.55); 1H-NMR (d6-DMSO): δ, 12.83 (1H, s, NH); 7.97 (1H, s, H-2); 7.90 (1H, s, H-8); 7.36-7.22 (13H, m, DMT); 6.80-6.77 (4H, m, H-arom.); 6.16 (1H, m, H-1’); 5.37 (1H, d, J(2’,3’) = 7.5 Hz, H-2’); 4.99 (1H, d, J(3’,2’) = 7.5 Hz, H-3’); 4.57 (1H, m, H-4’); 4.35 (2H, m, H2-5’); 3.78 (6H, s, 2 × OCH3); 3.33 (2H, m, CH2); 2.41 (3H, m, CH3-arom.); 1.85 (2H, m, CH2); 1.65 (3H, s, CH3-acetal); 1.29 (2H, m, CH2).
3-((3aS,4R,6R,6aS)-6-((bis(4-methoxyphenyl)(phenyl)methoxy)methyl)-2-(3-hydroxypropyl)-2-methyl-tetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-1H-purin-6(9H)-one, diastereoisomeric mixture [(R)-7 + (S)-7]
A diastereoisomeric mixture of (R)-6 + (S)-6 (772 mg, 1.0 mmol) was dissolved in 7M NH3 in MeOH (20 mL) and stirred for 72 h at ambient temperature. After evaporation of the solvent the raw product was chromatographed (silica gel 60, (i) CHCl3/MeOH, 97:3, (ii) CHCl3/MeOH, 9:1, v/v). From the main fraction the title compound [(R)-7 + (S)-7] was obtained as colourless solid (0.25 g, 38%). TLC (silica gel, CHCl3/MeOH, 9:1, v/v): Rf, 0.62; UV (MeOH): λmax, 240 nm (ε = 38,100 M-1cm-1; ε260 = 23,000 M-1cm-1); Anal. calcd. for C36H38N4O8 (654.709): C, 66.04%; H, 5.85%; N, 8.56%. Found: C, 66.09%; H, 5.91%; N, 8.41%. The diastereoisomeric mixture [(R)-7 + (S)-7] proved to be chromatographically separable only with difficulties; therefore, the precursor diastereoisomers (R)-6 and (S)-6 were de-toluoylated separately and worked up as described above.
(R)-7: 1H-NMR (d6-DMSO): δ, 12.34 (s, br., 1H, NH); 8.22 (s, 1H, H-2); 7.93 (s, 1H, H-8); 7.286-6.763 (4m, 13H, H-aromat.); 6.20 (d, 1H, J(1’,2’) = 1.0 Hz), H-1’); 5.39 (dd, 1H, J(2’,1’) = 1.0 Hz, J(2’,3’) = 6.5 Hz), H-2’); 4.91 (dd, 1H, J(3’,2’) = 6.5 Hz, J(3’,4’) = 3.0 Hz, H-3’); 4.50 (1, 1H, J = 5.5 Hz, OH); 4.32 (pq, 1H, J(4’,5’) = 3.0 Hz, H-4’); 3.73, 3.72 (2s, 2 × 3H, 2 OCH3); ~3.4 (m, CH2); 3.22, 3.04 (2m, 2H, H2-5’); 1.78 (m, 2H, CH2); 1.61 (m, 2H, CH2); 1.27 (s, 3H, CH3-ketal).
(S)-7: 12.4 (s, br. 1H, NH); 8.24 (s, 1H, H-2); 7.95 (s, 1H, H-8); 7.287-6.762 (4m, 13H, H-aromat.); 6.25 (d, 1H, H-1’); 5.34 (d, 1H, J(2’,1’) = 5.0 Hz, H-2’); 4.92 (m, 1H, H-3’); 4.50 (m, 1H, OH); 4.35 (m, 1H, H-4’); 3.45 (m, 2H, CH2); 3.724, 3.723 (2s, 2 × 3H, 2 OCH3); 3.23, 3.07 (2 m, 2H, H2-5’); 1.59 (m, 2H, CH2); 1.44 (m, 2H, CH2); 1.49 (s, 3H, CH3-ketal).
3-((3aS,4R,6R,6aS)-4-((bis(4-methoxyphenyl)(phenyl)-methoxy)methyl)-2-methyl-6-(6-oxo-1,6-dihydropurin-9-yl)-tetrahydrofuro[3,4-d][1,3]dioxol-2-yl)propyl 2-cyanoethyldiisopropyl-phosphoramidite, diastereoisomeric mixture [(R)-8 + (S)-8]
The diastereoisomeric mixture (R)-7 + (S)-7 (115 mg, 0.175 mmol) was suspended in abs. CH2Cl2 (7 mL) and N-ethyldiisopropylamine (345 µL, 1.97 mmol) as well as chloro-(2-cyanoethoxy)-N,N-diisopropylethylaminophosphane (340 µL, 1.53 mmol) were added. After 3 h at ambient temperature (N2 atmosphere). The reaction mixture was chromatographed (silica gel 60, 6 × 10 cm, CHCl3/MeOH, 9:1, v/v), and the content of the main fraction was pooled. Evaporation of the solvent gave the title phosphoramidites as colourless foam (0.10 g, 67%). TLC (silica gel, CHCl3/MeOH, 9:1, v/v): Rf, 0.50, 0.54, 0.58, 0.61 (4 diastereoisomers); 31P-NMR (CDCl3): δ, 147.44, 147.36, 139.79, 139.74.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

