Extraction and isolation
Ground dry plant material (100 g) was extracted three times with ethanol (1:10 w/V) for 60 min with sonication. The extract was filtered and evaporated to dryness under reduced pressure. The residue (1.97 g) was dissolved in EtOAc (10 mL) and partitioned three times against water (100 mL) to remove inactive polar compounds. The combined organic phases were evaporated and the residue (0.885 g) fractionated on a VLC column (7x10 cm i.d.) with solvent mixtures of increasing polarity, to afford six fractions. Fraction 1 (165 mg) was eluted with toluene (200 mL); fraction 2 (585 mg) was eluted with toluene/EtOAc (80:20, 200 mL); fraction 3 was eluted with toluene/EtOAc (50:50, 200 mL); fraction 4 (47 mg) was eluted with eluted with toluene/EtOAc (20:80, 200 mL); fraction 5 was eluted with EtOAc (200 mL) and fraction 6 (21 mg) was eluted with MeOH (400 mL). Fraction 2, which was the most active in the bioassay, was submitted to reversed-phase HPLC (HPLC-system I). This fractionation afforded five fractions. Further HPLC fractionation (HPLC-system II) of the active fraction 3 (32.2 mg) on an analytic column led to the isolation and identification of
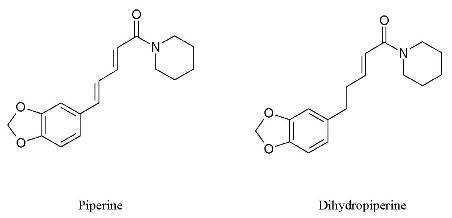
1 (5.3 mg) and
2 (2.6 mg) (
Figure 1) as the active components.
Piperine (1): 1H-NMR δ (ppm): 7.40 (ddd, J = 15.0 Hz, J = 1.6 Hz, J = 8.0 Hz, 1H), 6.98 (d, J = 1.6 Hz, 1H), 6.89 (dd, J = 8.0 Hz, J = 1.6 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 6.77 (d, J = 15.0 Hz, 1H), 6.76 (dd, J = 8.5 Hz, J = 15 Hz, 1H), 6.44 (d, J = 15.0 Hz, 1H), 5.98 (s, 2H); 3.63 (br s, 2H), 3.52 (br s, 2H), 1.55-1.70 (m, 6H; MS: m/z 285.14 [M]+.
4,5-Dihydropiperine (2): 1H-NMR δ (ppm): 6.70 (dt, J = 15.1 Hz, J = 7.0 Hz, 1H), 6.65 (d, J = 7.8 Hz, 1H), 6.60 (d, J = 1.7 Hz, 1H), 6.50 (dd, J = 7.8 Hz, J = 1.7 Hz, 1H), 6.18 (dt, J = 15.0 Hz, J = 1.5 Hz, 1H), 5.80 (s, 2H), 3.55 (br s, 2H), 3.35 (br s, 2H), 2.60 (t, J = 7.6 Hz, 2H), 2.40 (dt, J = 7.3 Hz, J = 1.3 Hz, 2H), 1.55 (m, 6H); MS: m/z 287.15 [M]+.
