Thioacetyl-Terminated Ferrocene-Anthraquinone Conjugates: Synthesis, Photo- and Electrochemical Properties Triggered by Protonation-Induced Intramolecular Electron Transfer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction


2. Results and Discussion
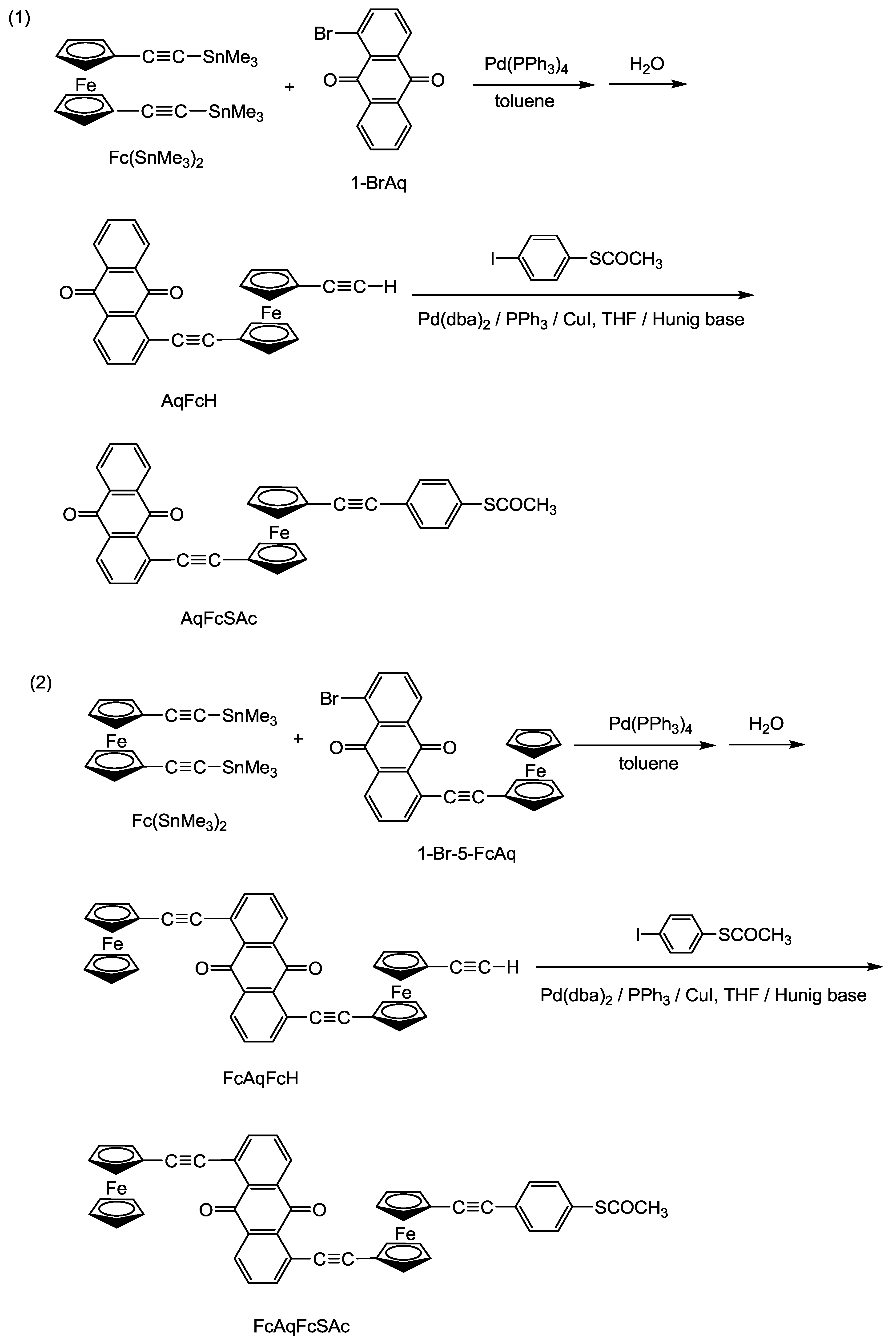
2.1. Synthesis
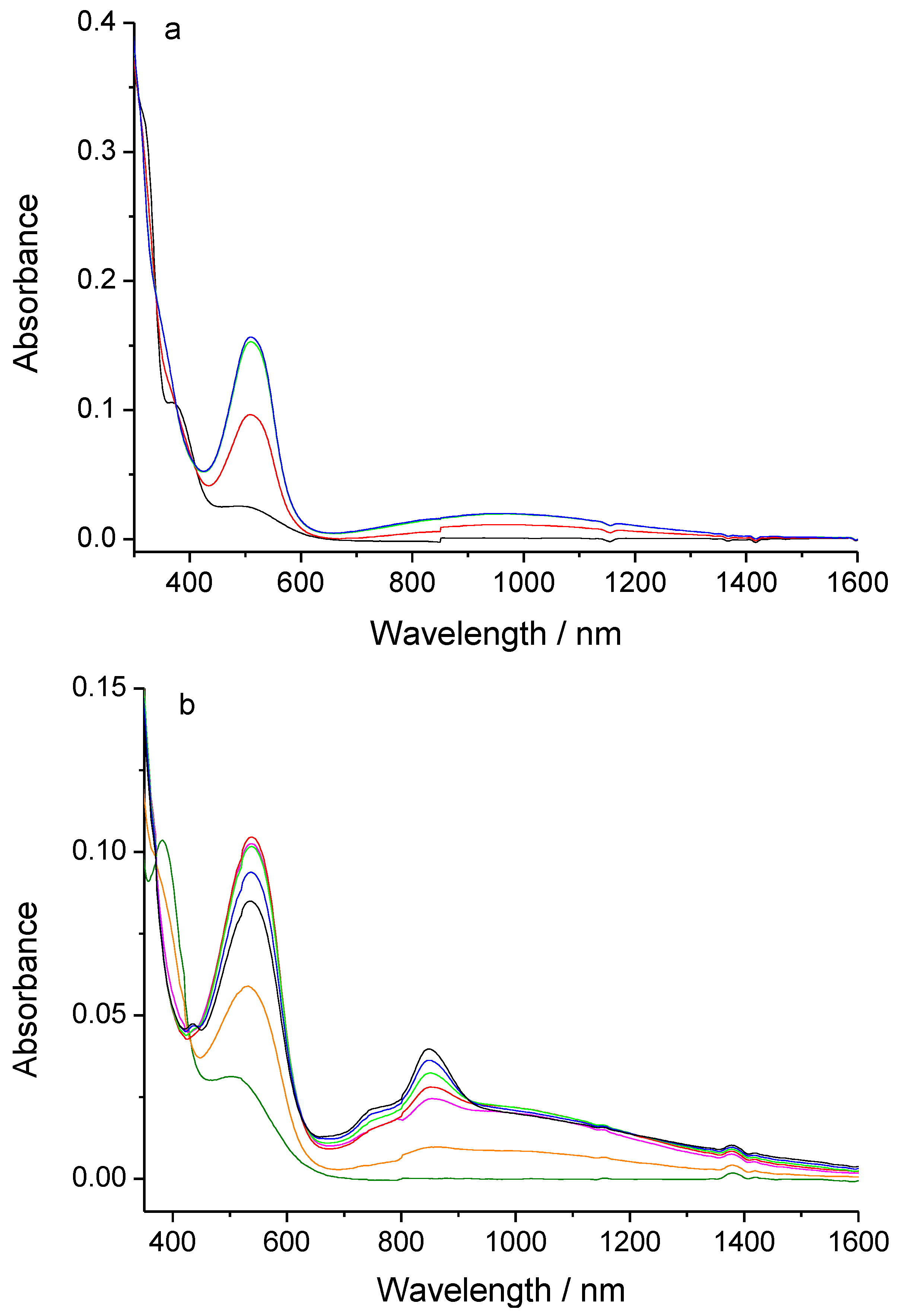
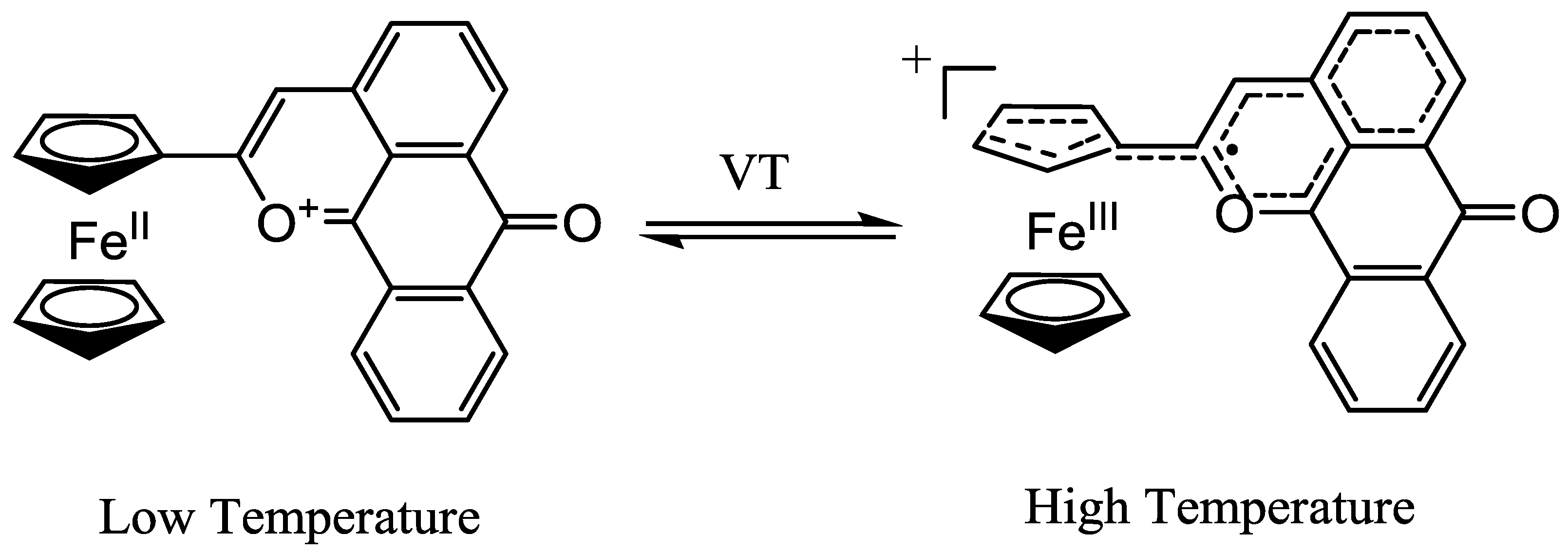
2.2. Electronic properties

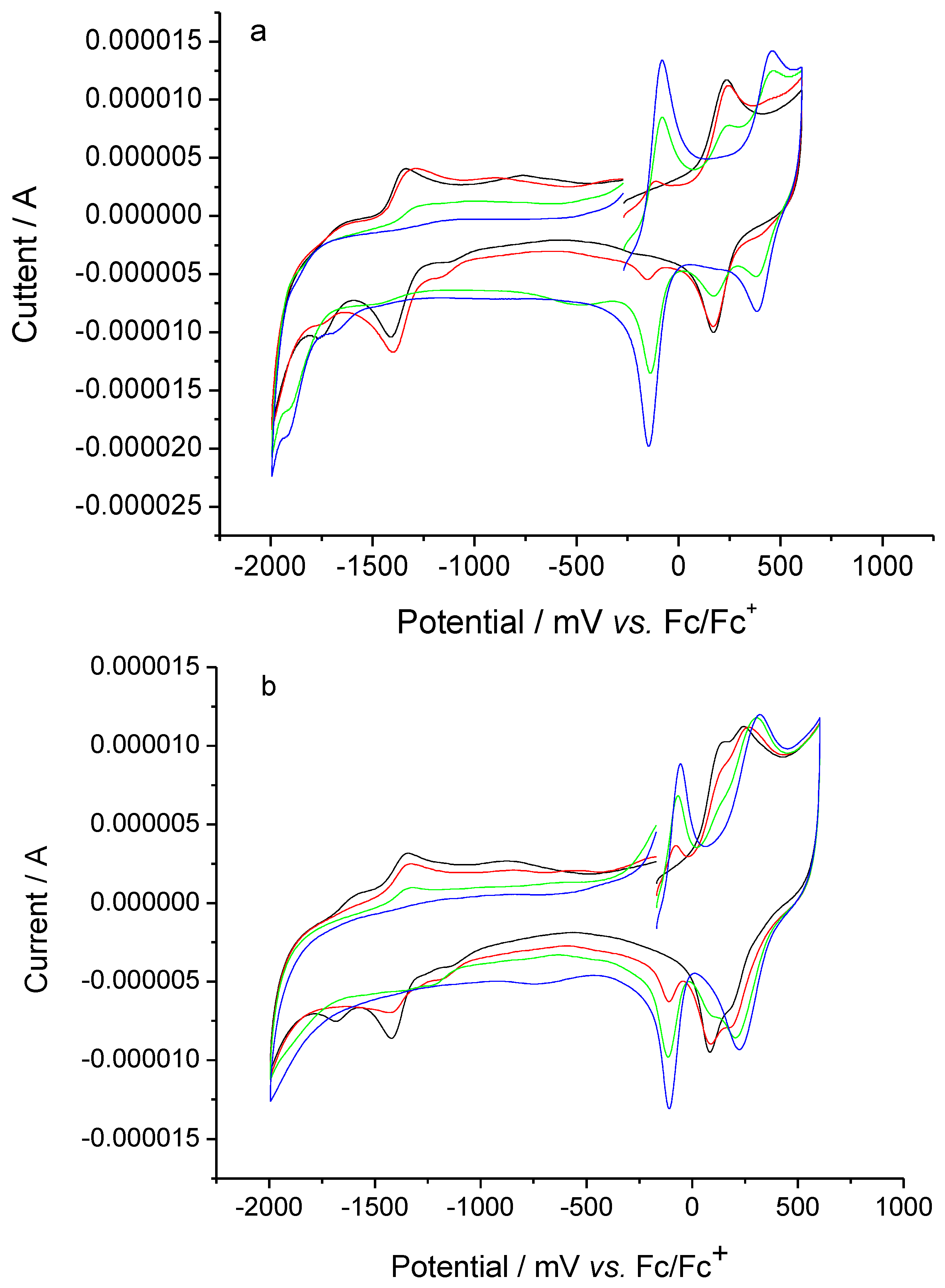
2.3. Electrochemical properties

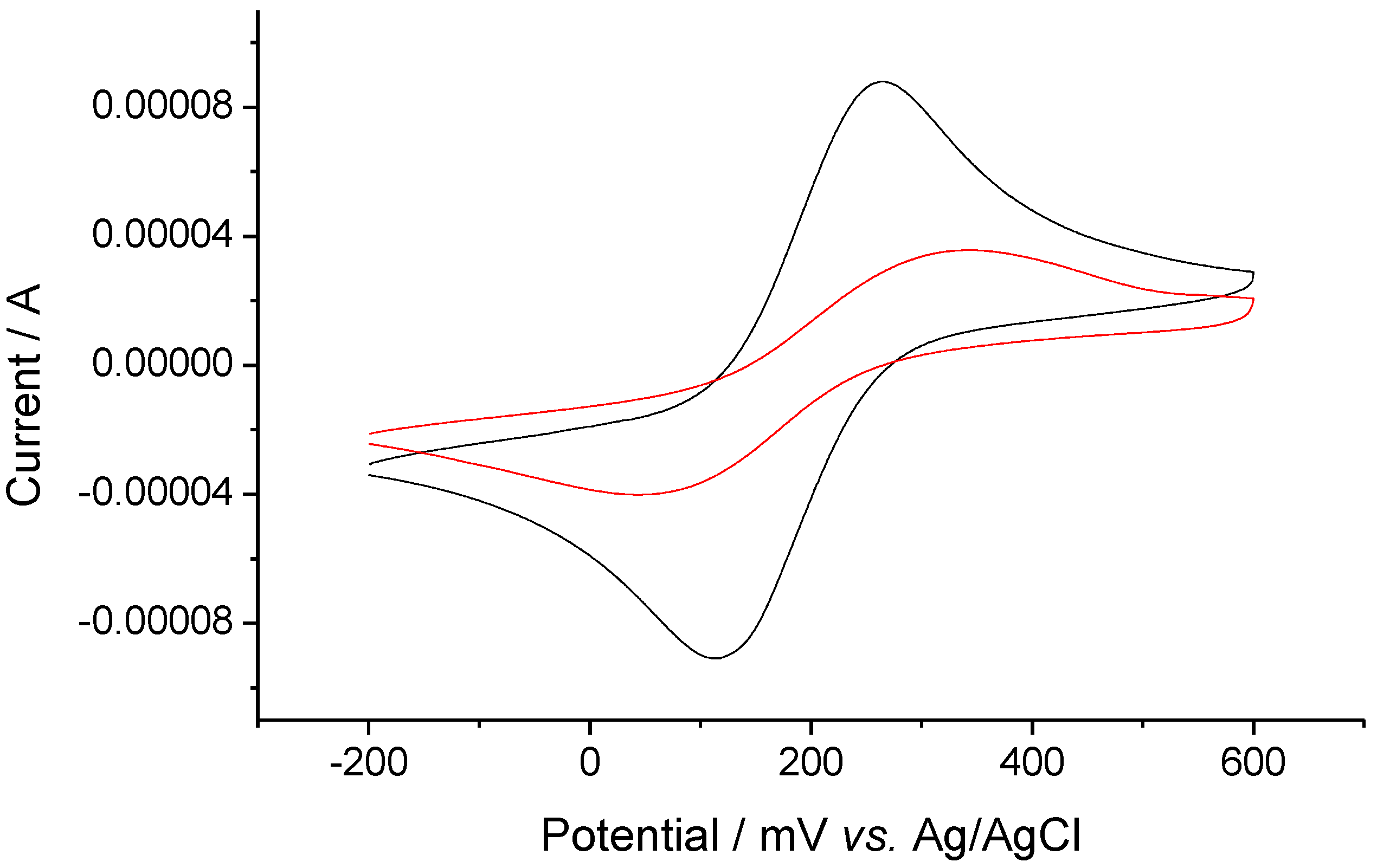
2.4. SAMs on gold

3. Experimental
3.1. General methods
3.2. Spectroscopy
3.3. Electrochemical measurements
3.4. Synthesis
3.5. SAMs preparation
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the Fc-Aq compounds are available from the authors.
References and Notes
- Wasielewski, M.R. Photoinduced Electron transfer in supramolecular systems for artificial photosynthesis. Chem. Rev. 1992, 92, 435–461. [Google Scholar] [CrossRef]
- Kurreck, H.; Huber, M. Model reactions for photosynthesis - Photoinduced charge and energy transfer between covalently linked porphyrin and quinone units. Angew. Chem. Int. Ed. Engl. 1995, 34, 849–866. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L. Mimicking photosynthetic solar energy transduction. Acc. Chem. Res. 2001, 34, 40–48. [Google Scholar] [CrossRef]
- Holten, D.; Bocian, D.F.; Lindsey, J.S. Probing electronic communication in covalently linked multiporphyrin arrays. A guide to the rational design of molecular photonic devices. Acc. Chem. Res 2002, 35, 57–69. [Google Scholar] [CrossRef]
