Temozolomide with Radiation Therapy in High Grade Brain Gliomas: Pharmaceuticals Considerations and Efficacy;A Review Article

Abstract
:Introduction
Materials and Methods
Identification of Eligible Studies
Data Extraction
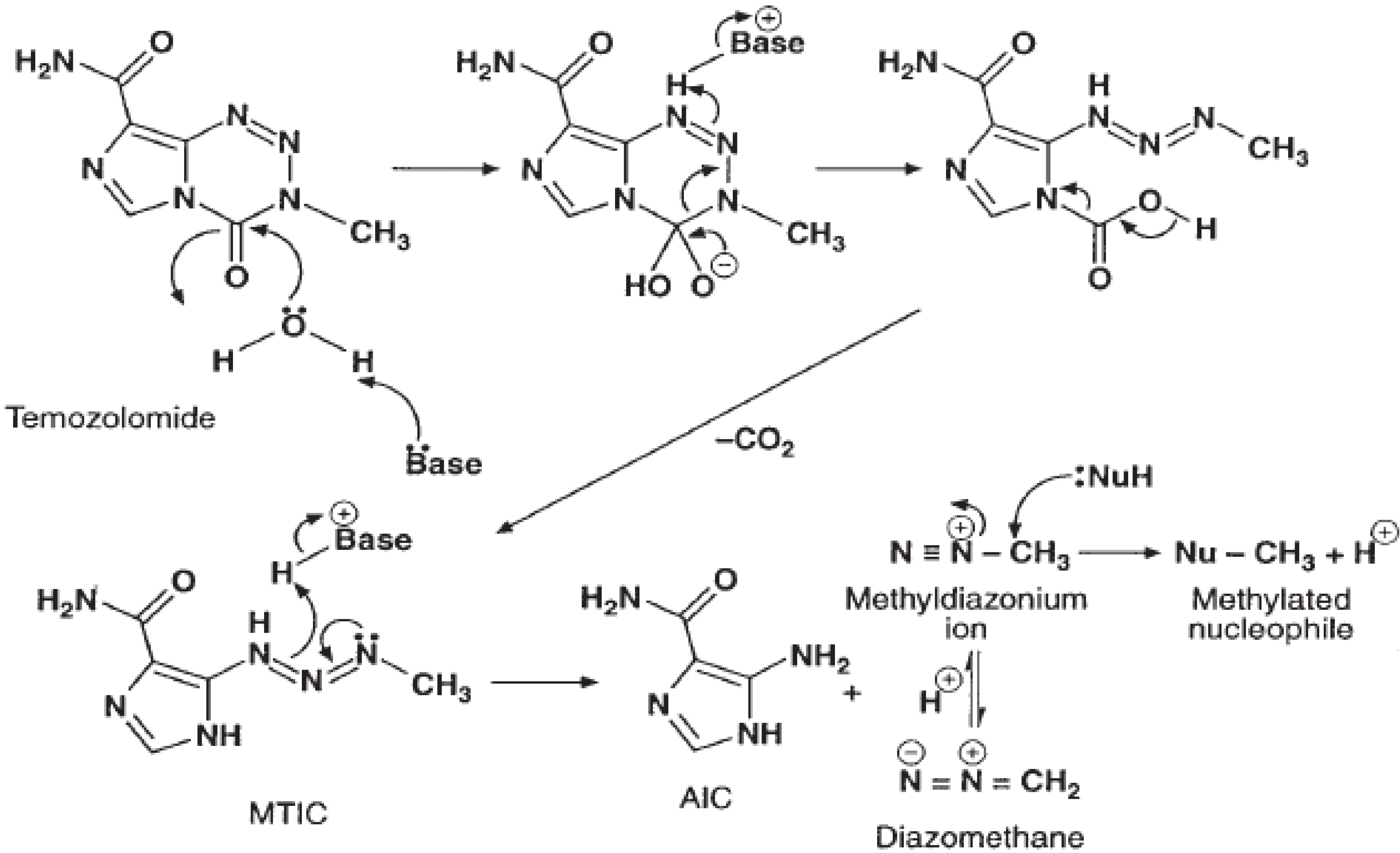
Chemical structure and mechanism of action

Mechanisms of Resistance
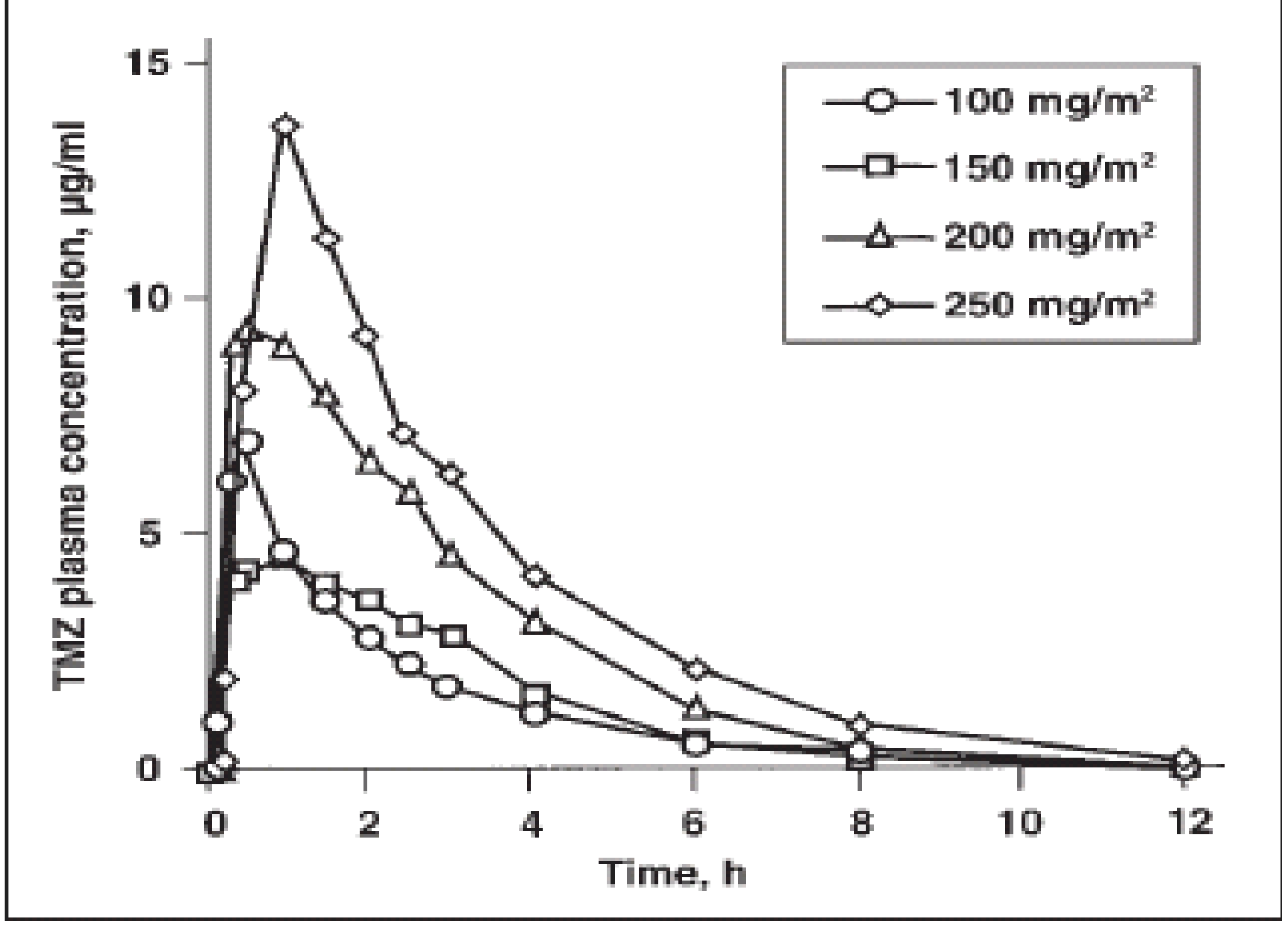
Clinical pharmacology


{kind=link}
{kind=link}
| Oral dose | ||
|---|---|---|
| Parameter | 150 mg/m2 (n=12) | 200 mg/m2 (n=6) |
| Cmax | 7.75 μg/mL | 10.7 μg/mL |
| Tmax | 0.85 h | 0.89 h |
| AUC | 22.6 μg·h/mL | 29.7 μg·h/mL |
| T1/2 | 1.81 h | 1.84 h |
| Clearance | 3.05 mL/kg/min | 2.85 mL/kg/min |
| Volume of distribution | 0.48 L/kg | 0.45 L/kg |
Temozolomide in Malignant Gliomas
Combination of Temozolomide with Radiation Therapy
Eligible trials
| Primary therapy | ||||
|---|---|---|---|---|
| Author | Year published Type of trial | Number of patients | Treatment schecule | Outcomes |
| Athanasiou [66] | 2005, RCT | 130 | Concomitant TMZ daily with RT for 6 weeks. Adjuvant TMZ on days 1-5 and 15-19 for less than 6 cycles. | Primary: Survival; PFS. Secondary: Safety |
| Stupp [64] | 2005, RCT | 573 | Concomitant daily TMZ (75mg/m2/day) during RT (<7weeks). Adjuvant TMZ for first 5 days out of 28 for 6 or fewer cycles. RT was 60Gy focally to the tumour and a 2-3cm margin over 30 sessions and 6 weeks. | Primary: Survival. Seconday: PFS, Safety, QoL |
| Recurrent disease | ||||
| Yung [67] | 2000, RCT | 225 | Temozolomide: 200mg/m2/day (if chemotherapy naive) or 150mg/m2/day (if prior chemotherapy) for 5 days out of a 28 day cycle. Procarbazine 150mg/m2/day (chemotherapy naive) or 125mg/m2/day (if prior chemotherapy) for 28 consecutive days in a 56 day cycle | Objective response, sixmonth PFS, median PFS, survival, adverse events |
Results
Conclusions
References and Notes
- O’Reilly, S.M.; Newlands, E.S.; Glaser, M.G.; Brampton, M.; Rice-Edwards, J.M.; Illingworth, R.D.; Richards, P.G.; Kennard, C.; Colquhoun, I.R.; Lewis, P. Temozolomide: A new cytotoxic agent with promising activity against primary brain tumors. Eur. J. Cancer 1993, 29, 940–942. [Google Scholar] [CrossRef]
