3-(2-Aminophenyl)-4-methyl-1,3-thiazole-2(3H)-thione as an Ecofriendly Sulphur Transfer Agent to Prepare Alkanethiols in High Yield and High Purity

Abstract
:Introduction
Results and Discussion
Experimental
General
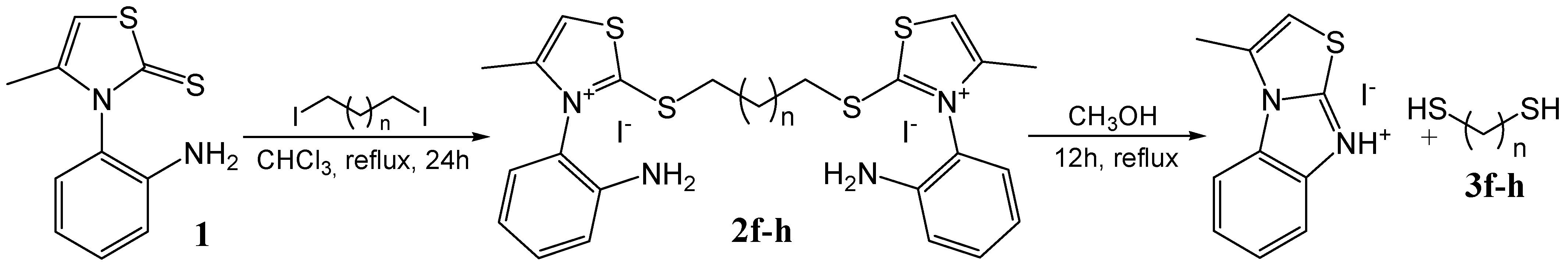
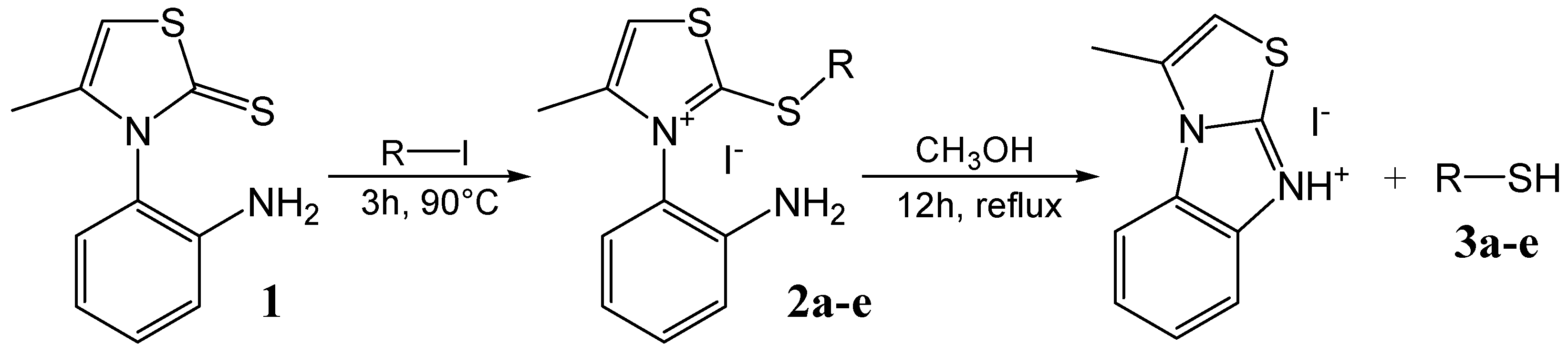
General procedure for the synthesis of monothiazolium iodides 2a-e
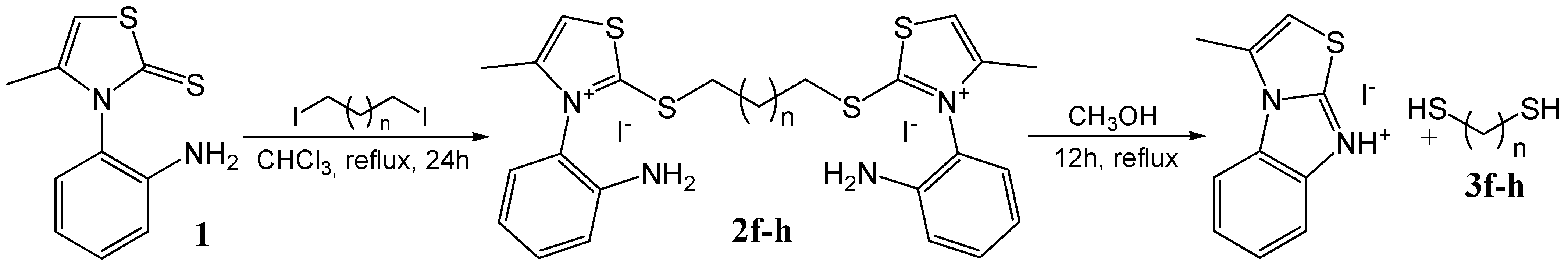
Procedure for the synthesis of bis-thiazolium diiodides 2f and 2g
Procedure for the synthesis of bis-thiazolium diiodide 2h
General procedure for the synthesis of n-alkanethiols
General procedure for the synthesis of α,ω-alkanedithiols
Conclusions
Acknowledgements
References and Notes
- Zamborini, F.P.; Campbell, J.K.; Crooks, R.M. Spectroscopic, voltammetric, and electrochemical scanning tunneling microscopic study of underpotentially deposited Cu corrosion and passivation with self-assembled organomercaptan monolayers. Langmuir 1998, 14, 640–647. [Google Scholar] [CrossRef]
- He, H.X.; Zhang, H.; Li, Q.G.; Zhu, T.; Li, S.F.Y.; Liu, Z.F. Fabrication of designed architectures of Au nanoparticles on solid substrate with printed self-assembled monolayers as templates. Langmuir 2000, 16, 3846–3851. [Google Scholar] [CrossRef]
- Woehrle, G.H.; Warner, M.G.; Hutchison, J.E. Ligand exchange reactions yield subnanometer, thiol-stabilized gold articles with defined optical transitions. J. Phys. Chem. B 2002, 106, 9979–9981. [Google Scholar] [CrossRef]
- Shelley, E.J.; Ryan, D.; Johnson, S.R.; Couillard, M.; Fitzmaurice, D.; Nellist, P.D.; Chen, Y.; Palmer, R.E.; Preece, J.A. Dialkyl sulfides: Novel passivating agents for gold nanoparticles. Langmuir 2002, 18, 1791–1795. [Google Scholar] [CrossRef]
- Snow, A.W.; Ancona, M.G.; Kruppa, W.; Jernigan, G.G.; Foos, E.E.; Park, D. Self-assembly of gold nanoelectronic substrates. J. Mater. Chem. 2002, 12, 1222–1230. [Google Scholar] [CrossRef]
- Woehrle, G.H.; Hutchison, J.E. Thiol-functionalized undecagold clusters by ligand exchange: Synthesis, mechanism, and properties. Inorg. Chem. 2005, 44, 6149–6158. [Google Scholar] [CrossRef] [PubMed]
- Rucareanu, S.; Gandubert, V.J.; Lennox, R.B. 4-(N,N-dimethylamino)pyridine-protected Au nanoparticles: Versatile precursors for water- and organic-soluble gold nanoparticles. Chem. Mater. 2006, 18, 4674–4680. [Google Scholar] [CrossRef]
- Cha, S.-H.; Kim, J.-U.; Kim, K.-H.; Lee, J.-C. Preparation and photoluminescent properties of gold(I)-alkanethiolate complexes having highly ordered supramolecular structures. Chem. Mater. 2007, 19, 6297–6303. [Google Scholar] [CrossRef]
- Frank, R.L.; Smith, P.V. The preparation of mercaptans from alcohols. J. Am. Chem. Soc. 1946, 68, 2103–2104. [Google Scholar] [CrossRef]
