The Reaction of Diethyl Bromomalonate with p-tert-Butylthia-calix[4]arene: An Approach to Asymmetrical Derivatives

Chemistry Department, Sohag University, Sohag, 82524, Egypt
Molecules 2009, 14(5), 1755-1761; https://doi.org/10.3390/molecules14051755
Submission received: 7 April 2009
/
Revised: 20 April 2009
/
Accepted: 6 May 2009
/
Published: 7 May 2009
Abstract
:New dissymmetric and asymmetric p-tert-butylthiacalix[4]arene derivatives were prepared as a result of the reaction of p-tert-butylthiacalix[4]arene with diethyl bromomalonate in the presence of different alkali metals (Cs, K and Na) in refluxing acetone for 7 days. The structures of the prepared compounds were investigated by IR, 1H-NMR and MALDI-TOF mass spectroscopy.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Introduction
It is well proven that the new member of calixarenes [1,2,3,4,5] family, thiacalixarenes [6,7] (in which the methylene bridged groups has been replaced by sulfur atoms) surpass classical calixarenes as new building blocks because of the novel features offered by the presence of the sulfur atoms. Thiacalixarenes have generated increasing interest both in fundamental and applied chemistry. Many researchers are investigating the chemical behavior of thiacalixarenes as well as using them in different applications via different chemical studies. These studies include: i) modification of the thiacalixarene skeleton through derivatization of the upper and/or lower rims, as well as its sulfur bridged atoms and ii) investigation of the complexation properties of different thiacalixarenes with different chemical species. In this paper, the interaction of thiacalixarene 1 with diethyl bromomalonate in the presence of different alkali metal carbonates (Cs, K and Na) is reported. The functionalization of p-tert-butylthiacalixarene 1, with tetracarbonyl fragments and the remarkable binding ability towards different metals has been investigated [8,9,10]. The idea of this work was to see if we can increase the number of coordinating carbonyl groups by reacting p-tert-butylthiacalixarene 1 with diethyl bromomalonate. This might increase the binding ability of the new derivatives.
Results and Discussion
Previous reported procedures [8,9,10], were used for the first reaction, where p-tert-butylthia-calixarene 1 was reacted with diethyl bromomalonate in refluxing acetone in the presence of K2CO3 as catalyst. Surprisingly, an unprecedented selective synthesis of monosubstituted p-tert-butylthiacalixarene 2 containing one ethoxycarbonyl fragment (in 60 % yield) and the reported compound 3 [11] resulted (Scheme 1). When the reaction was carried out using Cs2CO3 as a base catalyst instead of K2CO3, a similar result was obtained, with a slight change in the yield of 2 (which increased to 70%). Different spectroscopic analysis (IR, 600 MHz 1H-NMR, and MALDI-TOF MS) were used to identify the structure of p-tert-butylthiacalixarene 2. The 1H-NMR spectrum exhibits three But group singlets with a relative ratio of 1:1:2, two doublets and two singlets in the aromatic region, one triplet for the CH3 group, one quartet for the OCH2 group and two singlets of OH group with a relative ratio 2:1, respectively. MALDI-TOF MS shows an m/z peak at 832 (M+K). IR shows bands at 1758 and 3380 cm-1 for the CO and OH groups, respectively.