- Newlands, E.S.; O’Reilly, S.M.; Glaser, M.G.; Bower, M.; Evans, H.; Brock, C.; Brampton, M.H.; Colquhoun, I.; Lewis, P.; Rice-Edwards, J.M.; Illingworth, R.; Richards, P.G. The Charing Cross Hospital experience with temozolomide in patients with gliomas. Eur. J. Cancer 1996, 32A, 2236–2241. [Google Scholar]
- Bower, M.; Newlands, E.S.; Bleehen, N.M.; Brada, M; Begent, R.J.; Calvert, H.; Colpuhoun, I.; Lewis, P.; Brampton, M.H. Multicentre CRC phase II trial of temozolomide in recurrent orprogressive highgrade glioma. Cancer Chemother. Pharmacol. 1997, 40, 484–488. [Google Scholar] [CrossRef]
- Yung, W.K.; Prados, M.D.; Yaya-Tur, R.; Rosenfeld, S.S.; Brada, M.; Friedman, H.S.; AlbrightR.; Olson, J.; Chang, S.M.; O’Neill, A.M.; Friedman, A.H.; Bruner, J.; Yue, N.; Dugan, M.; Zaknoen, S.; Levin, V.A. Multicenter phase II trial of temozolomide in patients with anaplastic astrocytoma or anaplastic oligoastrocytoma at first relapse. J. Clin. Oncol. 1999, 17, 2762–2771. [Google Scholar]
- Newlands, E.S.; Blackledge, G.R.; Slack, J.A.; Rustin, G.J.; Smith, D.B.; Stuart, N.S.; Quarterman, C.P.; Hoffman, R.; Stevens, M.F.; Brampton, M.H. Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 1992, 65, 287–291. [Google Scholar]
- Bleehen, N.M.; Newlands, E.S.; Lee, S.M.; Can Thatcher, N.; Selby, P.; Calvert, A.H.; Rustin, G.J.; Brampton, M.; Stevens, M.F. Cancer Research Campaign phase II trial of temozolomide in metastatic melanoma. J. Clin. Oncol. 1995, 13, 910–913. [Google Scholar]
- Middleton, M.R.; Lunn, J.M.; Morris, C.; Rustin, G.; Wedge, S.R.; Brampton, M.H.; Lind, M.J.; Lee, S.M.; Newell, D.R.; Bleehen, N.M.; Newlands, E.S.; Calvert, A.H.; Margison, G.P.; Thatcher, N. O6-Methylguanine- DNA methyltransferase in pretreatment tumour biopsies as a predictor of response to temozolomide in melanoma. Br. J. Cancer 1998, 78, 1199–1202. [Google Scholar] [CrossRef]
- Stevens, M.F.; Hickman, J.A.; Stone, R.; Gibson, N.W.; Baig, G.U.; Lunt, E.; Newton, C.G. Antitumor imidazotetrazines. 1. Synthesis and chemistry of 8-carbamoyl-3-(2- chloroethyl)imidazo[5,1-d]-1,2,3,5-tetrazin-4(3 H)-one, a novel broad-spectrum antitumor agent. J. Med. Chem. 1984, 27, 196–201. [Google Scholar] [CrossRef]
- Newlands, E.S.; Stevens, M.F.G.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Patel, M.; McCully, C.; Godwin, K. Plasma and cerebrospinal fluid pharmacokinetics of temozolomide. Proc. Am. Soc. Clin. Oncol. 1995, 14, 461a. [Google Scholar] [CrossRef]