- Wardell, J.L. The Chemistry of the Thiol Group; Patai, S., Ed.; Wiley: London, UK, 1974; p. 179. [Google Scholar]
- Yamada, M.; Sotoya, K.; Sakakibara, T.; Takamoto, T.; Sudoh, R. Studies on N-alkyl-2(1H)-pyridothione. 1. A new synthetic method for thiols. J. Org. Chem. 1977, 42, 2180–2182. [Google Scholar] [CrossRef]
- Hu, J.; Fox, M.A. A convenient trimethylsilylthioxy-dehalogenation reaction for the preparation of functionalised thiols. J. Org. Chem. 1999, 64, 4959–4961. [Google Scholar] [CrossRef] [PubMed]
- Bandgar, B.P.; Sadavarte, V.S.; Uppalla, L.S. Remarkably fast direct synthesis of thiols from alcohols under mild conditions. Chem. Lett. 2000, 1304–1305. [Google Scholar] [CrossRef]
- Zhan, Z.-P.; Lang, K.; Liu, F.; Hu, L.-M. Water effects on SmI2 reduction: A novel method for the synthesis of alkyl thiols by SmI2-promoted reductions of sodium alkyl thiosulfates and alkyl thiocyanates. Synth. Commun. 2004, 34, 3203–3208. [Google Scholar] [CrossRef]
- Molina, P.; Alajarin, M.; Vilaplana, M.J. One pot conversion of alkyl halides into thiols under mild conditions. Tetrahedron Lett. 1985, 26, 469–472. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Takizawa, S.; Nanjo, M.; Mochida, K. Cleavage of a p-cyanobenzyl group from protected alcohols, amines, and thiols using triethylgermyl sodium. Chem. Lett. 2002, 1032–1033. [Google Scholar] [CrossRef]
- Lin, C.-E.; Richardson, S.K.; Garvey, D.S. L-Cysteine as a water-soluble cation scavenger in the removal of the 2,4,6-trimethoxybenzyl group from thiols. Tetrahedron Lett. 2002, 43, 4531–4533. [Google Scholar] [CrossRef]
- Behloul, C.; Guijarro, D.; Yus, M. Desilylation procedure via a naphthalene-catalysed lithiation reaction. Tetrahedron 2005, 61, 6908–6915. [Google Scholar] [CrossRef]
- Behloul, C.; Guijarro, D.; Yus, M. Deallyloxy- and debenzyloxycarbonylation of protected alcohols, amines and thiols via a naphthalene-catalysed lithiation reaction. Tetrahedron 2005, 61, 9319–9324. [Google Scholar] [CrossRef]
- Holmes, B.T.; Snow, A.W. Aliphatic thioacetate deprotection using catalytic tetrabutylammonium cyanide. Tetrahedron 2005, 61, 12339–12342. [Google Scholar] [CrossRef]
- Vanthuyne, N.; Andreoli, F.; Fernandez, S.; Roman, M.; Roussel, C. Synthesis, chiral separation, barrier to rotation and absolute configuration of N-(o-functionalized-aryl)-4-alkyl-thiazolin-2-one and thiazoline-2-thione atropisomers. Lett. Org. Chem. 2005, 2, 433–443. [Google Scholar] [CrossRef]