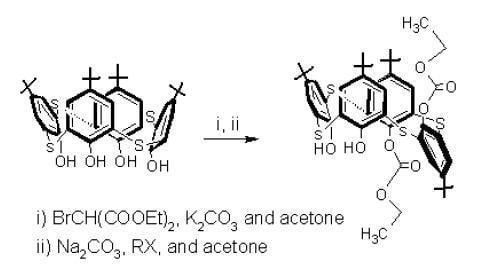
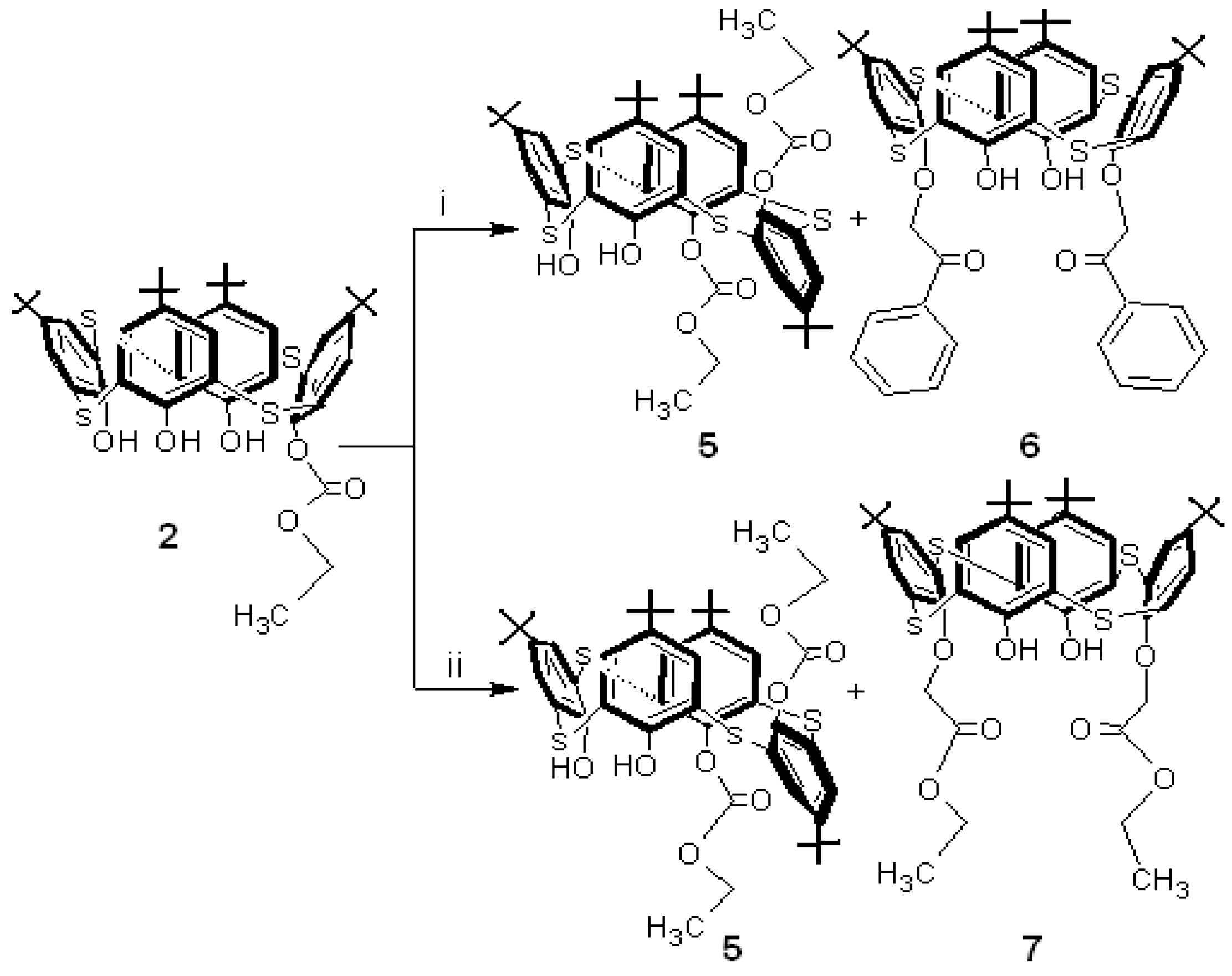
Since the ethoxycarbonyl fragment in p-tert-butylthiacalixarene 2 could be introduced to p-tert-butylthiacalixarene 1, through the reaction with ethyl chloroformate, and to investigate the selectivity of the monosubstituted p-tert-butylthiacalixarene 2, ethyl chloroformate was reacted with p-tert-butylthiacalixarene 1 in the presence of K2CO3 in acetone by using the same reaction conditions employed with diethyl bromomalonate. The tetrasubstituted p-tert-butylthiacalixarene 4, was obtained in 80 % yield (Scheme 1; separated by partial cone conformer) which is in agreement with the literature [8,9,10]. The structure of 4 was characterized using IR, 1H-NMR and MALDI-TOF-MS spectroscopy. The above selective synthesis of the monosubstitued thiacalixarene 2 may be explained by the generation of the ethoxycarbonyl fragment throughout the decomposition of the diethyl bromomalonate in the basic medium or by the attack of the phenolate-metal ion pairs at one of the carbonyl groups of the diethyl bromomalonate and metal ion template effect (Cs and K). This implies that the selective monosubstitution can be attributed to the reaction of thiacalixarene 1 with diethyl bromomalonate. For the preparation of asymmetrical p-tert-butylthiacalixarenes, introducing four different groups at the lower rims, monosubstituted thiacalixarene 2 was used for the second step in this study – the reaction of 2 with phenacyl bromide in the presence of Na2CO3 in acetone (Scheme 2).