- Imahori, H.; Mori, Y.; Matano, Y. Nanostructured artificial photosynthesis. J. Photochem. Photobiol. C - Photo. 2003, 4, 51–83. [Google Scholar] [CrossRef]
- Brabec, C.J.; Sariciftci, N.S.; Hummelen, J.C. Plastic solar cells. Adv. Funct. Mater. 2001, 11, 15–26. [Google Scholar] [CrossRef]
- Wienk, M.W.; Kroon, J.M.; Verhees, W.J.H.; Knol, J.; Hummelen, J.C.; van Hal, P.A.; Janssen, R.A.J. Efficient methano[70]fullerene/MDMO-PPV bulk heterojunction photovoltaic cells. Angew. Chem. Int. Ed. 2003, 42, 3371–3375. [Google Scholar] [CrossRef]
- Winder, C.; Sariciftci, N.S. Low bandgap polymers for photon harvesting in bulk heterojunction solar cells. J. Mater. Chem. 2004, 14, 1077–1086. [Google Scholar] [CrossRef]
- Li, G.; Shrotriya, V.; Huang, J.; Yao, Y.; Moriarty, T.; Emery, K.; Yang, Y. High-efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends. Nat. Mater. 2005, 4, 864–868. [Google Scholar] [CrossRef]
- Ma, W.; Yang, C.; Gong, X.; Lee, K.; Heeger, A.J. Thermally Stable, efficient polymer solar cells with nanoscale control of the interpenetrating network morphology. Adv. Funct. Mater. 2005, 15, 1617–1622. [Google Scholar] [CrossRef]
- Atienza, C.M.; Fernández, G.; Sánchez, L.; Martín, N.; Dantas, I.S.; Wienk, M.M.; Janssen, R.A.J.; Rahman, G.M.A.; Guldi, D.M. Light harvesting tetrafullerene nanoarray for organic solar cells. Chem. Commun. 2006, 514–516. [Google Scholar]
