[2+2+2] Cycloaddition Reactions of Macrocyclic Systems Catalyzed by Transition Metals. A Review

Abstract
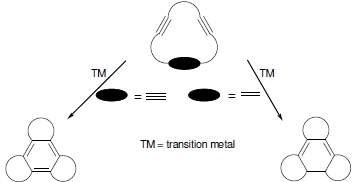
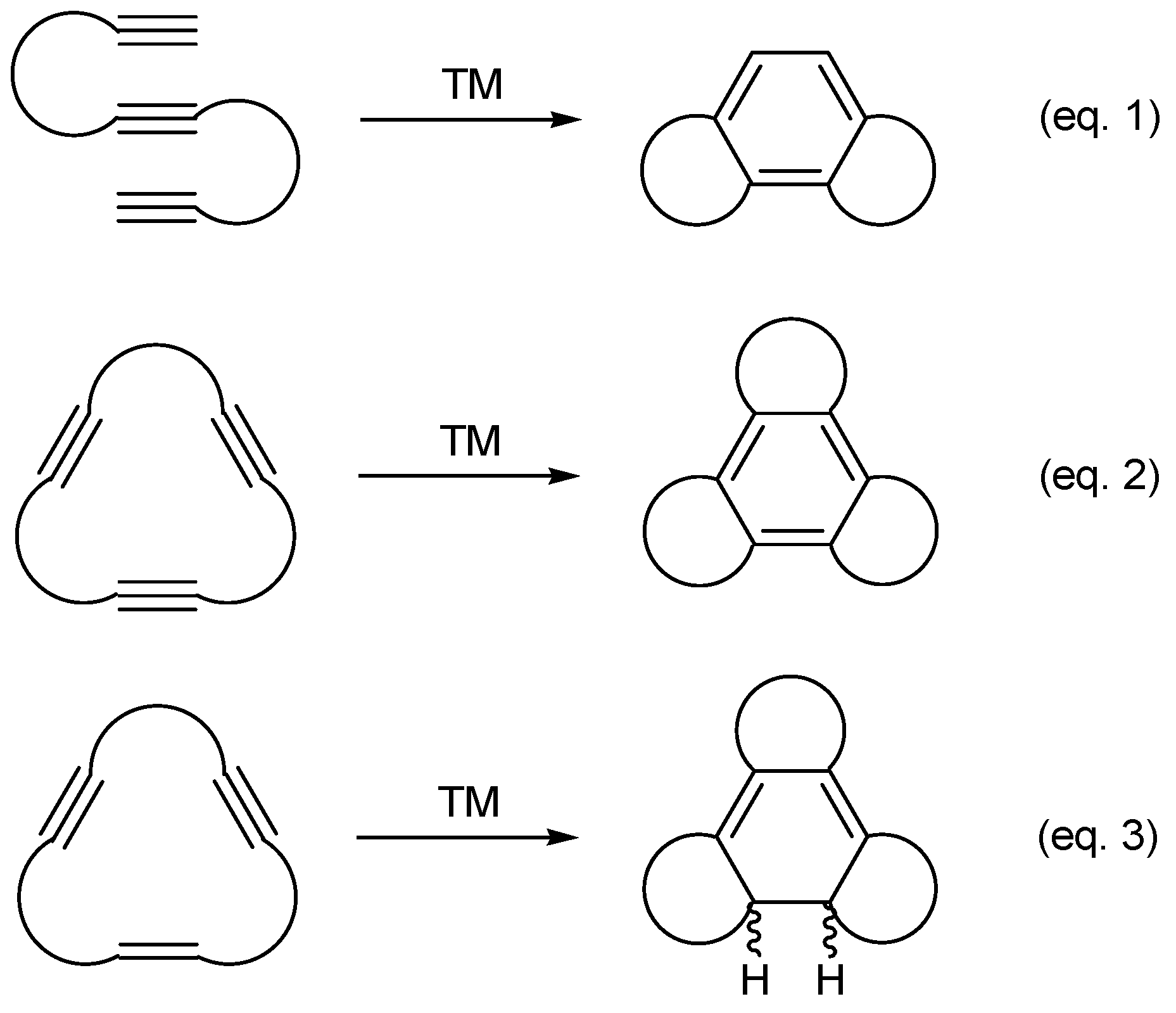
:
1. Introduction


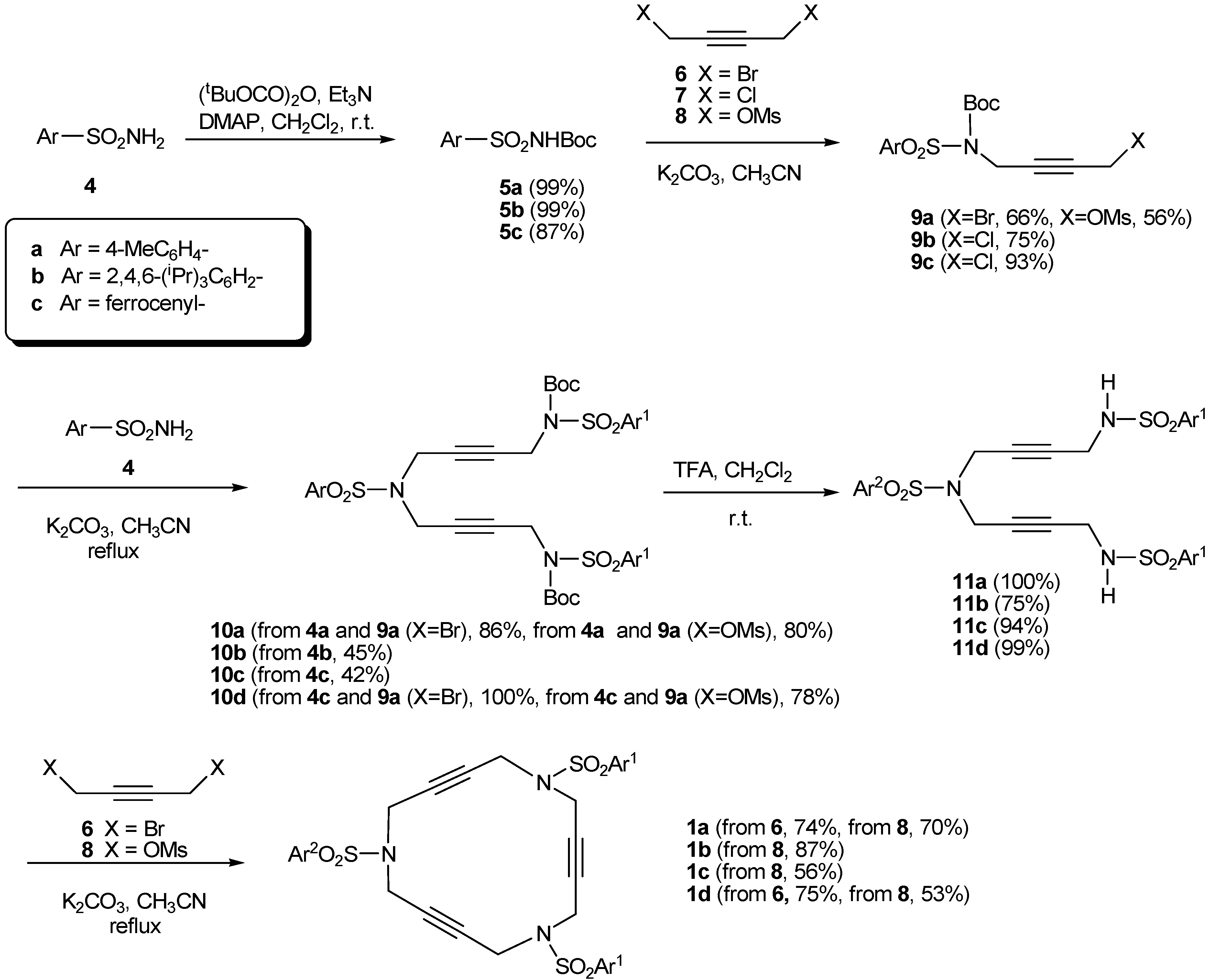