The reaction gave anti-1,2-disubstituted p-tert-butylthiacalixarene 5 [12] in 40% yield (dissymmetrical thiacalixarene has effective C2 symmetry) and the reported disubstituted p-tert-butylthiacalixarene 6 [13].
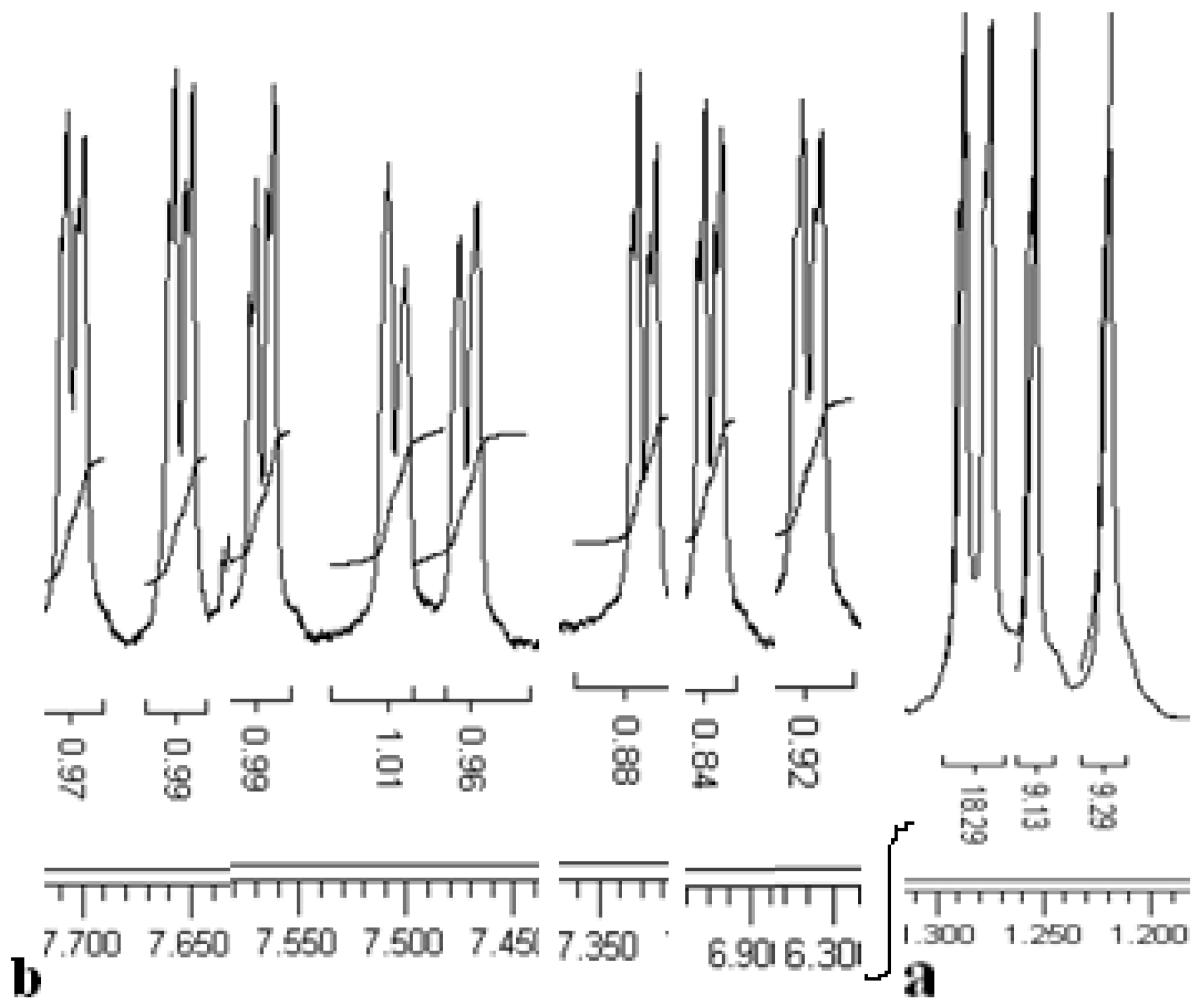
The 1H-NMR spectrum of compound 5 (Figure 1) shows asymmetrical signals for all protons which have been used for the structure determination: four But group singlets, eight doublets with characteristic aromatic proton coupling constants (J ~ 2.3 to 2.4 Hz), two OH group singlets, two CH3 group triplets, a quartet for the CH2, shifted upfield by the aromatic rings and one CH2 group multiplet in between two different groups (OH and Ar groups). To confirm the production of p-tert-butylthiacalixarene 5, p-tert-butylthiacalixarene 2 was reacted with another alkylation reagent, ethyl bromoacetate and p-tert-butylthiacalixarene 5 was formed along with the reported p-tert-butyl-thiacalixarene 7 [14] (Scheme 2).