- Sierra, M.; Herranz, M.Á.; Zhang, S.; Sánchez, L.; Martín, N.; Echegoyen, L. Self-assembly of C-60 π-extended tetrathiafulvalene (exTTF) dyads on gold surfaces. Langmuir 2006, 22, 10619–10624. [Google Scholar] [CrossRef]
- Delgado, J.L.; Espíldora, E.; Liedtke, M.; Sperlich, A.; Rauh, D.; Baumann, A.; Deibel, C.; Dyakonov, V.; Martín, N. Fullerene dimers (C60/C70) for energy harvesting. Chem. Eur. J. 2009, 15, 13474–13482. [Google Scholar]
- Okamoto, K.; Imahori, H.; Fukuzumi, S. Metal ion-promoted intramolecular electron transfer in a ferrocene-naphthoquinone linked dyad. Continuous change in driving force and reorganization energy with metal ion concentration. J. Am. Chem. Soc. 2003, 125, 7014–7021. [Google Scholar] [CrossRef]
- Thompson, A.L.; Ahn, T.-S.; Thomas, K.R.J.; Thayumanavan, S.; Martínez, T.J.; Bardeen, C.J. Using Meta conjugation to enhance charge separation versus charge recombination in phenylacetylene donor−bridge−acceptor complexes. J. Am. Chem. Soc. 2005, 127, 16348–16349. [Google Scholar]
- Perepichka, D.F.; Bryce, M.R. Molecules with exceptionally small HOMO-LUMO Gaps. Angew. Chem. Int. Ed. 2005, 44, 5370–5373. [Google Scholar] [CrossRef]
- Chernick, E.T.; Mi, Q.; Kelley, R.F.; Weiss, E.A.; Jones, B.A.; Marks, T.J.; Ratner, M.A.; Wasielewski, M.R. Electron donor-bridge-acceptor molecules with bridging nitronyl nitroxide radicals: influence of a third spin on charge- and spin-transfer dynamics. J. Am. Chem. Soc. 2006, 128, 4356–4364. [Google Scholar]