- Newlands, E.S.; Blackledge, G.R.P.; Slack, J.A.; Rustin, G.J.S.; Smith, D.B.; Stuart, N.S.A.; Quarterman, C.P.; Hoffman, R.; Stevens, M.F.G.; Brampton, M.H.; Gibson, A.C. Phase I Trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 1992, 65, 287–291. [Google Scholar] [CrossRef]
- Bleehen, N.M.; Newlands, E.S.; Lee, S.M.; Thatcher, N.; Selby, P.; Calvert, A.H.; Rustin, G.J.S.; Brampton, M.; Stevens, M.F.G. Cancer Research Campaign Phase II trial of temozolomide in metastatic melanoma. J. Clin. Oncol. 1995, 1, 910–913. [Google Scholar]
- O’Reilly, S.M.; Newlands, E.S.; Glaser, M.G.; Brampton, M.; Rice-Edwards, J.M.; Illingworth, R.D.; Richards, P.G.; Kennard, C.; Colquhoun, I.R.; Lewis, P.; Stevens, M.F.G. Temozolomide: a new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumours. Eur. J. Cancer 1993, 29A, 940–942. [Google Scholar]
- Bower, M.; Newlands, E.S.; Bleehen, N.M.; Brada, M.; Begent, R.J.H.; Calvert, H.; Colquhoun, I.; Lewis, P.; Brampton, M.H. Multicentre CRC Phase II trial of temozolomide in recurrent or progressive high-grade glioma. Cancer Chemother. Pharmacol. 1997, 40, 484–488. [Google Scholar] [CrossRef]
- Paulsen, F.; Hoffmann, W.; Becker, G.; Belka, C.; Weinmann, M.; Classen, J.; Kortmann, R.D.; Bamberg, M. Chemotherapy in the treatment of recurrent glioblastoma multiforme: ifosfamide versus temozolomide. J. Cancer Res. Clin. Oncol. 1999, 125, 411–418. [Google Scholar] [CrossRef]
- Estlin, E.J.; Lashford, L.; Ablett, S.; Price, L.; Gowing, R.; Gholkar, A.; Kohler, J.; Lewis, I.J.; Morland, B.; Pinkerton, C.R.; Stevens, M.C.G.; Mott, M.; Stevens, R.; Newell, D.R.; Walker, D.; Dicks- Mireaux, C.; McDowell, H.; Reidenberg, P.; Statkevich, P.; Marco, A.; Batra, V.; Dugan, M.; Pearson, A.D.J. Phase I study of temozolomide in paediatric patients with advanced cancer. United Kingdom Children’s Cancer Study Group. Br. J. Cancer. 1998, 78, 652–661. [Google Scholar] [CrossRef]
- Nicholson, H.S.; Krailo, M.; Ames, M.M.; Seibel, N.L.; Reid, J.M.; Liu-Mares, W.; Vezina, L.G.; Ettinger, A.G.; Reaman, G.H. Phase I study of temozolomide in children and adolescents with recurrent solid tumors: a report from the Children’s Cancer Group. J. Clin. Oncol. 1998, 16, 3037–3043. [Google Scholar]
