The Use of MoStBioDat for Rapid Screening of Molecular Diversity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
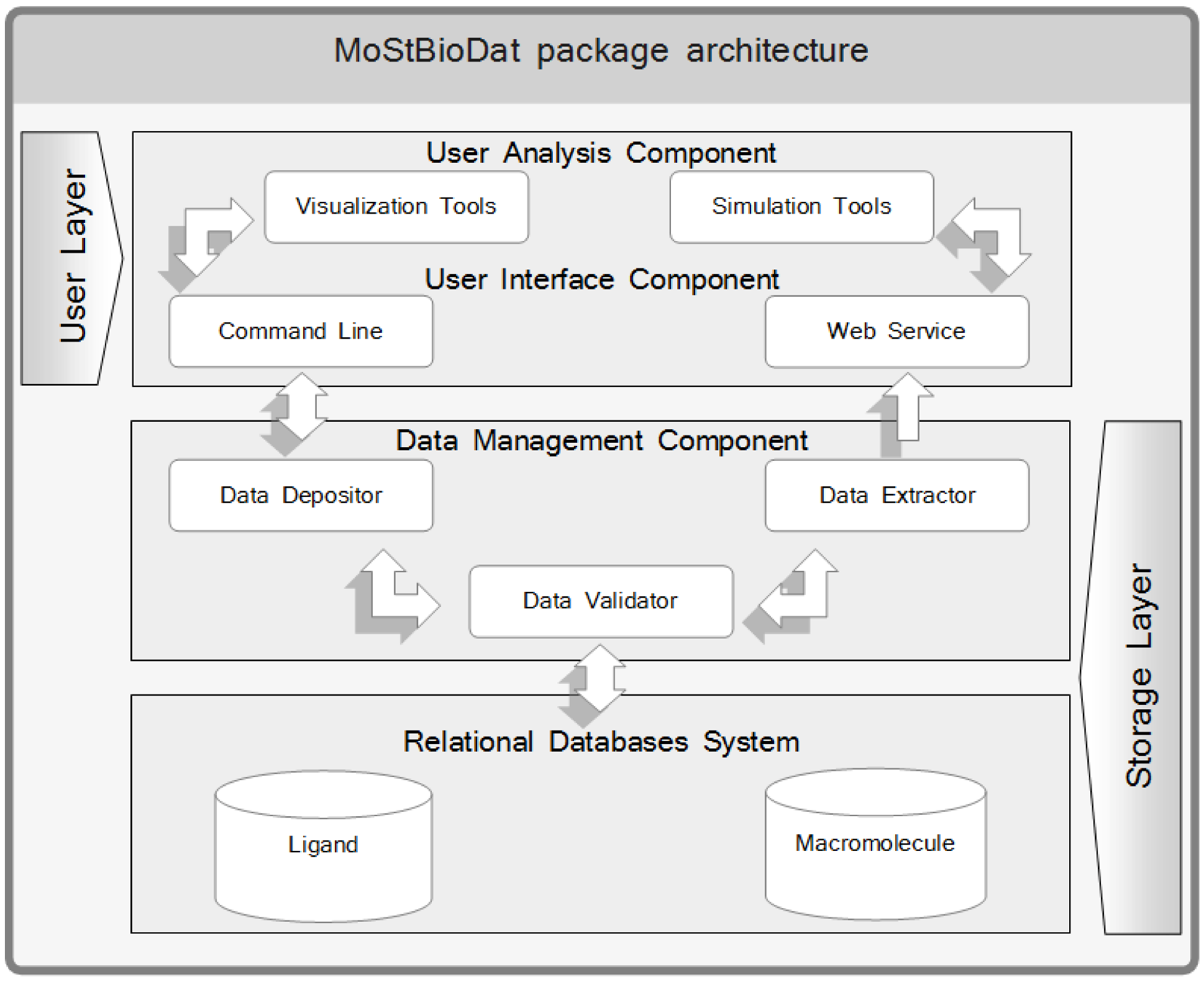
2.1. MoStBioDat architecture


2.2. ZINC database
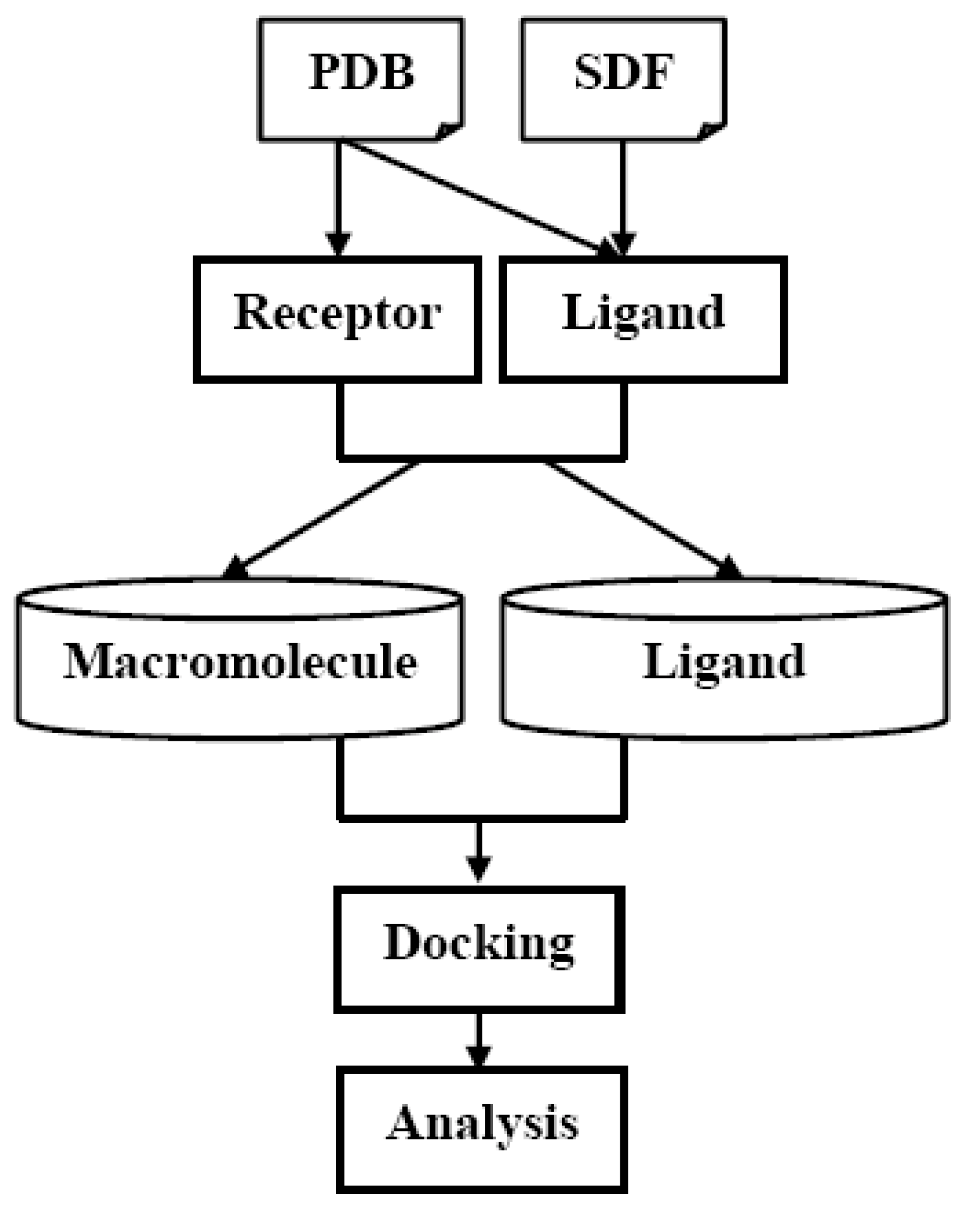
2.3. Rapid screening of the ZINC database subset

- nxy - the number of bits set into 1 shared in the fingerprint of molecule x and y
- nx - the number of bits set into 1 in the molecule x
- ny - the number of bits set into 1 in the molecule y
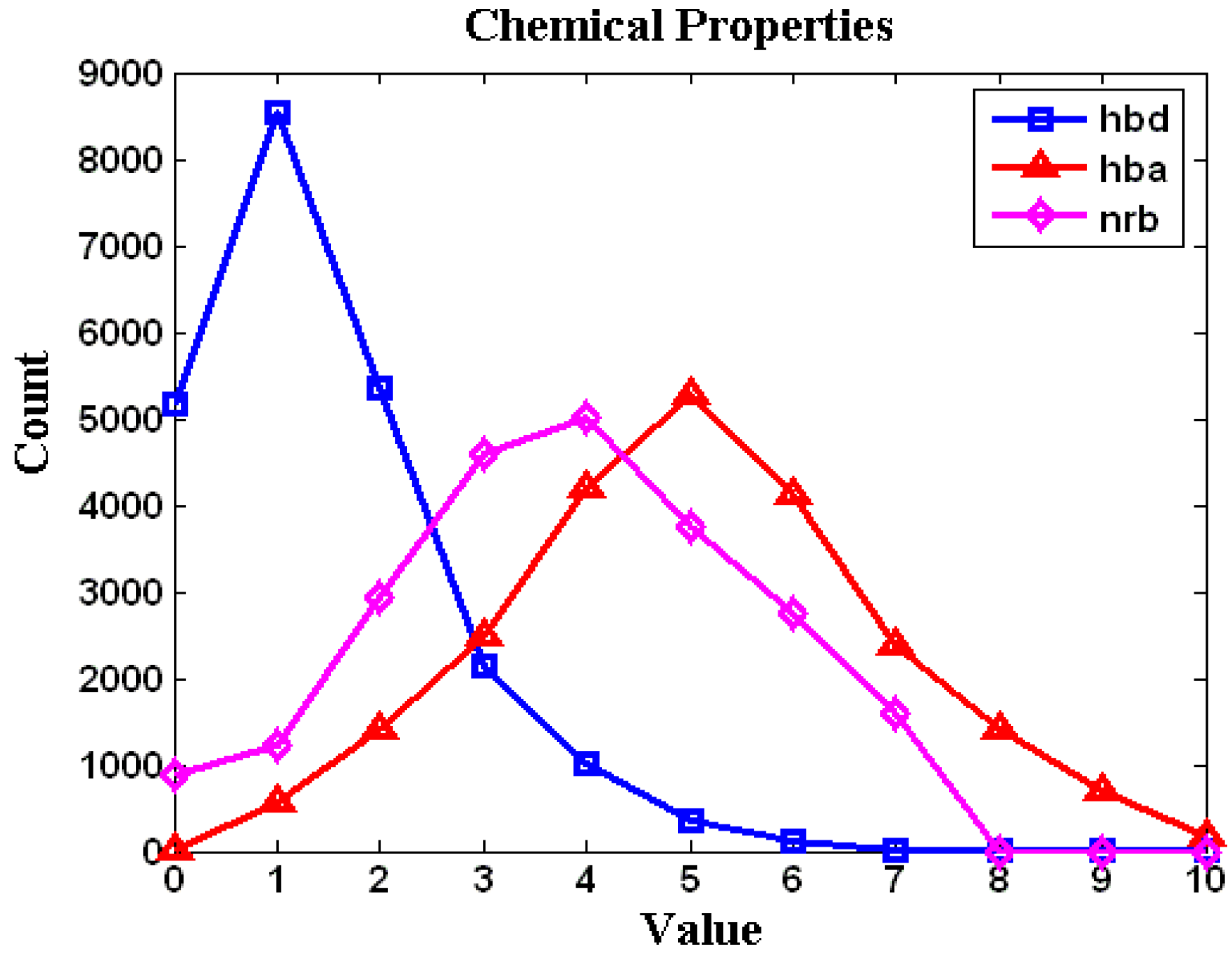
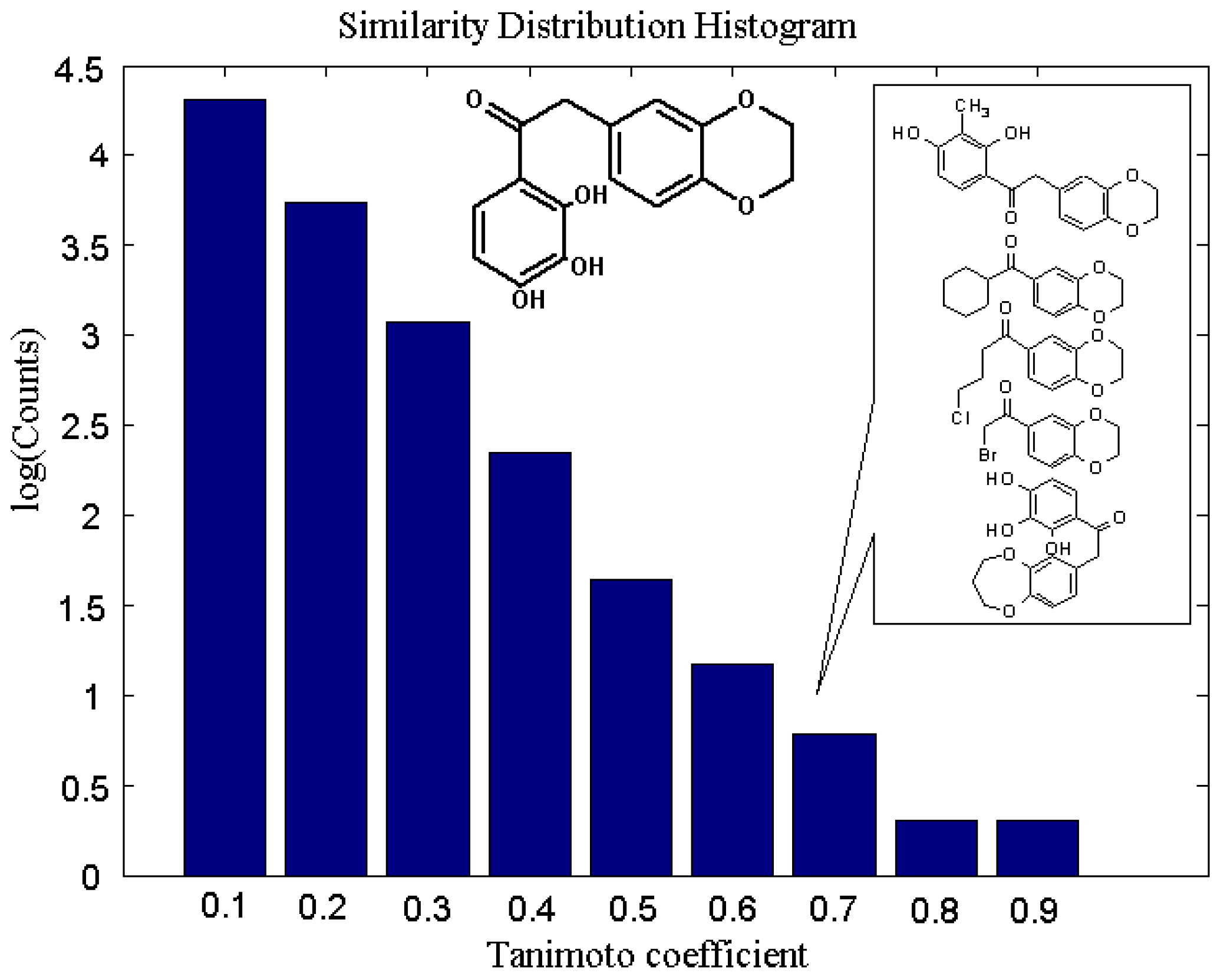
2. Results and Discussion



3. Experimental Section
3.1. Distribution
3.2. Installation and operation of the MoStBioDat package