- Kim, O.K.; Je, J.; Melinger, J.S. One-dimensional energy/electron transfer through a helical channel. J. Am. Chem. Soc. 2006, 128, 4532–4533. [Google Scholar] [CrossRef]
- Murphy, A.R.; Frechet, J.M.J. Organic semiconducting oligomers for use in thin film transistors. Chem. Rev. 2007, 107, 1066–1096. [Google Scholar] [CrossRef]
- Kulkarni, A.P.; Zhu, Y.; Babel, A.; Wu, P.T.; Jenekhe, S.A. New ambipolar organic semiconductors. 1. Synthesis, single-crystal structures, redox properties, and photophysics of phenoxazine-based donor−acceptor molecules. Chem. Mater. 2008, 20, 4200–4211. [Google Scholar] [CrossRef]
- Zhang, W.W.; Mao, W.L.; Hu, Y.X.; Tian, Z.Q.; Wang, Z.L.; Meng, Q.J. Phenothiazine-anthraquinone donor-acceptor molecules: Synthesis, electronic properties and DFT-TDDFT computational study. J. Phys. Chem.A 2009, 113, 9997–10004. [Google Scholar] [CrossRef]
- Murata, M.; Yamada, M.; Fujita, T.; Kojima, K.; Kurihara, M.; Kubo, K.; Kobayashi, Y.; Nishihara, H. Structural conversion and spin separation in bis(ferrocenylethynyl)anthraquinones triggered by proton-coupled intramolecular electron transfer. J. Am. Chem. Soc. 2001, 123, 12903–12904. [Google Scholar]
- Murata, M.; Fujita, T.; Yamada, M.; Kurihara, M.; Nishihara, H. Novel protonation-induced structural conversion of an ethynylene-bridged ferrocene-anthraquinone complex. Chem. Lett. 2000, 1328–1329. [Google Scholar]