- Stevens, M.F.; Hickman, J.A.; Langdon, S.P.; Chubb, D.; Vickers, L.; Stone, R.; Baig, G.; Goddard, C.; Gibson, N.W.; Slack, J.A. Antitumoractivity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987, 47, 5846–5852. [Google Scholar]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar]
- Pegg, A.E.; Dolan, M.E.; Moschel, R.C. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog. Nucleic Acid Res. Mol. Biol. 1995, 51, 167–223. [Google Scholar] [CrossRef]
- D’Incalci, M.; Citti, L.; Taverna, P.; Catapano, C.V. Importance of the DNA repair enzyme O6-alkyl guanine alkyltransferase (AT) in cancer chemotherapy. Cancer Treat. Rev. 1988, 15, 279–292. [Google Scholar] [CrossRef]
- Pegg, A.E.; Byers, T.L. Repair of DNA containing O6-alkylguanine. FASEB J. 1992, 6, 2302–2310. [Google Scholar]
- Tisdale, M.J. Antitumor imidazotetrazines–XV. Role of guanine O6 alkylation in the mechanism of cytotoxicity of imidazotetrazinones. Biochem. Pharmacol. 1987, 36, 457–462. [Google Scholar] [CrossRef]
- Baer, J.C.; Freeman, A.A.; Newlands, E.S.; Watson, A.J.; Rafferty, J.A.; Margison, G.P. Depletion of O6-alkylguanine-DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. Br. J. Cancer 1993, 67, 1299–1302. [Google Scholar] [CrossRef]
- Redmond, S.M.; Joncourt, F.; Buser, K.; Ziemiecki, A.; Altermatt, H.J.; Fey, M.; Margison, G.; Cerny, T. Assessment of P-glycoprotein, glutathione-based detoxifying enzymes and O6-alkylguanine-DNA alkyltransferase as potential indicators of constitutive drug resistance in human colorectal tumors. Cancer Res. 1991, 51, 2092–2097. [Google Scholar]
- Franchi, A.; Papa, G.; D’Atri, S.; Piccioni, D.; Masi, M.; Bonmassar, E. Cytotoxic effects of dacarbazine in patients with acute myelogenous leukemia: a pilot study. Haematologica 1992, 77, 146–150. [Google Scholar]
- D’Atri, S.; Piccioni, D.; Castellano, A.; Tuorto, V.; Franchi, A.; Lu, K.; Christiansen, N.; Frankel, S.; Rustum, Y.M.; Papa, G.; Mandelli, F.; Bonmassar, E. Chemosensitivity to triazene compounds and O6-alkylguanine-DNA alkyltransferase levels: studies with blasts of leukaemic patients. Ann. Oncol. 1995, 6, 389–393. [Google Scholar]