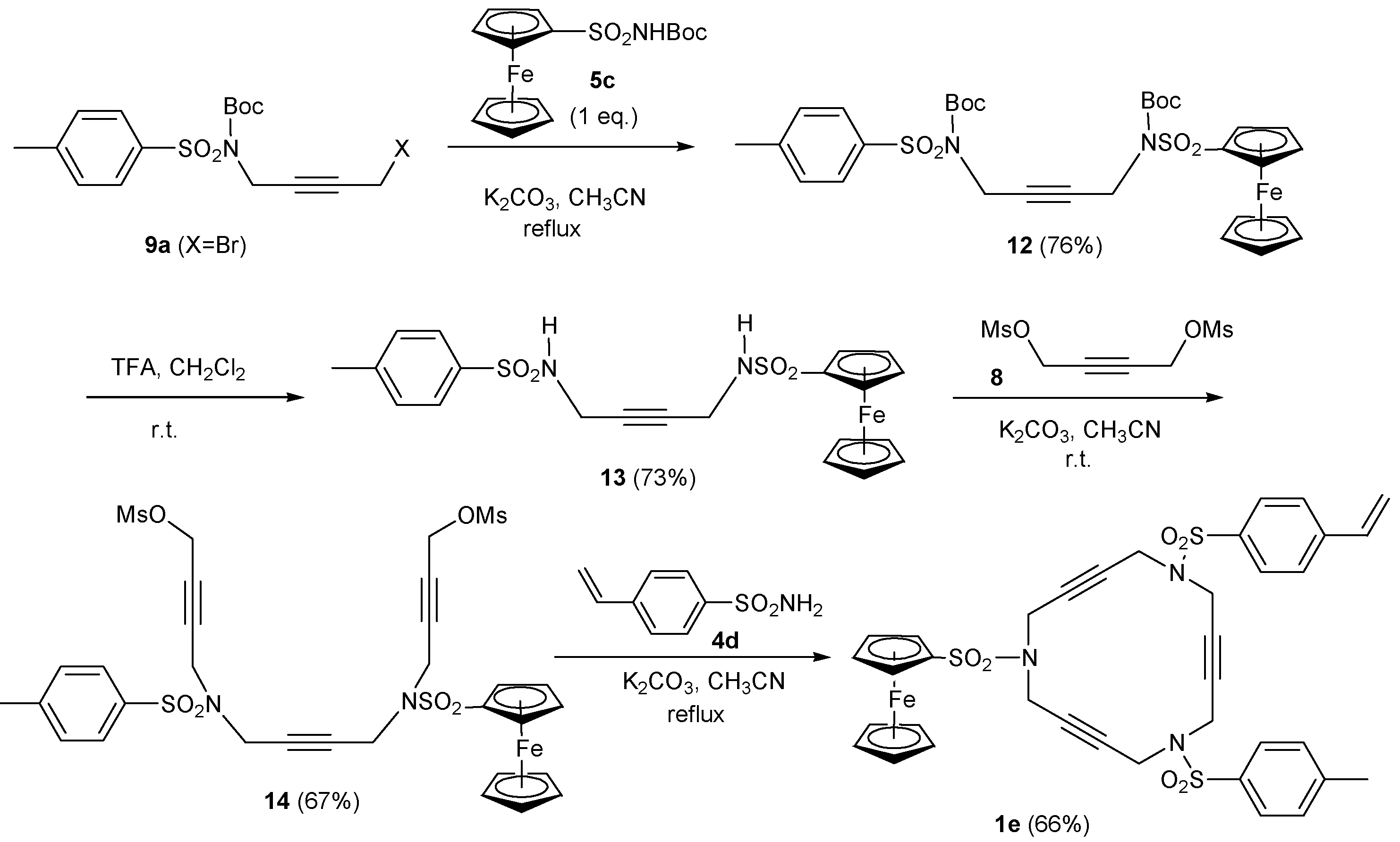
