Synthesis of γ-Nitro Aliphatic Methyl Esters Via Michael Additions Promoted by Microwave Irradiation

Abstract
:1. Introduction
2. Results and Discussion
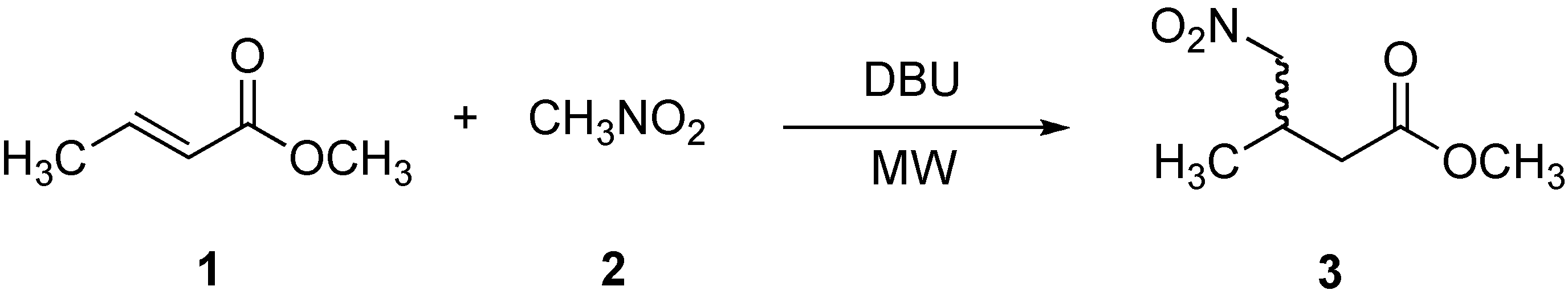
2.1. Preparation of methyl 3-methyl-4-nitrobutanoate (3)


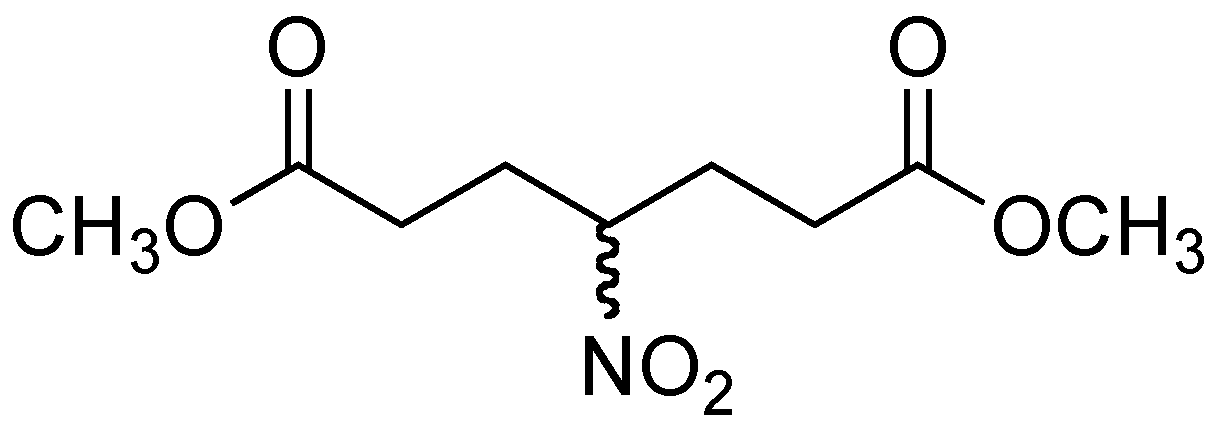{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Eq. DBU | Power (W) | Temperature (oC) | Time (min) | Yield 3 (%) |
|---|---|---|---|---|---|
| 1 | 1.1 | - | r. t. | 4320 | 72 |
| 7 | 0.01 | 50 | 70-75 | 5 | 10 c |
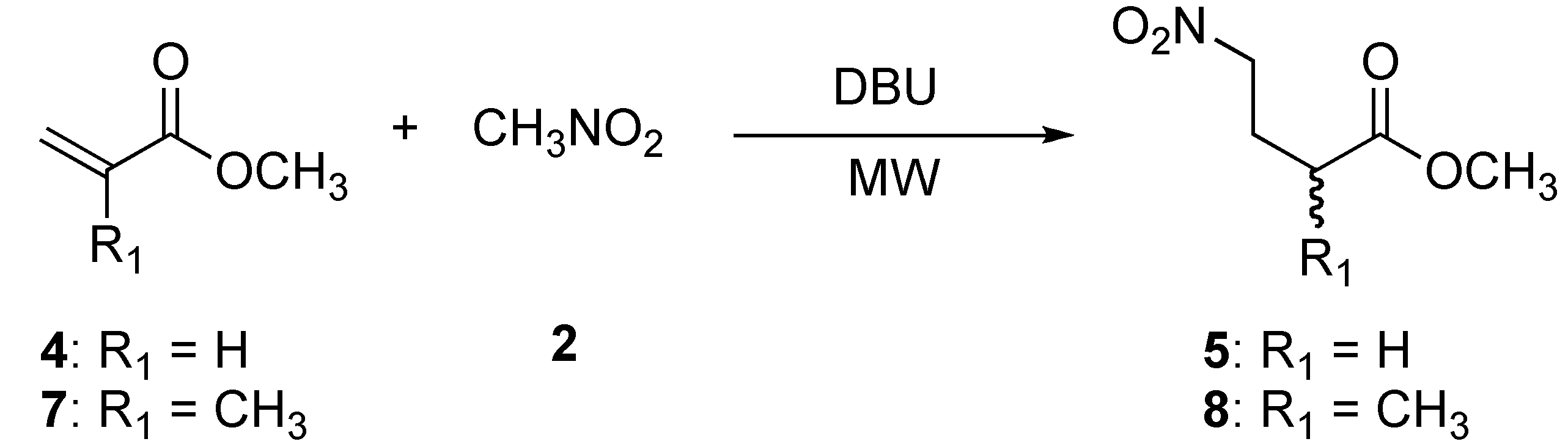
2.2. Preparation of methyl 4-nitrobutanoate (5) and methyl 2-methyl-4-nitrobutanoate (8)




| Entry | Eq. Base | Power (W) | Temperature (oC) | Time (min) | 8 / 9 / 7 (%) a |
|---|---|---|---|---|---|
| 1 | 1.1 DBU | - | r. t. | 4320 | 47 / 53 / 0 |
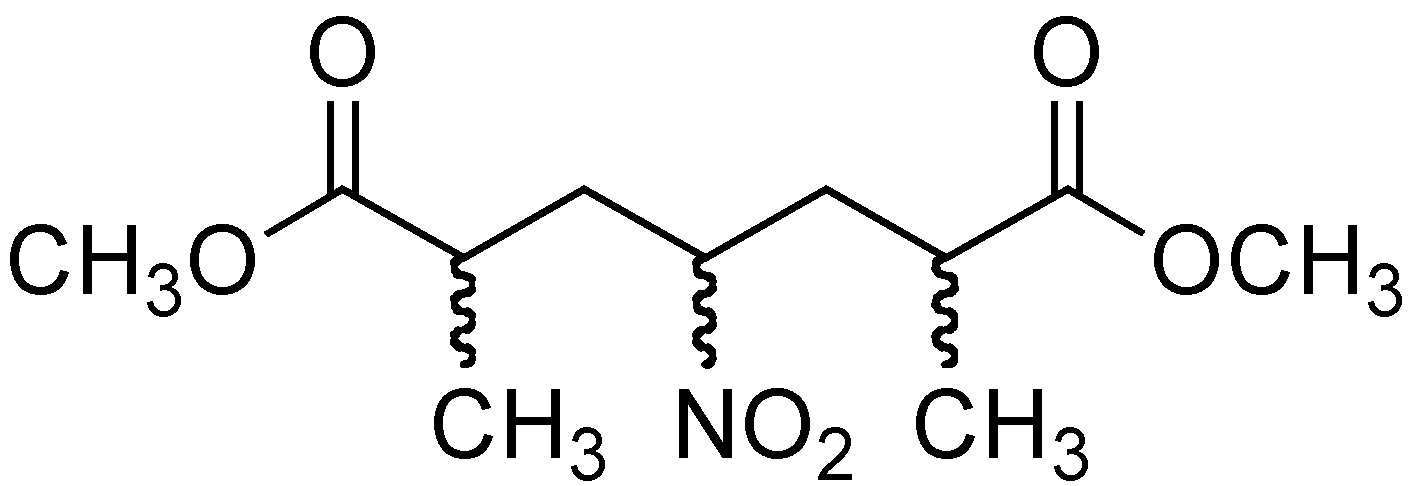
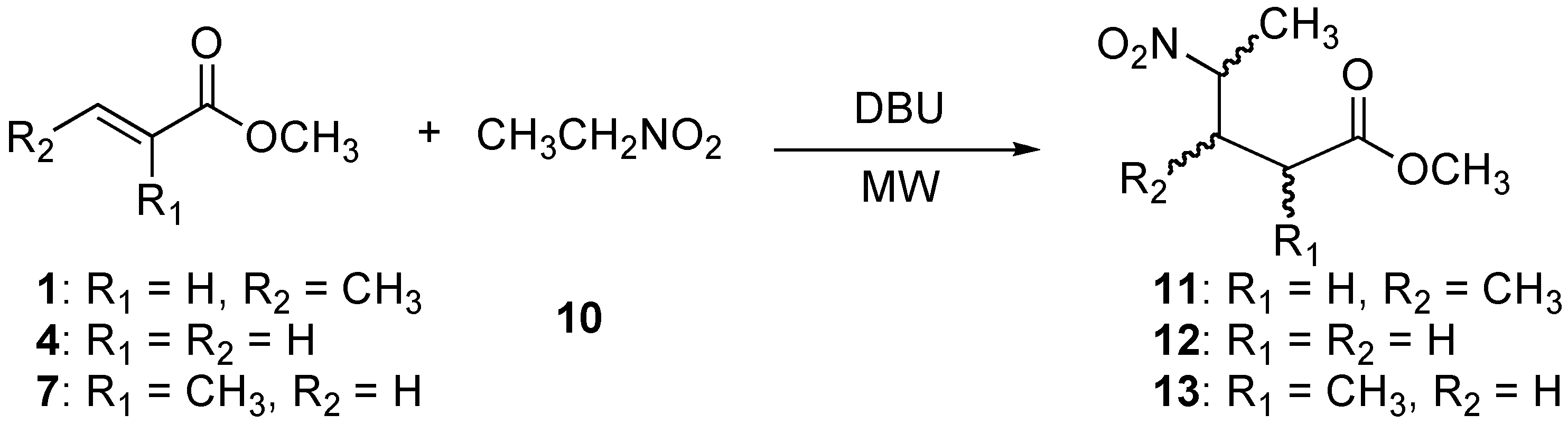
2.3. Preparation of methyl 3-methy-4-nitropentanoate (11), methyl 4-nitropentanoate (12), and methyl 2-methy-4-nitropentanoate (13)

| Entry | Methyl acrylates | Eq. DBU | Power (W) | Temperature (oC) | Time (min) | Product (%) a |
|---|---|---|---|---|---|---|
| 1 | 1 | 0.05 | 50 | 70-75 | 5 | 11 (98) b |
3. Conclusions
4. Experimental
4.1. General
4.2. General procedure: Synthesis of methyl 4-nitro alkyl esters via microwave irradiation
Acknowledgements
References and Notes
- Loupy, A.; Petit, A.; Hamelin, J.; Texier-Boullet, F.; Jacquault, P.; Mathe, D. New solvent-free organic synthesis using focused microwaves. Synthesis 1998, 9, 1213–1234. [Google Scholar]