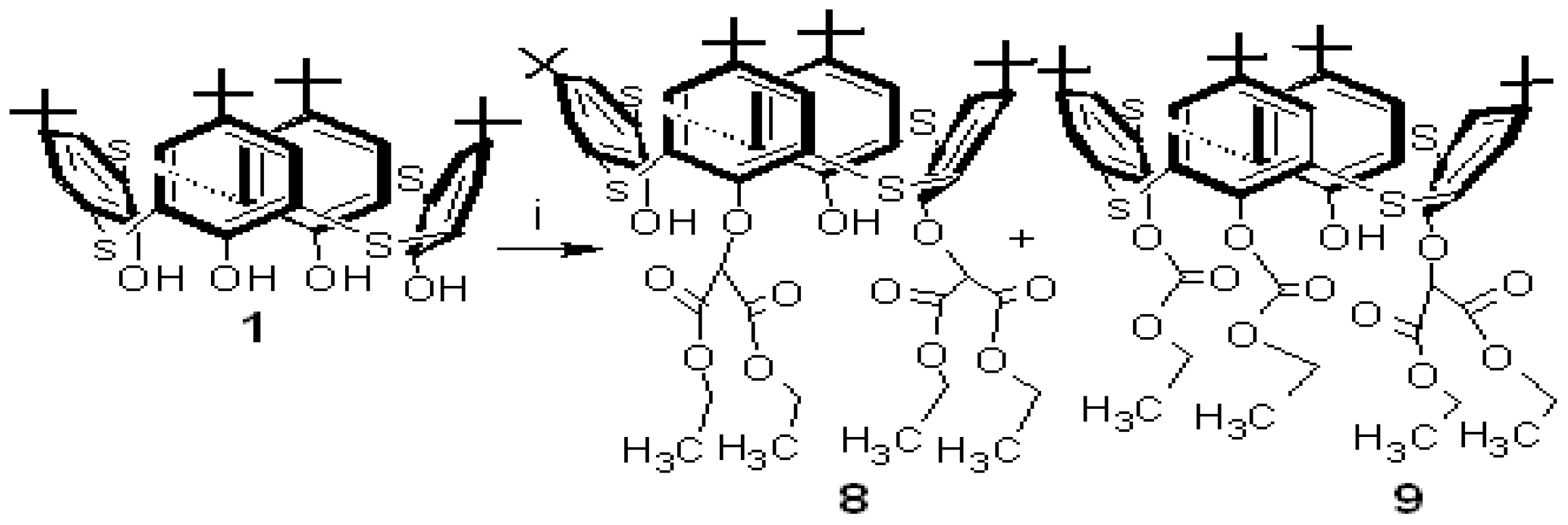
The reaction of p-tert-butylthiacalixarene 1 with diethyl bromomalonate in the presence of Na2CO3 in acetone gave two new p-tert-butylthiacalixarenes, 8 and 9, in 30% and 40% yields, respectively (Scheme 3).
Both p-tert-butylthiacalixarenes 8 and 9 were separated from the carbonate and the acetone layer, respectively. IR, MALDI-TOF-MS and 1H-NMR spectroscopic methods were used for determination of the their structures.