- Kondo, M.; Murata, M.; Nishihara, H.; Nishibori, E.; Aoyagi, S.; Yoshida, M.; Kinoshita, Y.; Sakata, M. Guest-induced instant and reversible crystal-to-crystal transformation of 1,4-bis(ferrocenylethynyl)anthraquinone. Angew. Chem. Int. Ed. 2006, 45, 5461–5464. [Google Scholar] [CrossRef]
- Kojima, K.; Zhang, W.W.; Kondo, M.; Uchikawa, M.; Namiki, K.; Fujita, T.; Murata, M.; Kobayashi, Y.; Nishihara, H. Synthesis of p-conjugated ferrocene-anthraquinone alternating polymers and their protonation reactions. J. Inorg. Organomet. Polym. Mater. 2007, 17, 135–141. [Google Scholar] [CrossRef]
- Kondo, M.; Uchikawa, M.; Zhang, W.W.; Namiki, K.; Kume, S.; Murata, M.; Kobayashi, Y.; Nishihara, H. Protonation-induced cyclocondensation of 1-aryl ethynylanthraquinones: expanding the p conjugation. Angew. Chem. Int. Ed. 2007, 46, 6271–6274. [Google Scholar] [CrossRef]
- Kondo, M.; Uchikawa, M.; Namiki, K.; Zhang, W.W.; Kume, S.; Nishibori, E.; Suwa, H.; Aoyagi, S.; Sakata, M.; Murata, M.; Kobayashi, Y.; Nishihara, H. Counterion-dependent valence tautomerization of ferrocenyl-conjugated pyrylium salts. J. Am. Chem. Soc. 2009, 131, 12112–12124. [Google Scholar]
- Kramer, S.; Fuierer, R.R.; Gorman, C.B. Scanning Probe lithography using self-assembled monolayers. Chem. Rev. 2003, 103, 4367–4418. [Google Scholar] [CrossRef]
- Smith, R.K.; Lewis, P.A.; Weiss, P.S. Patterning self-assembled monolayers. Prog. Surf. Sci. 2004, 75, 1–68. [Google Scholar] [CrossRef]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 2005, 105, 1103–1169. [Google Scholar] [CrossRef]