- Tentori, L.; Graziani, G.; Gilberti, S.; Lacal, P.M.; Bonmassar, E.; D’Atri, S. Triazene compounds induce apoptosis in O6-alkylguanine- DNA alkyltransferase deficient leukemia cell lines. Leukemia 1995, 9, 1888–1895. [Google Scholar]
- Walker, M.C.; Masters, J.R.W.; Margison, G.P. O6-alkylguanine-DNA-alkyltransferase activity and nitrosourea sensitivity in human cancer cell lines. Br. J. Cancer 1992, 66, 840–843. [Google Scholar] [CrossRef]
- Bobola, M.S.; Tseng, S.H.; Blank, A.; Berger, M.S.; Silber, J.R. Role of O6-methylguanine-DNA methyltransferase in resistance of human brain tumor cell lines to the clinically relevant methylating agents temozolomide and streptozotocin. Clin. Cancer Res. 1996, 2, 735–741. [Google Scholar]
- Liu, L.; Markowitz, S.; Gerson, S.L. Mismatch repair mutations override alkyltransferase in conferring resistance to temozolomide but not to 1,3-bis(2-chloroethyl)nitrosourea. Cancer Res. 1996, 56, 5375–5379. [Google Scholar]
- Friedman, H.S.; Johnson, S.P.; Dong, Q.; Schold, S.C.; Rasheed, B.K.A.; Bigner, S.H.; Ali- Osman, F.; Dolan, E.; Colvin, O.M.; Houghton, P.; Germain, G.; Drummond, J.T.; Keir, S.; Marcelli, S.; Bigner, D.D.; Modrich, P. Methylator resistance mediated bymismatch repair deficiency in a glioblastoma multiforme xenograft. Cancer Res. 1997, 57, 2933–2936. [Google Scholar]
- Wedge, S.R.; Porteous, J.K.; Newlands, E.S. 3-aminobenzamide and/or O6-benzylguanine evaluated as an adjuvant to temozolomide or BCNU treatment in cell lines of variable mismatch repair status and O6-alkylguanine–DNA alkyltransferase activity. Br. J. Cancer 1996, 74, 1030–1036. [Google Scholar] [CrossRef]
- Tisdale, M.J. Antitumour imidazotetrazines-XI: effect of 8-carbamoyl-3-methylimidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one [CCRG 81045; M and B 39831 NSC 362856] on poly(ADP-ribose) metabolism. Br. J. Cancer 1985, 52, 789–792. [Google Scholar] [CrossRef]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADPribose) n participates in DNA excision repair. Nature (Lond.) 1980, 283, 593–596. [Google Scholar]
- Boulton, S.; Pemberton, L.C.; Porteous, J.K.; Curtin, N.J.; Griffin, R.J.; Golding, B.T.; Durkacz, B.W. Potentiation of temozolomideinduced cytotoxicity: a comparative study of the biological effects of poly(ADP-ribose) polymerase inhibitors. Br. J. Cancer 1995, 72, 849–856. [Google Scholar] [CrossRef]