- Caddick, S. Microwave assisted organic reactions. Tetrahedron 1995, 51, 10403–10432. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Lidström, P.; Tierney, J.; Wathey, B.; Westman, J. Microwave assisted organic synthesis. Tetrahedron 2001, 57, 9225–9283. [Google Scholar] [CrossRef]
- Escalante, J.; Carrillo-Morales, M.; Linzaga, I. Michael Additions of Amines to Methyl Acrylates Promoted by Microwave Irradiation. Molecules 2008, 13, 340–347. [Google Scholar] [CrossRef]
- Larhed, M.; Hallberg, A. Microwave-promoted palladium catalyzed coupling reactions. J. Org. Chem. 1996, 61, 9582–9584. [Google Scholar] [CrossRef]
- Garg, N.; Larhed, M.; Hallberg, A. Heck arylation of 1,2-cyclohexanedione and 2-ethoxy-2-cyclohexenone. J. Org. Chem. 1998, 63, 4158–4162. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Wang, L.; Namboodiri, V.; Pagni, R.M. Rapid microwave-enhanced, solventless Sonogashira coupling on alumina. Tetrahedron Lett. 2000, 41, 5151–5154. [Google Scholar] [CrossRef]
- Blettner, C.G.; Köning, W.A.; Stenzel, W.; Schotten, T. Microwave-assisted aqueous Suzuki cross-coupling reactions. J. Org. Chem. 1999, 64, 3885–3890. [Google Scholar] [CrossRef]
- Larhed, M.; Hoshino, M.; Hadida, S.; Curran, D.P.; Hallberg, A. Rapid fluorous Stille coupling reactions conducted under microwave irradiation. J. Org. Chem. 1997, 62, 5583–5587. [Google Scholar] [CrossRef]
- Maleczka, R.E.; Lavis, J.M.; Clark, D.H.; Gallagher, W.P. Microwave-assisted one-pot hydrostannylation/Stille couplings. Org. Lett. 2000, 2, 3655–3658. [Google Scholar] [CrossRef]
- Loupy, A. (Ed.) Microwaves in Organic Synthesis, 2nd. Ed.; Wiley-VCH: Weinheim, Germany, 2006; Vol. 1, pp. 1–56.