- Whitesides, G.M. Nanoscience, nanotechnology, and chemistry. Small 2005, 1, 172–179. [Google Scholar] [CrossRef]
- Zotti, G.; Vercelli, B.; Berlin, A. Monolayers and multilayers of conjugated polymers as nanosized electronic components. Acc. Chem. Res. 2008, 41, 1098–1109. [Google Scholar]
- Shen, C.; Buck, M. Patterning of self-assembled monolayers based on differences in molecular conductance. Nanotechnology 2009, 20, 245306:1–245306:6. [Google Scholar]
- Hulme, B.E.; Land, E.J.; Phillips, G.O. Pulse radiolysis of 9,10-anthraquinones. J. Chem. Soc.Faraday Trans. I 1972, 68, 1992–2002. [Google Scholar] [CrossRef]
- Chambers, J.Q. Chemistry of Functional Groups. In The Chemistry of the Quinonoid Compounds; Patai, S., Ed.; Wiley-Interscience: New York, NY, USA, 1974; Chapter 14. [Google Scholar]
- Kubo, K.; Kondow, H.; Nishihara, H. Oxidative-decomposition and electron-transfer kinetics of self-assembled monolayers of biferrocene-terminated alkanethiol on gold. Electrochemistry 1999, 67, 1129–1131. [Google Scholar]
- Smalley, J.F.; Feldberg, S.W.; Chidsey, C.E.D.; Linford, M.R.; Newton, M.D.; Liu, Y.P. The kinetics of electron transfer through ferrocene-terminated alkanethiol monolayers on gold. J. Phys. Chem. 1995, 99, 13141–13149. [Google Scholar]