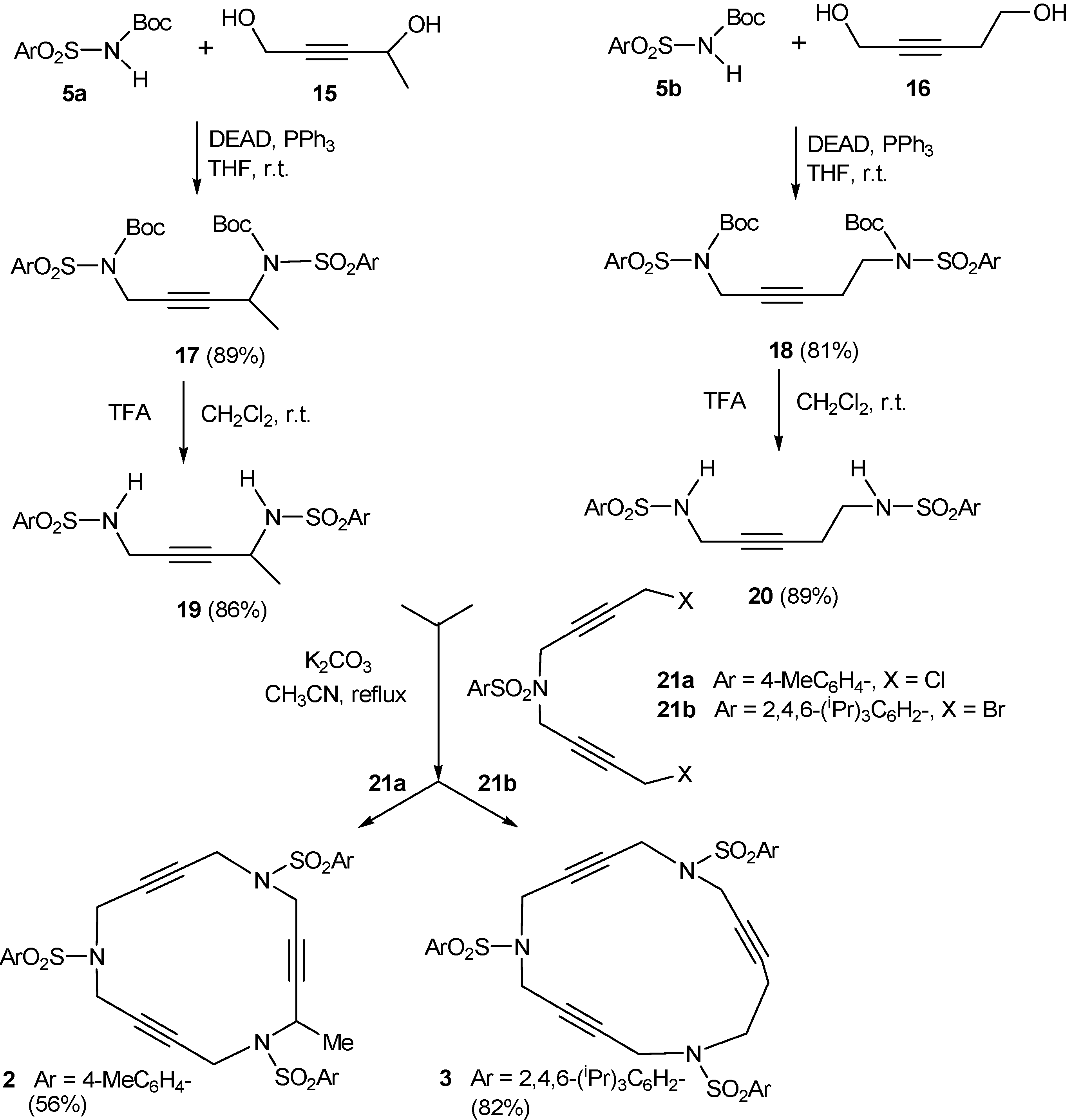
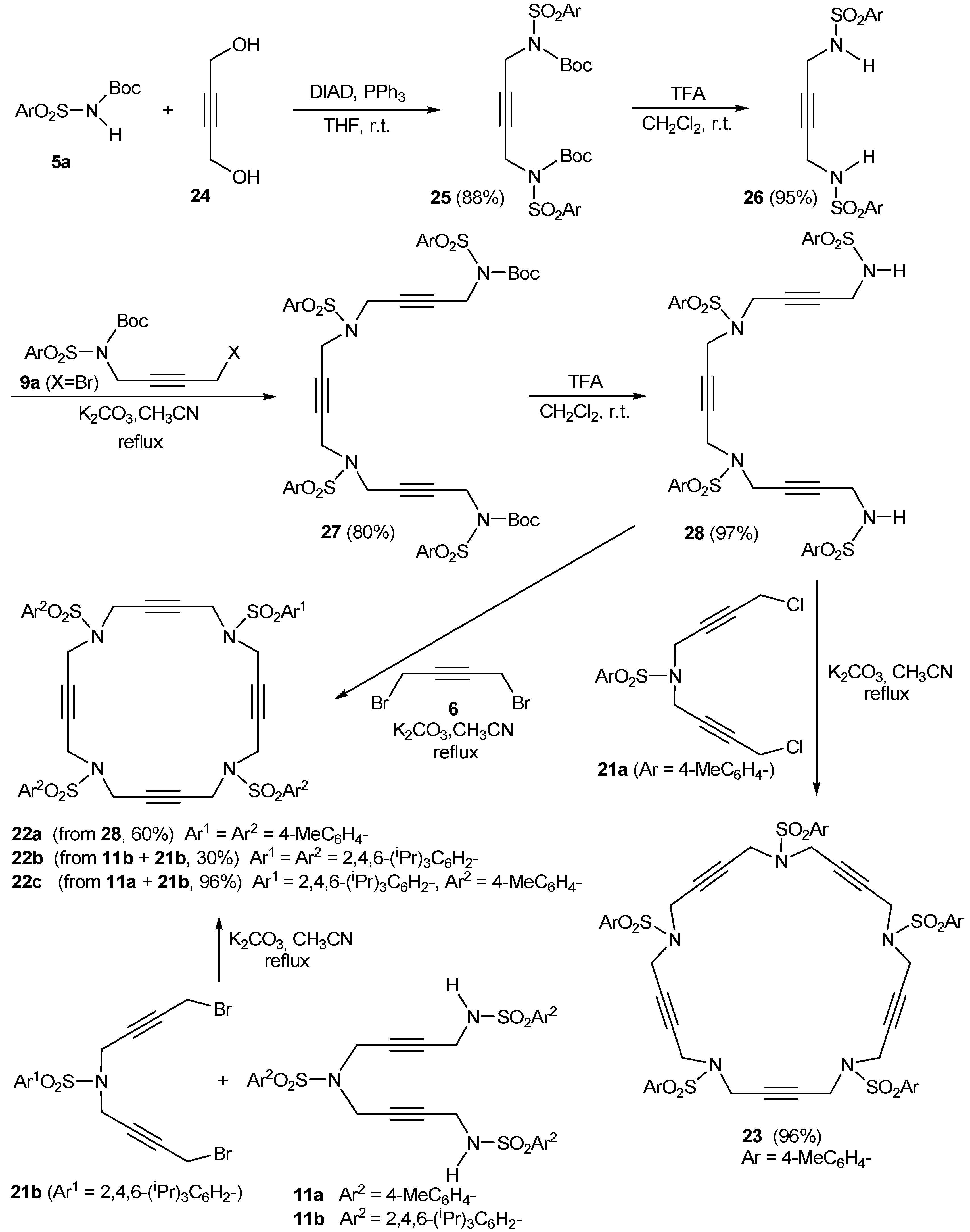