- Wu, Z.; Chan, C.L.; Eastman, A.; Bresnick, E. Expression of human O6-methylguanine-DNA methyltransferase in Chinese hamster ovary cells and restoration of cellular resistance to certain N-nitroso compounds. Mol. Carcinog. 1991, 4, 482–488. [Google Scholar] [CrossRef]
- Tentori, L.; Turriziani, M.; Franco, D.; Serafino, A.; Levati, L.; Roy, R.; Bonmassar, E.; Graziani, G. Treatment with temozolomide andpoly(ADP-ribose) polymerase inhibitors induces early apoptosis and increases base excision repair gene transcripts in leukemic cells resistant to triazene compounds. Leukemia 1999, 13, 901–909. [Google Scholar] [CrossRef]
- Liu, L.; Taverna, P.; Whitacre, C.M.; Chatterjee, S.; Gerson, S.L. Pharmacologic disruption of base excision repair sensitizes mismatch repair-deficient and -proficient colon cancer cells to methylating agents. Clin. Cancer Res. 1999, 5, 2908–2917. [Google Scholar]
- Catapano, C.V.; Broggini, M.; Erba, E.; Ponti, M.; Mariani, L.; Citti, L.; D’Incalci, M. In vitro and in vivo methazolastone-induced DNA damage and repair in L-1210 leukemia sensitive and resistant to chloroethylnitrosoureas. Cancer Res. 1987, 47, 4884–4889. [Google Scholar]
- Deans, B.; Tisdale, M.J. Antitumour imidazotetrazines XXVIII 3-methyladenine DNA glycosylase activity in cell lines sensitive and resistant to temozolomide. Cancer Lett. 1992, 63, 151–157. [Google Scholar] [CrossRef]
- Imperatori, L.; Damia, G.; Taverna, P.; Garattini, E.; Citti, L.; Boldrini, L.; D’Incalci, M. 3T3 NIH murine fibroblasts and B78 murine melanoma cells expressing the Escherichia coli N3-methyladenine-DNA glycosylase I do not become resistant to alkylating agents. Carcinogenesis (Lond.) 1994, 15, 533–537. [Google Scholar] [CrossRef]
- Reidenberg, P.; Statkevich, P.; Judson, I. Effect of food on the oral bioavailability of temozolomide, a new chemotherapeutic agent. Proc. Am. Soc. Clin. Pharmacol. Ther. 1996, 59, 199a. [Google Scholar] [CrossRef]
- Brock, C.S.; Newlands, E.S.; Wedge, S.R.; Bower, M.; Evans, H.; Colquhoun, I.; Roddie, M.; Glaser, M.; Brampton, M.H.; Rustin, G.J. Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res. 1998, 58, 4363–4367. [Google Scholar]
- Kenilworth, N.J. Data on File; Schering-Plough Corporation: New Jersey, USA, 1998.