Synthesis of piperine analogs
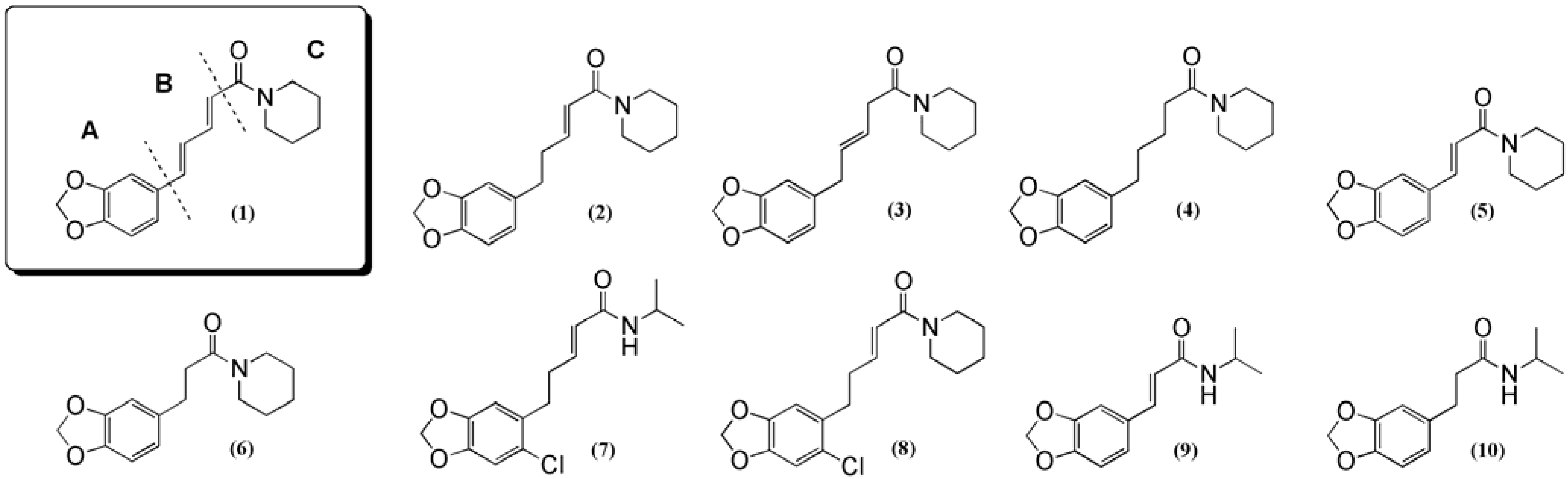
To assess the structure-activity relationships a small library of ten synthesized analogs was tested in the bioassay (
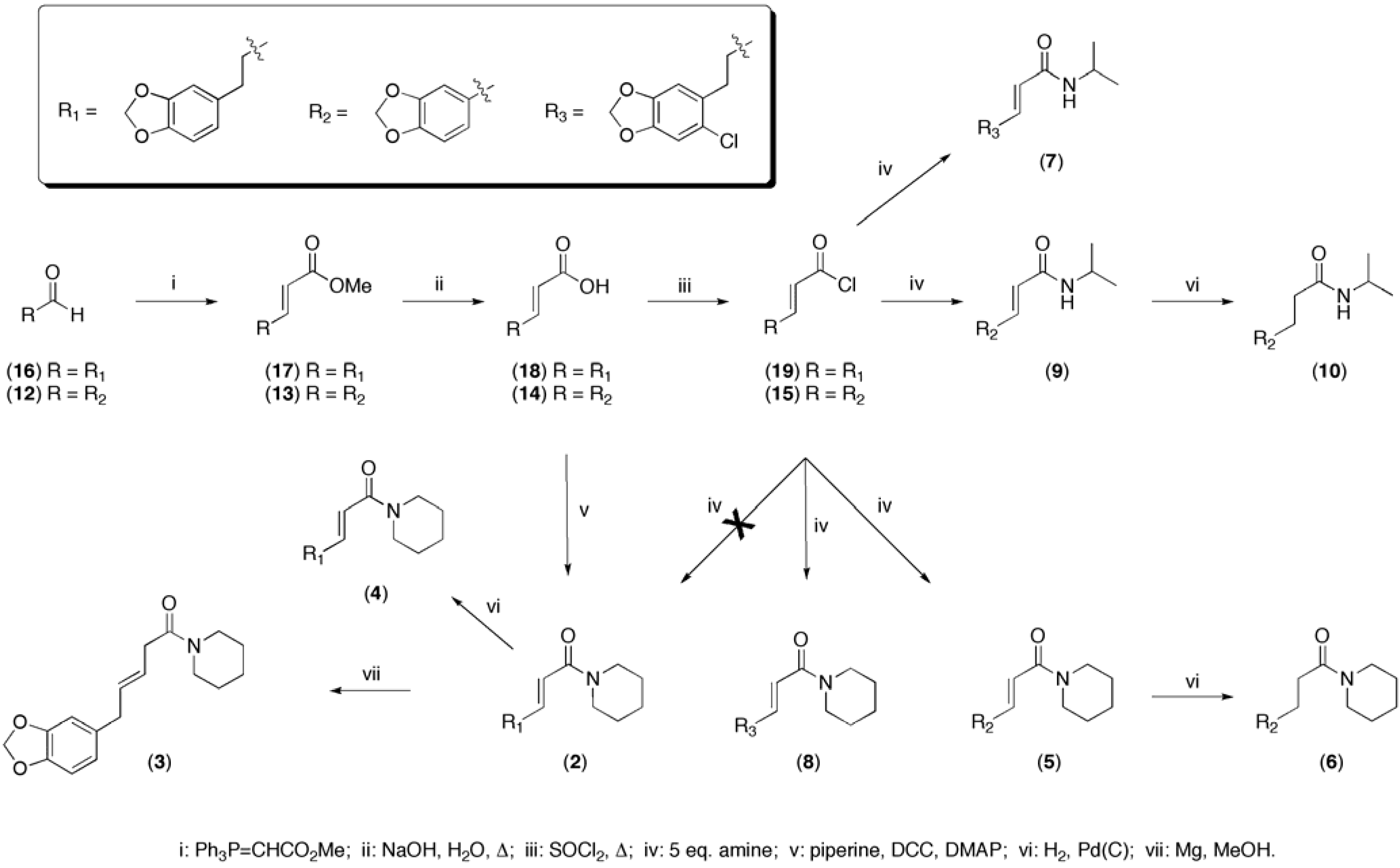
Scheme 1). Wittig reaction between commercially available piperonal (
12) and methyl triphenylphosphoranylidene acetate afforded the α,β-unsaturated ester
13 in high yield. The methyl ester was hydrolysed with 1 M NaOH and converted to the corresponding acid chloride
15 using thionyl chloride. The two amides
5 + 10 (
Figure 1) were synthesized from
15. The last three steps from ester to amide were performed in one pot, without purification. In order to obtain sufficient material for rigorous testing, a synthesis of
2 was performed using same method as described above (
Scheme 1), starting with the homologous aldehyde
16 as starting material. Unexpectedly, the last step yielded the chlorinated analog
8. The chlorinated isopropyl analog
7 was likewise synthesized using the method described above. Dihydropiperine
2 was subsequently synthesized through a DCC coupling of the acid with piperidine. The last four analogs were synthesized directly from other synthesized test compounds by reduction of the double bonds under standard (H
2/Pd) conditions (Figure 2) and the transposed dihydropiperine
3 was synthesized by reduction of piperine with magnesium in methanol [
23].
Scheme 1.
Synthesis of compounds 2-10.
Scheme 1.
Synthesis of compounds 2-10.