- A=DB2SmiDict(host=’localhost’,db=’ligand’,user=’’,passwd=’’,path=’/tmp/Log’,filename=’db2smi’)B=SubStructSearch(smidict=A.readb(logdebug=False),path=’/tmp/Log’,filename=’smisubsearch’)
- Tanimoto Search ###B.TanimotoSearch(refsmile=’Cc1cccccc1’,coeff=0.7, outfile=’/tmp/TanimotoSearch.txt’)
- Lipinski’s Rule of File Search ###B.RO5Search(outfile=’/tmp/RO5Search.txt’,MolWT=500,HBA=10,HBD=5,LogP=5)
3.3. Data format
3.4. Substructure searches
4. Conclusions
Acknowledgements
- Sample availability: Not available.
References and Notes
- Polanski, J. Receptor dependent multidimensional QSAR for modeling drug-receptor interactions. Curr. Med. Chem. 2009, in press. [Google Scholar]
- Ekonomiuk, D.; Su, X.C.; Ozawa, K.; Bodenreider, C.; Lim, S.P.; Otting, G.; Huang, D.; Caflisch, A. Flaviviral protease inhibitors identified by fragment-based library docking into a structure generated by molecular dynamics. J. Med. Chem. 2009, in press. [Google Scholar]
- Kolb, P.; Caflisch, A. Automatic and efficient decomposition of two-dimensional structures of small molecules for fragment-based high-throughput docking. J. Med. Chem. 2006, 49, 7384–7392. [Google Scholar] [CrossRef]
- Moriaud, F.; Doppelt-Azeroual, O.; Martin, L.; Oguievetskaia, K.; Koch, K.; Vorotyntsev, A.; Adcock, S.; Delfaud, F. Computational fragment-based approach at PDB scale by protein local similarity. J. Chem. Inf. Model. 2009, 49, 280–294. [Google Scholar] [CrossRef]
- Irwin, J.; Shoichet, B. ZINC - A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- ZINC database; Shoichet Laboratory, Department of Pharmaceutical Chemistry, University of California, San Francisco, 2009; No. 3_p0.0.sdf. 2009. Available online: http://zinc.docking.org/ (accessed 29 April 2009).
- Chen, J.; Swamidass, J.S.; Dou, Y.; Bruand, J.; Baldi, P. ChemDB: A public database of small molecules and related chemoinformatics resources. Bionformatics 2005, 21, 4133–4139. [Google Scholar] [CrossRef]
- ChemDB database project. Available online: http://cdb.ics.uci.edu/ (accessed 4 September 2009).