- Roussel, C.; Andreoli, F.; Roman, M.; Hristova, M.; Vanthuyne, N. New route to 3-alkylthiazolo[3,2-a]benzimidazole derivatives. Molecules 2005, 10, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Roussel, C.; Roman, M.; Andreoli, F.; Del Rio, A.; Faure, R.; Vanthuyne, N. Non-racemic atropisomeric (thio)ureas as neutral enantioselective anion receptors for amino-acid derivatives: Origin of smaller Kass with thiourea than urea derivatives. Chirality 2006, 18, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Steele, R.M.; Monti, C.; Gennari, C.; Piarulli, U.; Andreoli, F.; Vanthuyne, N.; Roussel, C. Enantioselective cyanosilylation of aldehydes catalysed by a diastereomeric mixture of atropisomeric thioureas. Tetrahedron: Asymmetry 2006, 17, 999–1006. [Google Scholar] [CrossRef]
- Roussel, C.; Kaid-Slimane, R.; Andreoli, F.; Renaudin, M.; Vanthuyne, N. Synthesis, chiral separation, and absolute configuration of bis-(N-aryl) atropisomeric triads: 1,2-bis-[4-methyl-2-(thi)oxo-2,3-dihydrothiazol-3-yl]-benzene. Chirality 2009, 21, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Bellec, N.; Lorcy, D.; Robert, A. Towards functionalised quasi-planar dithiadiazafulvalenes: Synthesis of various precursors. Synthesis 1998, 10, 1442–1446. [Google Scholar] [CrossRef]
- National Institute of Advanced Industrial Science and Technology. For comparison of spectroscopic data see: SDBS Web http://riodb01.ibase.aist.go.jp/sdbs/ accessed on 27 October 2009.
- For comparison of spectroscopic data see: SIGMA-ALDRICH website. http://www.sigmaaldrich.com/ accessed on 13 November 2009.
Sample Availability: Samples of compounds 2a-h and 3a-h are available from the authors. |


{kind=link}
{kind=link}
| Reagent | Thiazolium salt | Yield (%) | Thiol | Yield (%) |
|---|---|---|---|---|
| CH3(CH2)6I | 2a | 96 | 3a | 90 |
| CH3(CH2)8I | 2b | 96 | 3b | 92 |
| CH3(CH2)9I | 2c | 94 | 3c | 94 |
| CH3(CH2)11I | 2d | 93 | 3d | 91 |
| CH3(CH2)17I | 2e | 98 | 3e | 92 |
| Reagent | Thiazolium salt | Yield (%) | Thiol | Yield (%) |
|---|---|---|---|---|
| I(CH2)3I | 2f | 94 | 3f | 91 |
| I(CH2)4I | 2g | 95 | 3g | 90 |
| I(CH2)5I | 2h | 89 | 3h | 92 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mehdid, M.A.; Djafri, A.; Roussel, C.; Andreoli, F. 3-(2-Aminophenyl)-4-methyl-1,3-thiazole-2(3H)-thione as an Ecofriendly Sulphur Transfer Agent to Prepare Alkanethiols in High Yield and High Purity. Molecules 2009, 14, 4634-4643. https://doi.org/10.3390/molecules14114634
Mehdid MA, Djafri A, Roussel C, Andreoli F. 3-(2-Aminophenyl)-4-methyl-1,3-thiazole-2(3H)-thione as an Ecofriendly Sulphur Transfer Agent to Prepare Alkanethiols in High Yield and High Purity. Molecules. 2009; 14(11):4634-4643. https://doi.org/10.3390/molecules14114634
Chicago/Turabian StyleMehdid, Mohammed Amine, Ayada Djafri, Christian Roussel, and Federico Andreoli. 2009. "3-(2-Aminophenyl)-4-methyl-1,3-thiazole-2(3H)-thione as an Ecofriendly Sulphur Transfer Agent to Prepare Alkanethiols in High Yield and High Purity" Molecules 14, no. 11: 4634-4643. https://doi.org/10.3390/molecules14114634