trans-3-Benzo[1,3]dioxol-5-yl-acrylic acid methyl ester (13): Piperonal (12; 1.5 g, 10 mmol) was dissolved in DCM (40 mL) and the mixture cooled in an ice-water bath to 5 ºC. Methyl triphenylphosphoranylidene acetate (5.04 g, 15 mmol), dissolved in DCM (50 mL), was added drop- wise and the mixture stirred for 24 hours at room temperature and evaporated under reduced pressure. The crude product was purified by flash-silica gel chromatography (EtOAc/heptane 1:1) to afford the title compound as colorless oil. (1.9 g; 86%; TLC Rf 0.50, EtOAc/heptane, 1:2). 1H-NMR δ (ppm): 7.56-7.62 (d, 1H), 7.02 (m, 1H), 6.98 (d, 1H), 6.79-6.82 (d, 1H), 6.25-6.30 (d, 1H), 6.00 (s, 2H), 3.79 (s, 3H).
trans-3-Benzo[1,3]dioxol-5-yl-acrylic acid (14): 3-Benzo[1,3]dioxol-5-yl-acrylic acid methyl ester (13; 1.9 g, 9.2 mmol) was dissolved in THF (50 mL). To the mixture was added 1M NaOH (50 mL) and the mixture stirred for 24 hours. THF was removed under reduced pressure and conc. HCl added (pH < 3). The slurry was filtered and the title compound obtained as white crystals, which were dried at reduced pressure and used directly in the following synthesis. Yield: (1.76 g, 99%).
trans-3-Benzo[1,3]dioxol-5-yl-acryloyl chloride (15): 3-Benzo[1,3]dioxol-5-yl-acrylic acid (14; 6.0 g, 31.2 mmol) was dissolved in thionyl chloride (3.7 g, 312 mmol). The reaction was heated at reflux for 1 hour and concentrated under reduced pressure to afford the title compound. The compound was used directly in the following reaction.