- Kenilworth, N.J. Data on File. Study CI93-114; Schering-Plough Corporation: New Jersey, USA, 1998.
- Nicholson, H.S.; Ames, M.M.; Krailo, M. Phase I and pharmacokinetic study of temozolomide (TEM) in children and adolescents. A report from the Children’s Cancer Group (CCG). Proc. Am. Soc. Clin. Oncol. 1995, 14, 449a. [Google Scholar]
- Dhodapkar, M.; Reid, J.; Ames, M.M. Phase I clinical trial and pharmacokinetics of temozolamide (NSC 362856). Proc. Am. Assoc. Cancer Res. 1994, 35, 245a. [Google Scholar]
- Eckardt, J.R.; Weiss, G.R.; Burris, H.A. Phase I clinical trial andpharmacokinetic trial of SCH52365 (temozolomide) given orally daily ´ 5 days. Proc. Am. Soc. Clin. Oncol. 1995, 14, 484a. [Google Scholar]
- Winger, M.J.; Winger, D.R.; Cairncross, J.G. Supratentorial anaplastic gliomas in adults. The prognostic importance of extent of resection and prior low-grade glioma. J. Neurosurg. 71, 487–493.
- Hart, M.G.; Grant, R.; Metcalfe, S.E. Biopsy versus resection for High Grade Glioma. Cochrane Database Sys. Rev. 2000, Issue 2. [Google Scholar]
- Walker, M.D.; Alexander, E., Jr.; Hunt, W.E.; MacCarty, C.S.; Mahaley, M.S., Jr.; Mealey, J., Jr. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliom. A cooperative clinical trial. J. Neurosurg. 1978, 49, 333–343. [Google Scholar] [CrossRef]
- Kaal, E.C.A.; Vecht, C.J. The management of brain edema in brain tumours. Curr. Opin. Oncol. 2004, 16, 593–600. [Google Scholar] [CrossRef]
- Rampling, R.; James, A.; Papanastassoiu, V. The Present and Future Management of Malignant Brain Tumours: Surgery, Radiotherapy, Chemotherapy. J. Neurol. Neurosurg. Psychiatry 2005, 75(Suppl II), 24–30. [Google Scholar]
- Glioma Meta-Analysts Trialists (GMT) Group. Chemotherapy for High Grade Glioma. Cochrane Database Sys. Rev. 2002, Issue 3. [Google Scholar]
- Cairncross, G.; Berkey, B.; Shaw, E.; Jenkins, R.; Scheithauer, B.; Brachman, D. Phase III Trial of Chemotherapy Plus Radiotherapy Compared With Radiotherapy Alone for Pure and Mixed Anaplastic Oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J. Clin. Oncol. 2006, 24, 18, 2707–2714. [Google Scholar]
- Van den Bent, M.J.; Carpentier, A.F.; Brandes, A.A.; Sanson, M.; Taphoorn, M.J.; Bernsen, H.J.; Renay, M.; Tijssen, C.C.; Grisold, W.; Sipos, L.; Haaxma-Reiche, H.; Kros, J.M.; van Kouwenhoven, M.C.; Vecht, C.J.; Allgeier, A.; Lacombe, D.; Gorlia, T. Adjuvant procarbazine, iomustine and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: A randomized european organisation for research and treatment of cancer phase III trial. J. Clin. Oncol. 2006, 24, 2715–2722. [Google Scholar] [CrossRef]
- Lanzetta, G.; Campanella, C.; Rozzi, A.; Nappa, M.; Costa, A.; Fedele, F.; Innocenzi, G.; Agliardi, F.M.; Salvati, M.; Minniti, G.; Frati, A.; Frati, L.; Vecchione, A. Temozolamide in radio-chemotherapy combined treatment for newly-diagnosed glioblastoma multiforme: Phase II clinical trial. Anticancer Res. 2003, 23, 5159–5164. [Google Scholar]
- Stupp, R.; Dietrich, P.Y.; Ostermann Kraljevic, S.; Pica, A.; Maillard, I.; Maeder, P.; Meuli, R.; Janzer, R.; Pizzolato, G.; Miralbell, R.; Porchet, F.; Regli, L.; de Tribolet, N.; Mirimanoff, R.O.; Leyvraz, S. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radioation plus temozolamide followed by adjuvant temozolamdide. J. Clin. Oncol. 2002, 20, 1375–1382. [Google Scholar] [CrossRef]