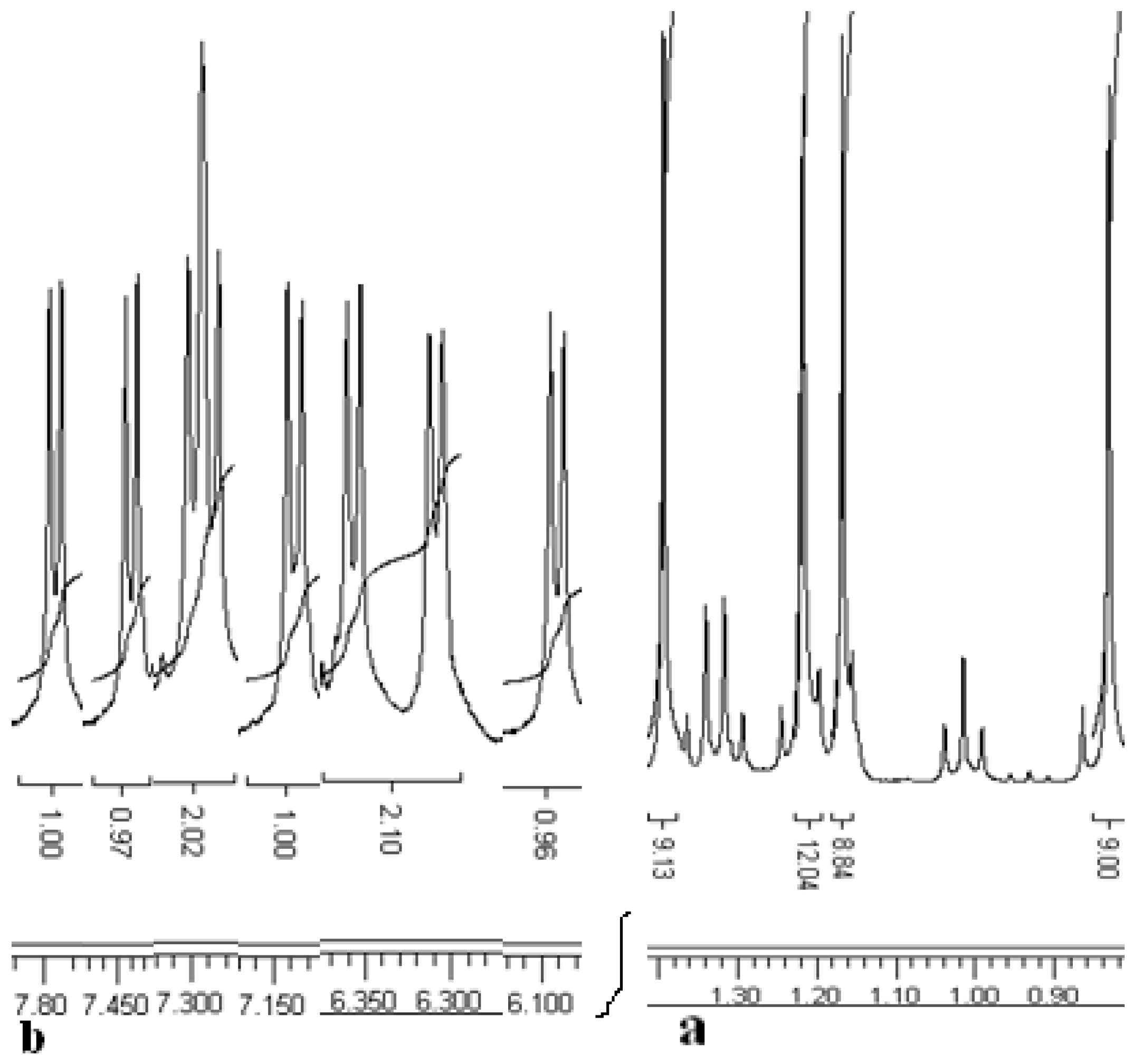
The 1H-NMR spectrum of compound 8 exhibits two But group singlets, four multiplets for the methylene groups and four doublets with characteristic meta coupling in the aromatic region (J ~ 2.4 Hz). The 1H-NMR spectrum of asymmetrical thiacalixarene 9 shows asymmetrical signals for all the types of protons, for instance, four singlets for the But groups, four CH3 group triplets, four CH2 group multiplets and eight doublets with characteristic coupling constants (J ~ 2.3 to 2.8 Hz) in the aromatic region (Figure 2).
Experimental
General
All NMR spectra were recorded on a Bruker DRX600 NMR spectrometer equipped with a triple-gradient TXI (H/C/N) probe operating at a magnetic field strength of 14.1 T., as well as on a Varian Mercury VX-300 NMR spectrometer. All melting points were determined on a Koffler melting point apparatus. IR spectra were obtained on a Nicolet 710 FT-IR spectrometer. Mass spectra were recorded on a MALDI-TOF MS REFLEX III (Bruker-Daltonics, Germany).
Synthesis of thiacalixarene 2
To a suspention of thiacalixarene 1 (2 g, 2.77 mmol) and anhydrous potassium carbonate (6.16 g, 44.32 mmol) and/or cesium carbonate (7.21 g, 22.16 mmol) in dry acetone (100 mL) diethylbromomalonate (8.2 mL, 44.32 mmol) was added. The mixture was refluxed for a week, then it was filtered and separated into two layers: carbonate and acetone. Compound 2 was separated from the carbonate layer after extraction by CH2Cl2 and obtained as a white solid. Compound 3 was separated from the acetone layer.