trans-3-Benzo[1,3]dioxol-5-yl-N-isopropyl-acrylamide (9): 3-Benzo[1,3]dioxol-5-yl-acryloyl chloride (15; 0.5 g; 2.38 mmol) was added to a solution of isopropylamine (0.85 g; 14 mmol) in DCM (20 mL). The reaction was stirred at room temperature for 1 hour and concentrated under reduced pressure. The crude product was dissolved in EtOAc (50 mL) and the organic phase washed with 4 M HCl and 1M NaOH. To the organic phase was added heptane (50 mL) and the mixture concentrated slowly, at reduced pressure, until crystals were seen. The reaction was cooled and the product collected by filtration leaving the title compound as white crystals. (305 mg, 55%; TLC Rf 0.6, EtOAc/heptane, 1:2). 1H-NMR δ (ppm): 7.53-7.48 (d, 1H), 6.94 (d+s, 2H), 6.76 (d, 1H), 6.22 (d, 1H), 5.97 (s, 2H), 5.55 (bs, 1H), 4.21 (m 1H), 1.21 (d. 6H); 13C-NMR: δ (ppm): 165.88, 148.40, 148.39, 140.66, 129.52, 123.99, 119.28, 108.73, 106.50, 101.63, 41.77, 23.09; MS: m/z 233.11 [M]+; HRMS (ESI+), M+Na+: 256.0944; calculated for C13H15NO3Na: 256.0950.
Antiepilepsirine (5): 3-Benzo[1,3]dioxol-5-yl-acryloyl chloride (15; 0.5 g; 2.38 mmol) was added to a solution of piperidine (0.85 g; 10 mmol) in DCM (20 mL). The reaction was stirred at rt. for 1 hour and concentrated under reduced pressure. The crude product was dissolved in 50 mL EtOAc and the organic phase washed with 4 M HCl and then 1M NaOH. The product was obtained as described for (9) leaving the title compound as white crystals. (85 mg; 14%; TLC Rf 0.63, EtOAc/heptane, 1:2).). 1H-NMR δ (ppm): (see above); 13C-NMR δ (ppm): 167.01, 149.03, 148.39, 142.18, 130.18, 123.83, 115.86, 108.70, 106.54, 101.62, 48.03, 44.01, 26.00, 25.50, 24.89; MS: m/z 259.12 [M]+; HRMS (ESI+), M+H+: 260.1295; calculated for C15H18NO3: 260.1287.
trans-5-Benzo[1,3]dioxol-5-yl-pent-2-enoic acid methyl ester (17): 3-Benzo[1,3]dioxol-5-yl-propionaldehyde (16; 0.48 g, 2.70 mmol) was dissolved in DCM (10 mL). The reaction mixture was cooled on an ice-water bath to 5 ºC. Methyl triphenylphosphoranylidene acetate (1.35 g, 4 mmol), dissolved in DCM (20 mL), was added drop wise to the reaction. The mixture was stirred for 24 hours at room temperature and evaporated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/heptane 1:1) to afford the title compound as oil. (410 mg, 65%; TLC Rf 0.63, EtOAc/heptane, 1:2) 1H-NMR: δ 6.91 (m, 1H), 6.68 (d, 1H), 6.60 (d, 1H), 6.50 (s, 1H), 6.95 (s, 2H), 5.82 (d, 1H), 3.67 (s, 3H), 2.67 (t, 2H), 2.45 (m, 2H); MS: m/z 234.09 [M]+.
trans-5-Benzo[1,3]dioxol-5-yl-pent-2-enoic acid (18): 5-Benzo[1,3]dioxol-5-yl-pent-2-enoic acid methyl ester (20; 0.61 mg, 2.6 mmol) was dissolved in THF (10 mL). To the reaction mixture was added 1M NaOH (10 mL) and the mixture stirred for 72 hours. THF was removed under reduced pressure and conc. HCl was added (pH < 3). The water phase was washed with EtOAc and the organic phases combined and concentrated under reduced pressure. The crude product was purified by flash chromatography (EtOAc/heptane 1:1) to afford the title compound as white crystals, which were dried under reduced pressure. (450 mg, 78%; TLC Rf 0.33, EtOAc/heptane, 1:2); 1H-NMR δ (ppm): 7.05 (m, 1H), 6.8-6.7 (m, 3H), 5.92 (s, 2H), 5.80 (d, 1H), 2.68 (m, 2H), 2.46 (m, 2H).