- Tour, J.M.; Jones, L., II; Pearson, D.L.; Lamba, J.J.S.; Burgin, T.P.; Whitesides, G.M.; Allara, D.L.; Parikh, A.N.; Atre, S.V. Self-assembled monolayers and multilayers of conjugated thiols, a,w-dithiols, and thioacetyl-containing adsorbates. Understanding Attachments between potential molecular wires and gold surfaces. J. Am. Chem. Soc. 1995, 117, 9529–9534. [Google Scholar]
- Chen, J.; Wang, W.; Reed, M.A.; Rawlett, A.M.; Price, D.W.; Tour, J.M. Room-temperature negative differential resistance in nanoscale molecular junctions. Appl. Phys. Lett. 2000, 77, 1224–1226. [Google Scholar] [CrossRef]
- Bumm, L.A.; Arnold, J.J.; Cygan, M.T.; Dunbar, T.D.; Burgin, T.P.; Jones, L.; Allara, D.L.; Tour, J.M.; Weiss, P.S. Are single molecular wires conducting? Science 1996, 271, 1705–1707. [Google Scholar]
- Cygan, M.T.; Dunbar, T.D.; Weiss, P.S. Insertion, conductivity, and structures of conjugated organic oligomers in self-assembled alkanethiol monolayers on Au{111}. J. Am. Chem. Soc. 1998, 120, 2721–2732. [Google Scholar]
- Shaporenko, A.; Rössler, K.; Lang, H.; Zharnikov, M. Self-assembled monolayers of ferrocene-substituted biphenyl ethynyl thiols on gold. J. Phys. Chem. B 2006, 110, 24621–24628. [Google Scholar] [CrossRef]
- Doyle, M.P.; Siegfried, B.; Dellaria, J.F., Jr. Alkyl nitrite-metal halide deamination reactions. 2. Substitutive deamination of arylamines by alkyl nitrites and Copper(II) halides. A direct and remarkably efficient conversion of arylamines to aryl halides. J. Org. Chem. 1977, 42, 2426–2431. [Google Scholar] [CrossRef]