5,11,17,23-Tetra-tert-butyl-25-[(ethoxycarbonyl)oxy]-26,27,28-trihydroxy-2,8,14,20-tetrathiacalix[4]- arene (cone) (2): Yield. 60-70%; Mp: 218 °C; IR (cm-1): 1758 (CO), 3380 (OH); MS m/z 832 (M+K); 1H-NMR (TMS, 300 MHz, CDCl3) δ 0.76 (9H, s, But ), 1.16 (9H, s, But), 1.28 (18H, s, But), 1.36 (3H, t, J =7.1, CH3), 4.46 (2H, q, J = 7.1, CH2), 7.04 (2H, s, ArH), 7.48 (2H, s, ArH), 7.60 (2H, d, J = 2.4, ArH), 7.73 (2H, d, J = 2.4, ArH), 8.20 (2H, s, OH), 9.10 (1H, s, OH).
Synthesis of thiacalixarene 4
The above method was used for the preparation of 4, obtained as a white solid.
5,11,17,23-Tetra-tert-butyl-25,26,27,28-tetrakis[(ethoxycarbonyl)oxy]-2,8,14,20-tetrathiacalix[4]-arene (partial cone) (4): Yield: 80%; Mp: 330 °C; IR (cm-1): 1756 (CO); MS m/z 1072 (MH+); 1H-NMR (TMS, 300 MHz, CDCl3) δ 1.09 (18H, s, But ), 1.38 (9H, s, But), 1.47 (9H, s, But), 1.33-1.50 (12H, m, CH3), 4.19 (2H, q, J = 7.0, CH2), 4.26-4.48 (6H, m, CH2), 7.18 (2H, d, J = 2.4, ArH), 7.44 (2H, s, ArH), 7.45 (2H, d, J = 2.4, ArH), 7.78 (2H, s, ArH).
Synthesis of thiacalixarenes 5, 6 and 7
Thiacalixarene 2 (0.6 g, 0.75 mmol) and Na2CO3 (0.1 g, 0.94 mmol) were suspended in acetone (50 mL) and phenacyl bromide (0.15 g, 0.75 mmol) or ethyl bromoacetate (0.1 mL, 0.9 mmol) were added. The mixture was refluxed for 24 hours. The reaction mixture was concentrated almost to dryness and the solid residue was separated by column chromatography using a 50/50 n-hexane-CH2Cl2 mixture as eluent, to separate the thiacalixarenes 5 and 6 from the reaction with phenacyl bromide; or thiacalixarenes 5 and 7 from the reaction with ethyl bromoacetate.
5,11,17,23-Tetra-tert-butyl-25,26-bis[(ethoxycarbonyl)oxy]-27,28-dihydroxy-2,8,14,20-tetrathiacalix-[4]arene (anti-1,2-alternate) (5) : Yield: 40%; yellow solid; Mp: 310 °C; IR (cm-1): 1754 (CO); MS m/z 889 (M+Na); 1H-NMR (TMS, 300 MHz, CDCl3) δ 1.07-1.17 (6H, m, CH3), 1.22 (9H, s, But), 1.26 (9H, s, But), 1.28 (9H, s, But), 1.29 (9H, s, But), 3.7 (2H, q, J = 3.7, CH2), 4.14-4.34 (2H, m, CH2), 6.31 (2H, d, J = 2.3, ArH), 6.92 (2H, q, J = 2.3, ArH), 7.33 (2H, d, J = 2.4, ArH), 7.39 (1H, s, OH), 7.47 (2H, d, J = 2.4, ArH), 7.51 (2H, d, J = 2.4, ArH), 7.57 (2H, d, J = 2.4, ArH), 7.66 (2H, d, J = 2.4, ArH), 7.71 (2H, d, J = 2.4, ArH), 7.81 (1H, s, OH).
Synthesis of thiacalixarenes 8 and 9
The same procedure for the preparation of thiacalixarene 2 was used. Thiacalixarene 8 was separated from the carbonate layer and thiacalixarene 9 was separated from the acetone layer.