- Kappe, C.; Stadler, A.; Mannhold, R.; Kubinyi, H.; Folkers, G. Methods and Principles in Medicinal Chemistry: Microwaves in Organic and Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2005; Volume 25, pp. 9–24. [Google Scholar]
- Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light; CEM Publishing: Mattehews, N.C., USA, 2002; pp. 11–26. [Google Scholar]
- Kappe, O.C.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nature Reviews 2006, 5, 51–64. [Google Scholar]
- Mavandadi, F.; Lidströn, P. Microwave–Assisted Chemistry in Drug Discovery. Current Topics in Medicinal Chemistry 2004, 4, 773–792. [Google Scholar] [CrossRef]
- Shipe, W.D.; Wolkenberg, S.E.; Lidsley, C.W. Accelerating lead development by microwave–enhanced medicinal chemistry. Drug Discovery Today: Technologies 2005, 2, 155–161. [Google Scholar] [CrossRef]
- Mavandadi, F.; Piotti, A. The impact of microwave–assisted organic synthesis in drug discovery. DDT-Reviews 2006, 11, 165–174. [Google Scholar] [CrossRef]
- Kappe, O.C. Microwave dielectric heating in synthetic organic chemistry. Chem. Soc. Rev. 2008, 37, 1127–1139. [Google Scholar] [CrossRef]
- Perreux, L.; Loupy, A. A tentative rationalization of microwave effects in organic synthesis according to the reaction medium, and mechanistic considerations. Tetrahedron 2001, 57, 9199–9223. [Google Scholar] [CrossRef]
- Dandia, A.; Sati, M.; Arya, K.; Sharma, R.; Loupy, A. Facile one pot microwave induced solvent-free synthesis and antifungal, Antitubercular screening of spiro [1,5]-benzothiazepin-2,3´[3´H]indol-2[1´H]-ones. Chem. Pharm. Bull. 2003, 51, 1137–1141. [Google Scholar] [CrossRef]