trans-5-Benzo[1,3]dioxol-5-yl-pent-2-enoyl chloride (19): 5-Benzo[1,3]dioxol-5-yl-pent-2-enoic acid (18; 385 mg, 1.75 mmol) was dissolved in thionyl chloride (1.2 g, 10.4 mmol). The reaction was heated to reflux for 1 hour and, then concentrated under reduced pressure to afford the title compound quantitatively, which was used directly in the following reaction.
trans-5-(6-Chlorobenzo[1,3]dioxol-5-yl)-N-isopropylpent-2-enamide (7): 5-Benzo[1,3]dioxol-5-yl-pent- 2-enoyl chloride (19; 208 mg; 0.88 mmol) was added to a solution of isopropylamine (0.85 g; 14 mmol) in DCM (10 mL). The reaction was stirred at room temperature for 1 hour and concentrated under reduced pressure. The crude product was dissolved in EtOAc (20 mL) and the organic phase washed with 4 M HCl and 1M NaOH. The product was obtained as described for (10) and further purified using HPLC-system II leaving the product as white crystals. 1H-NMR δ (ppm): 6.75 (s, 1H), 6.60 (s, 1H), 6.60 (d, J = 1.7 Hz, 1H), 6.18 (dt, J = 15.0 Hz, J = 1.5 Hz, 1H), 5.85 (s, 2H), 4.10 (m, 1H), 2.60 (t, J = 7.3 Hz, 2H), 2.40 (dt, J = 7.3 Hz, J = 1.3 Hz, 2H), 1.20 (s, 6H); MS: m/z 295.10 [M]+; HRMS (ESI+), M+H+: 296.1052; calculated for C15H19NO3Cl: 296.1053.
trans-5-(6-Chlorobenzo[1,3]dioxol-5-yl)-1-(piperidin-1-yl)pent-2-en-1-one (8): 5-Benzo[1,3]di-oxol-5-yl-pent-2-enoyl chloride (19; 208 mg; 0.88 mmol) was added to a solution of piperidine (0.85 g; 10 mmol) in DCM (10 mL). The reaction was stirred at room temperature for 1 hour and concentrated under reduced pressure. The crude product was dissolved in EtOAc (20 mL) and the organic phase washed with 4M HCl and then 1M NaOH. The product was obtained as described for (10) and further purified using HPLC-system II leaving the product as white crystals. 1H-NMR δ (ppm): 6.75 (s, 1H), 6.60 (s, 1H), 5.85 (s, 2H), 3.56 (br s, 2H), 3.35 (br s, 2H), 2.60 (t, J = 7.6 Hz, 2H), 2.40 (dt, J = 7.3 Hz, J = 1.3 Hz, 2H), 1.56 (m, 6H); MS: m/z 321.11 [M]+; HRMS (ESI+), M+Na+: 344.1034; calculated for C17H20NO3NaCl: 344.1029.
4,5-Dihydropiperine (2): A mixture of the carboxylic acid (21; 440 mg; 1 mol), piperidine (1.9 g; 22 mol), dicyclohexylcarbodiimide (DCC, 412 mg; 2 mmol), 4-(dimethylamino)pyridine (DMAP, 244 mg; 2 mmol) in dichloromethane (10 mL) was stirred at room temperature for 2 h. The clear solution became turbid after 5 minutes. The precipitated dicyclohexylurea was filtered off and washed with dichloromethane. The combined organic phases were extracted with a saturated aqueous citric acid solution and water, dried (MgSO4) and evaporated to give the corresponding amide which was purified by flash chromatography (EtOAc/heptane 1:1) to afford the title compound, which was re-crystallized (EtOAc/heptane) to afford 2 as white crystals. (80 mg; 14%; TLC Rf 0.58, EtOAc/heptane, 1:2) 1H-NMR (CDCl3) (data: see above).