5,11,17,23-Tetra-tert-butyl-25,26-bis[(1,1-diethoxycarbonyl)methoxy]-27,28-dihydroxy-2,8,14,20-tetrathiacalix[4]arene (cone) (8): Yellow solid; Mp: 330 °C; Yield: 30%; IR (cm-1): 1751 (CO); MS m/z 1061 (M+Na); 1H-NMR (TMS, 300 MHz, CDCl3) δ 0.96 (6H, t, J = 7.1, CH3), 1.18 (6H, t, J = 7.1, CH3), 1.21 (18H, s, But ), 1.23 (18H, s, But), 3.72 (2H, m, CH2), 3.79 (2H, m, CH2), 3.93-4.04 (6H, m, CH2 + OCH), 6.05 (2H, q, J = 2.4, ArH), 6.87 (2H, d, J = 2.4, ArH), 7.27 (2H, s, OH), 7.44 (2H, d, , J = 2.4, ArH), 7.65 (2H, d, J = 2.4, ArH).
5,11,17,23-Tetra-tert-butyl-25-[(1,1-diethoxycarbonyl)methoxy]-26,28-bis[(ethoxycarbonyl)oxy-26-hydroxy-2,8,14,20-tetrathiacalix[4]arene (cone) (9): Yellow solid; Yield: 40%; Mp: 275 °C; IR (cm-1): 1758 (CO); MS m/z 1047 (M+Na); 1-NMR (TMS, 300 MHz, CDCl3) δ = 0.84 (9H, s, But ), 1.01 (6H, t, J = 7.1, CH3) 1.17 (9H, s, But) 1.22 (9H, s, But), 1.22 (6H, t, J = 7.1, CH3), 1.33 (12H, m, CH3), 1.4 (9H, s, But), 3.98- 4.50 (10H, m, CH2 + OCH, OH), 6.10 (2H, d, J = 2.3, ArH), 6.31 (2H, q, J = 2.3, ArH), 6.36 (2H, d, J = 2.3, ArH), 7.14 (2H, d, J = 2.5, ArH), 7.29 (2H, d, J = 2.8, ArH), 7.30 (2H, d, J = 2.8, ArH), 7.44 (2H, d, J = 2.5, ArH), 7.80 (2H, d, J = 2.5, ArH).
Conclusions
p-tert-Butylthiacalix[4]arenes mono-, di-, tri- and tetra-substituted at the lower rim in different conformers have been prepared as a result of the reaction of p-tert-butylthiacalix[4]arene with diethyl bromomalonate in the presence of different alkali metals (Cs, K and Na) in acetone. Two of these derivatives are dissymmetrical and asymmetrical and could have potential use for chiral discrimination.
Supplementary Files
Supplementary File 1Acknowledgements
Financial support by the Al-Qassim University (grant No. SR-S-007-043) is gratefully acknowledged. Dr. Rienhard Wimmer at Aalborg University is acknowledged for the 1HNMR spectra.
References
- Asfari, Z.; Bohmer, V.; Harrowfield, J.; Vicens, J. (Eds.) Calixarenes 2001; Kluwer Academic: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Mandolini, L.; Ungaro, R. (Eds.) Calixarenes in Action; Imperial College: London, UK, 2000. [Google Scholar]
- Gutsche, C.D. Calixarenes revisited: Monographs in Supramolecular Chemistry; Stoddart, J.F., Ed.; The Royal Society of Chemistry: Cambridge, USA, 1998; Vol. 6. [Google Scholar]
- Vicens, J.; Asfari, Z.; Harrowfield, J.M. (Eds.) Calixarenes 50th Anniversary: Commemorative Issue; Academic: Dordrecht, The Netherlands, 1994. [Google Scholar]
- Vicens, J.; Bohmer, V. (Eds.) Calixarenes: A Versatile Class of Macrocyclic Compounds; Kluwer Academic: Dordrecht, The Netherlands, 1991. [Google Scholar]
- Morohashi, N.; Narumi, F.; Iki, N.; Hattori, T.; Miyano, S. Thiacalixarenes. Chem. Rev. 2006, 106, 5291–5316. [Google Scholar] [CrossRef] [PubMed]
- Lhotak, P. Chemistry of Thiacalixarenes. Eur. J. Org. Chem. 2004, 1675–1692. [Google Scholar] [CrossRef]
- Iki, N.; Narumi, F.; Fujimoto, T.; Morohashi, N.; Miyano, S. Selective Synthesis of Three Conformational Isomers of Tetrakis[(ethoxycarbonyl)methoxy]thiacalix[4]arene and Their Complexation Properties Towards Alkali Metal Ions. J. Chem. Soc. Perkin Trans. 2 1998, 2, 2745–2750. [Google Scholar] [CrossRef]