- Dandia, A.; Sati, M.; Arya, K.; Loupy, A. A. One-pot dry media synthesis of new tetracyclic 1,5-benzothiazepines under microwave activation. Heterocycles 2003, 60, 563–569. [Google Scholar] [CrossRef]
- Chasar, D.W. An improved Synthesis of 4-nitrocarboxilic Acid Esters. Synthesis Communications 1982, 841–842. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. Facile and Inexpensive Synthesis of 4-Oxoalkanoic Acids from Primary Nitroalkanes and Acrolein. Synthesis 1986, 1024–1026. [Google Scholar] [CrossRef]
- Chetia, A.; Saikia, Ch. J.; Lekhok, K.C.; Boruah, R.C. A facile synthesis of 1,7-dicarbonyl compounds via three-component Michael addition reactions. Tetrahedron Letters 2004, 45, 2649–2651. [Google Scholar] [CrossRef]
- Chamakh, A.; M´hirsi, M.; Villieras, J.; Lebreton, J.; Amri, H. A Simplified Route to (E)-2-Alkylidene-1,4-diketones. Synthesis 2000, 295–299. [Google Scholar]
- Ballini, R.; Barboni, L.; Fiorini, D.; Palmieri, A.; Petrini, M. Nitro compounds as useful reagents for the synthesis of dicarbonyl derivates. Arkivoc 2006, (vi), 127–152. [Google Scholar]
- Tucker, J.A.; Clayton, T.L.; Mordas, D.M. Alkylation of Ketene Silyl Acetals with Nitroolefins Mediated by Sterically Encumbered Lewis Acids. J. Org. Chem. 1997, 62, 4370–4375. [Google Scholar] [CrossRef]
- Busch, K.; Groth, U.M.; Kühnle, W.; Schöllkopf, U. Asymmetric Synthesis of Diastereomerically and Enantiomerically Pure α-Amino-γ-Nitro Carboxylic Esters via Michael Addition of the Titanated Bislactim Ether of Cyclo (-L-Val-Gly-) to Nitroolefines. Tetrahedron 1992, 48, 5607–5618. [Google Scholar] [CrossRef]