- Wedge, S.R.; Porteous, J.K.; Glaser, M.G; Marcus, K.; Newlands, E.S. In vitro evaluation of temozolomide combined with X-irradiation. Anticancer Drugs 1997, 8, 92–97. [Google Scholar] [CrossRef]
- Hirose, Y.; Berger, S.M.; Russell, O.P. p53 Effects Both the Duration of G2/M Arrest and the Fate of Temozolomide-treated Human Glioblastoma Cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Chakravarti, A.; Erkkinen, G.M.; Nestler, U.; Stupp, R.; Mehta, M.; Aldape, K.; Gilbert, R.M.; Black, P.; Loeffler, S.J. Temozolomide-Mediated Radiation Enhancement in Glioblastoma: A Report on Underlying Mechanisms. Clin. Cancer Res. 2006, 12, 4738–4746. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; Hau, P.; Brandes, A.A.; Gijtenbeek, J.; Marosi, C.; Vecht, C.J.; Mokhtari, K.; Wesseling, P.; Villa, S.; Eisenhauer, E.; Gorlia, T.; Weller, M. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet. Oncol. 2009, Mar 6. [Epub ahead of print]. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; Curschmann, J.; Janzer, R.C.; Ludwin, S.K.; Gorlia, T.; Allgeier, A.; Lacombe, D.; Cairncross, J.G.; Eisenhauer, E.; Mirimanoff, R.O. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastom. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Taphoorn, M.J.; Stupp, R.; Coens, C.; Osoba, D.; Kortmann, R.; van den Bent, M.J.; Mason, W.; Mirimanoff, R.O.; Baumert, B.G.; Eisenhauer, E.; Forsyth, P. Bottomley A Health-related quality of life in patients with glioblastoma: A randomised controlled trial. Lancet. Oncol. 2005, 6, 937–944. [Google Scholar] [CrossRef]
- Athanassiou, H.; Synodinou, M.; Maragoudakis, E.; Paraskevaidis, M.; Verigos, C.; Misailidou, D.; Antonadou, D.; Saris, G.; Beroukas, K.; Karageorgis, P. Randomised Phase II study of Temozolomide and Radiotherapy Compared with Radiotherapy Alone in Newly Diagnosed Glioblastoma Multiforme. J. Clin. Oncol. 2005, 23, 2372–2377. [Google Scholar] [CrossRef]
- Yung, A.; Levin, V.A.; Albright, A. Randomized trial of temodal (TEM) vs. procarbazine (PCB) in glioblastoma multiforme (GBM) at first relapse. Proc. Am. Soc. Clin. Oncol. 2000, 18, 139. [Google Scholar]
- Sample availability: Not available.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Koukourakis, G.V.; Kouloulias, V.; Zacharias, G.; Papadimitriou, C.; Pantelakos, P.; Maravelis, G.; Fotineas, A.; Beli, I.; Chaldeopoulos, D.; Kouvaris, J. Temozolomide with Radiation Therapy in High Grade Brain Gliomas: Pharmaceuticals Considerations and Efficacy;A Review Article. Molecules 2009, 14, 1561-1577. https://doi.org/10.3390/molecules14041561
Koukourakis GV, Kouloulias V, Zacharias G, Papadimitriou C, Pantelakos P, Maravelis G, Fotineas A, Beli I, Chaldeopoulos D, Kouvaris J. Temozolomide with Radiation Therapy in High Grade Brain Gliomas: Pharmaceuticals Considerations and Efficacy;A Review Article. Molecules. 2009; 14(4):1561-1577. https://doi.org/10.3390/molecules14041561
Chicago/Turabian StyleKoukourakis, Georgios V., Vassilios Kouloulias, Georgios Zacharias, Christos Papadimitriou, Panagiotis Pantelakos, George Maravelis, Andreas Fotineas, Ivelina Beli, Demetrios Chaldeopoulos, and John Kouvaris. 2009. "Temozolomide with Radiation Therapy in High Grade Brain Gliomas: Pharmaceuticals Considerations and Efficacy;A Review Article" Molecules 14, no. 4: 1561-1577. https://doi.org/10.3390/molecules14041561