- Bak, A.; Polanski, J.; Stockner, T.; Kurczyk, A. MoStBioDat - Molecular and structural bioinformatics database. Comb. Chem. High Throughput Screen. 2009, in press. [Google Scholar]
- MoStBioDat project. Available online: http://www.chemoinformatyka.us.edu.pl/mostbiodat/ (accessed 7 September 2009).
- Python programming language. Available online: http://www.python.org/ (accessed 7 September 2009).
- Wu, B.; Tai, K.; Murdock, S.; Ng, M.; Johnston, S.; Fangohr, H.; Jeffreys, P.; Cox, S.; Essex, J.; Sansom, M. BioSimGrid: A distributed database for biomolecular simulations. In Proceedings of the UK e-Science All Hands Meeting, Nottingham, UK, 2-4th September, 2003; pp. 412–419.
- Ng, M.; Johnston, S.; Murdock, S.; Wu, B.; Tai, K.; Fangohr, H.; Cox, S.; Essex, J.; Sansom, M.; Jeffreys, P. Efficient data storage and analysis for generic biomolecular simulation data. In Proceedings of UK e-Science All Hands Meeting, Nottingham, UK, 31st August - 3rd September 2004; pp. 443–450.
- MySQL database project. Available online: http://www.mysql.com/ (accessed 11 March 2009).
- Molecular Modeling Toolkit. Available online: http://sourcesup.cru.fr/projects/mmtk/ (accessed 7 September 2009).
- Visual Molecular Dynamics. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed 7 September 2009).
- R for Statistical Computing and Graphics. Available online: http://www.r-project.org/ (accessed 7 September 2009).
- Swamidass, S.; Baldi, P. Bounds and algorithms for fast exact searches of chemical fingerprints in linear and sub-linear time. J. Chem. Inf. Model. 2007, 47, 302–317. [Google Scholar] [CrossRef]
- Stahl, M.; Mauser, H. Database clustering with a combination of fingerprint and maximum common substructure methods. J. Chem. Inf. Model. 2005, 45, 542–548. [Google Scholar] [CrossRef]
- Daylight Theory Manual. Available online: http://www.daylight.com/ (accessed 20 January 2009).
- Flower, D. On the properties of bit-string based measures of chemical similarity. J. Chem. Inf. Comput. Sci. 1998, 38, 379–386. [Google Scholar] [CrossRef]
- Podolyan, Y.; Karypis, G. Common pharmacophore identification using frequent clique detection algorithm. J. Chem. Inf. Model. 2009, 49, 13–21. [Google Scholar] [CrossRef]
- Cao, Y.; Jiang, T.; Girke, T. A maximum common substructure-based algorithm for searching and predicting drug-like compounds. Bioinformatics 2008, 24, 366–374. [Google Scholar]
- Lipinski, C.; Lombardo, F.; Dominy, B.; Feeney, P. Experimental and computational approaches to estimate solubility and permeability in drug Discovery and development settings. Adv. Drug Del. Res. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Olah, M.; Bologa, C.; Oprea, T. Strategies for Compound Selection. Curr. Drug Discov. Tech. 2004, 1, 211–220. [Google Scholar] [CrossRef]
- Fialkowski, M.; Bishop, K.J.; Chubukov, V.A.; Campbell, C.J.; Grzybowski, B.A. Architecture and evolution of organic chemistry. Angew. Chem. Int. Ed. Engl. 2005, 44, 7263–7269. [Google Scholar]
- Hann, M.; Oprea, T. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef]
- Open Source Chemistry Toolbox. Available online: http://openbabel.org/ (accessed 3 April 2009).
- OpenEye Scientific Software. Available online: http://www.eyesopen.com/ (accessed 14 May 2009).
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bak, A.; Polanski, J.; Kurczyk, A. The Use of MoStBioDat for Rapid Screening of Molecular Diversity. Molecules 2009, 14, 3436-3445. https://doi.org/10.3390/molecules14093436
Bak A, Polanski J, Kurczyk A. The Use of MoStBioDat for Rapid Screening of Molecular Diversity. Molecules. 2009; 14(9):3436-3445. https://doi.org/10.3390/molecules14093436
Chicago/Turabian StyleBak, Andrzej, Jaroslaw Polanski, and Agata Kurczyk. 2009. "The Use of MoStBioDat for Rapid Screening of Molecular Diversity" Molecules 14, no. 9: 3436-3445. https://doi.org/10.3390/molecules14093436