- Stoikov, I.I.; Omran, O.A.; Solovieva, S.E.; Latypov, S.K.; Enikeev, K.M.; Gubaidullin, A.T.; Antipin, I.S.; Konovalov, A.I. The Synthesis of Tetracarbonyl Derivatives of Thiacalix[4]arene in Different Conformations and Their Complexation Properties Towards Alkali Metal Ions. Tetrahedron 2003, 59, 1469–1476. [Google Scholar] [CrossRef]
- Solovieva, S.E.; Gruner, M.; Omran, A.O.; Gubaidullin, A.T.; Litvinov, I.A.; Habicher, W.D.; Antipin, I.S.; Konovalov, A.I. Synthesis, Structure, and Complexation Properties of Tetraamide Derivatives of Thiacalix[4]arene in Different Conformations. Russ. Chem. Bull. 2005, 9, 2104–2112. [Google Scholar] [CrossRef]
- Hori, Y.; Nagano, Y.; Uchiyama, H.; Yamada, Y.; Taniguchi, H. Bromination of Active Hydrogen Compounds by Bromotrichloromethane and 1,8-Diazabicyclo[5.4.0]Undecene-7. A Convenient Synthesis for Bromides and Olefins. Chem. Lett. 1978, 73–76. [Google Scholar] [CrossRef]
- Bomer, V.; Kraft, D.; Tabatabai, M. Inherently Chiral Calixarenes. J. Incl. Phenom. Mol. Recogn. 1994, 19, 17–39. [Google Scholar] [CrossRef]
- Omran, A.O.; Larsen, K.; Wimmer, R.; Yu, D. Synthesis, Separation and Characterization of Thiacalix[4]arenes Diastereomers. Phosphorus Sulfur Silicon 2008, 183, 155–160. [Google Scholar] [CrossRef]
- Dudic, M.; Lhota´k, P.; Stibor, I.; Dvorˇa´kova´, H.; Lang, K. Synthesis and Spectroscopic Properties of Porphyrin-(thia)calix[4]arene Conjugates. Tetrahedron 2002, 58, 5475–5482. [Google Scholar] [CrossRef]
Sample Availability: Samples are available from the author. |
Scheme 1.
Synthesis of monosubstituted p-tert-thiacalix[4]arene 2 (60-70% yield) and tetrasubstituted p-tert-butylthiacalix[4]arene 4 (80 % yield).
Scheme 1.
Synthesis of monosubstituted p-tert-thiacalix[4]arene 2 (60-70% yield) and tetrasubstituted p-tert-butylthiacalix[4]arene 4 (80 % yield).

Reagents and conditions: (i) BrCH(COOEt)2/K2CO3, acetone; (ii) BrCH(COOEt)2/Cs2CO3, acetone; (iii) ClCOOEt/K2CO3, acetone.
Scheme 2.
Synthesis of dissymmetric thiacalix[4]arene 5 (40 % yield).

Reagents and conditions: (i) PhCOCH2Br/Na2CO3, acetone; (ii) BrCH2COOEt/Na2CO3, acetone.
Figure 1.
Sections of the 1H NMR spectrum of thiacalix[4]arene 5 of But (a) and ArH protons (b).

Scheme 3.
Synthesis of thiacalix[4]arene 8 (30% yield) and asymmetric thiacalix[4]arene 9 (40% yield).
Scheme 3.
Synthesis of thiacalix[4]arene 8 (30% yield) and asymmetric thiacalix[4]arene 9 (40% yield).

Reagent and conditions: (i) BrCH(COOEt)2/Na2CO3, acetone.
Figure 2.
Sections of the But (a) and ArH protons (b)in the1H-NMR spectrum of thiacalixarene 9.

© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Omran, O.A. The Reaction of Diethyl Bromomalonate with p-tert-Butylthia-calix[4]arene: An Approach to Asymmetrical Derivatives. Molecules 2009, 14, 1755-1761. https://doi.org/10.3390/molecules14051755
AMA Style
Omran OA. The Reaction of Diethyl Bromomalonate with p-tert-Butylthia-calix[4]arene: An Approach to Asymmetrical Derivatives. Molecules. 2009; 14(5):1755-1761. https://doi.org/10.3390/molecules14051755
Chicago/Turabian StyleOmran, Omran Abdellah. 2009. "The Reaction of Diethyl Bromomalonate with p-tert-Butylthia-calix[4]arene: An Approach to Asymmetrical Derivatives" Molecules 14, no. 5: 1755-1761. https://doi.org/10.3390/molecules14051755