- Long, N.J.; Martin, A.J.; Vilar, R.; White, A.J.P.; Williams, D.J.; Younus, M. Synthesis, characterization, and theoretical studies of new alkynylferrocene and -biferrocene ligands and their platinum-containing dimers and oligomers. Organometallis 1999, 18, 4261–4269. [Google Scholar] [CrossRef]
- Hsung, R.P.; Chidsey, C.E.D.; Sita, L.R. Synthesis and characterization of unsymmetric ferrocene-terminated phenylethynyl oligomers Cp2Fe-[C°C-C6H4]n-X (X = SH, SMe, SOMe, and SO2Me). Organometallics 1995, 14, 4808–4815. [Google Scholar] [CrossRef]
2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, W.-W.; Kondo, M.; Fujita, T.; Namiki, K.; Murata, M.; Nishihara, H. Thioacetyl-Terminated Ferrocene-Anthraquinone Conjugates: Synthesis, Photo- and Electrochemical Properties Triggered by Protonation-Induced Intramolecular Electron Transfer. Molecules 2010, 15, 150-163. https://doi.org/10.3390/molecules15010150
Zhang W-W, Kondo M, Fujita T, Namiki K, Murata M, Nishihara H. Thioacetyl-Terminated Ferrocene-Anthraquinone Conjugates: Synthesis, Photo- and Electrochemical Properties Triggered by Protonation-Induced Intramolecular Electron Transfer. Molecules. 2010; 15(1):150-163. https://doi.org/10.3390/molecules15010150
Chicago/Turabian StyleZhang, Wen-Wei, Mio Kondo, Takako Fujita, Kosuke Namiki, Masaki Murata, and Hiroshi Nishihara. 2010. "Thioacetyl-Terminated Ferrocene-Anthraquinone Conjugates: Synthesis, Photo- and Electrochemical Properties Triggered by Protonation-Induced Intramolecular Electron Transfer" Molecules 15, no. 1: 150-163. https://doi.org/10.3390/molecules15010150