- Hayashi, T.; Senda, T.; Ogasawara, M. Rhodium-Catalized Asymmetric Conjugate Addition of Organoboronic Acids to Nitroalkenenes. J. Am. Chem. Soc. 2000, 122, 10716–10717. [Google Scholar] [CrossRef]
- Flores-Sánchez, P.; Escalante, J.; Castillo, E. Enzymatic resolution of N-protected-β3-amino methyl esters, using lipase B from Candida antarctica. Tetrahedron-Asymmetry 2005, 16, 629–634. [Google Scholar] [CrossRef]
- Phiashivongsa, P.; Samoshin, V.V.; Gross, P.H. Henry condensations with 4,6-O-benzylidenylated and non-protected D-glucose and L-fucose via DBU-catalysis. Tetrahedron Lett. 2003, 44, 5495–5498. [Google Scholar] [CrossRef]
- Andruszkiewicz, R.; Silverman, R. A convenient synthesis of 3-alkyl-4-aminobutanoic acids. Synthesis 1989, 19, 953–955. [Google Scholar] [CrossRef]
- Available online: http://www.chem.wisc.edu/areas/reich/pkatable/index.htm (accessed March 30, 2009).
- Sample Availability: Samples are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Escalante, J.; Díaz-Coutiño, F.D. Synthesis of γ-Nitro Aliphatic Methyl Esters Via Michael Additions Promoted by Microwave Irradiation. Molecules 2009, 14, 1595-1604. https://doi.org/10.3390/molecules14041595
Escalante J, Díaz-Coutiño FD. Synthesis of γ-Nitro Aliphatic Methyl Esters Via Michael Additions Promoted by Microwave Irradiation. Molecules. 2009; 14(4):1595-1604. https://doi.org/10.3390/molecules14041595
Chicago/Turabian StyleEscalante, Jaime, and Francisco D. Díaz-Coutiño. 2009. "Synthesis of γ-Nitro Aliphatic Methyl Esters Via Michael Additions Promoted by Microwave Irradiation" Molecules 14, no. 4: 1595-1604. https://doi.org/10.3390/molecules14041595