(E)-5-(Benzo[1,3]dioxol-5-yl)-1-(piperidin-1-yl)pent-3-en-1-one (3): To piperine 1 (1 g, 3.5 mmol) in methanol (20 mL) was added magnesium powder (91 mg, 3.5 mmol) and the mixture stirred at room temperature for 2 hrs. The reaction mixture was filtered and the methanol removed under reduced pressure. The residue was neutralized with a 1 M NH4Cl solution (25 mL) and extracted with chloroform. The extract was concentrated to a gummy mass, which was purified by VLC over silica gel using hexane/EtOAc (1:1) as eluant to afford 3 as a syrup. (0.95 g, 95%; TLC Rf 0.55, EtOAc/heptane, 1:2). 1H-NMR δ (ppm): 6.75 (s, 1H), 6.60 (s, 1H), 6.60 (d, J = 1.7 Hz, 1H), 6.18 (dt, J = 15.0 Hz, J = 1.5 Hz, 1H), 5.85 (s, 2H), 5.60 (m, 4H), 3.5 (m, 4H), 3.3 (d, 2H), 3.1 (d, 2H), 1.56 (m, 6H); MS: m/z 287.15 [M]+; HRMS (ESI+), M+H+: 288.1600; calculated for C17H22NO3: 288.1600.
Tetrahydropiperidine (4): Piperine (1, 1g, 3.5 mmol) was dissolved in MeOH (20 mL) and Pd(C) was added (50 mg). The flask was closed and put under a H2 atmosphere for 6 hours. The reaction mixture was filtered and the organic phase concentrated under reduced pressure to afford the title compound as orange oil, which was purified by VLC over silica gel using hexane/EtOAc (1:1) as eluant to afford 3. (0.95 g, 95%; TLC Rf 0.60, EtOAc/heptane, 1:2). 1H-NMR δ (ppm): 6.75 (s, 1H), 6.60 (s, 1H), 6.60 (d, J = 1.7 Hz, 1H), 6.18 (dt, J = 15.0 Hz, J = 1.5 Hz, 1H), 5.85 (s, 2H), 3.5 (m, 4H), 2.6 (t, 2H), 2.4 (t, 3H), 1.6 (m, 12H); MS: m/z 289.17 [M]+; HRMS (ESI+), M+H+: 290.1748; calculated for C17H24NO3: 290.1756.
3-Benzo[1,3]dioxol-5-yl-1-piperidin-1-yl-propan-1-one (6): The amide 5 (1 g, 3.86 mmol) was dissolved in MeOH (20 mL), Pd(C) (50 mg) was added and the flask closed and put under a H2 atmosphere for 4 hours. The reaction mixture was filtered and the organic phase concentrated under reduced pressure to afford the title compound as brown crystals, which were purified using HPLC-system II. (0.99 g, 95%; TLC Rf 0.50, EtOAc/heptane, 1:2). 1H-NMR δ (ppm): 6.75 (s, 1H), 6.60 (s, 1H), 6.60 (d, J = 1.7 Hz, 1H), 6.18 (dt, J = 15.0 Hz, J = 1.5 Hz, 1H), 5.85 (s, 2H), 3.5 (m, 4H), 2.6 (t, 2H), 2.4 (t, 3H), 1.6 (m, 12H); MS: m/z 261.14 [M]+; HRMS (ESI+), M+Na+: 284.1273; calculated for C15H19NO3Na: 284.1263.
3-Benzo[1,3]dioxol-5-yl-N-isopropyl-propionamide (10): Compound 9 (1g, 4.02 mmol) was dissolved in MeOH (20 mL) and Pd(C) was added (50 mg). The flask was closed and put under a H2 atmosphere for 4 hours. The reaction mixture was filtered and the organic phase concentrated under reduced pressure to afford the title compound as tan crystals, which were purified using HPLC-system II. (0.93 g, 94%; TLC Rf 0.45, EtOAc/heptane, 1:2). 1H-NMR δ (ppm): 6.75 (s, 1H), 6.60 (s, 1H), 6.60 (d, J = 1.7 Hz, 1H), 6.18 (dt, J = 15.0 Hz, J = 1.5 Hz, 1H), 5.85 (s, 2H), 4.0 (m, 1H), 2.8 (t, 2H), 2.4 (t, 2H), 1.1 (m, 6H); MS: m/z 235.12 [M]+: HRMS (ESI+), M+Na+: 258.1106; calculated for C13H17NO3Na: 258.1106.



{kind=link}
{kind=link}
{kind=link}