2. Synthesis of Polyunsaturated Azamacrocycles
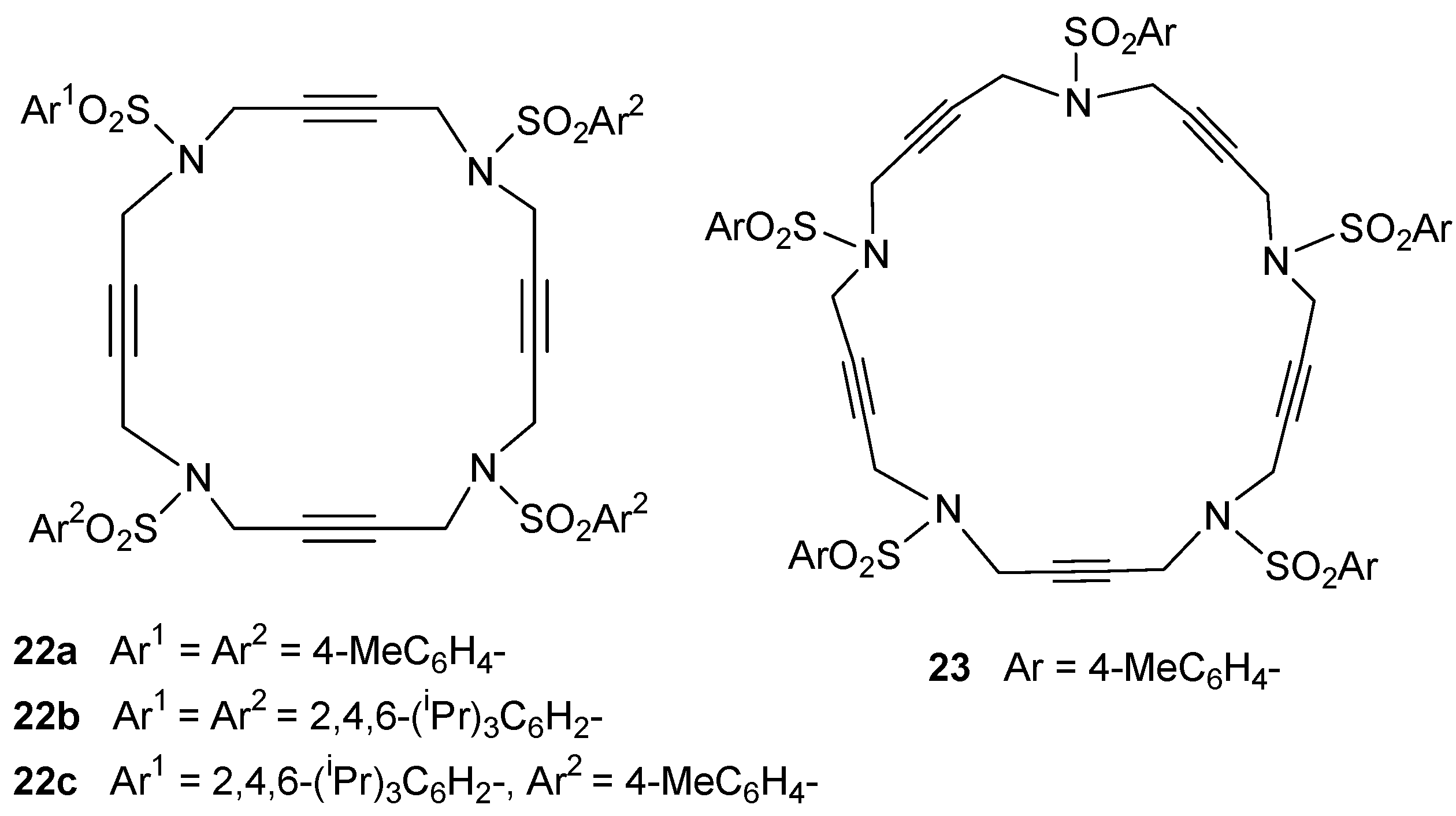
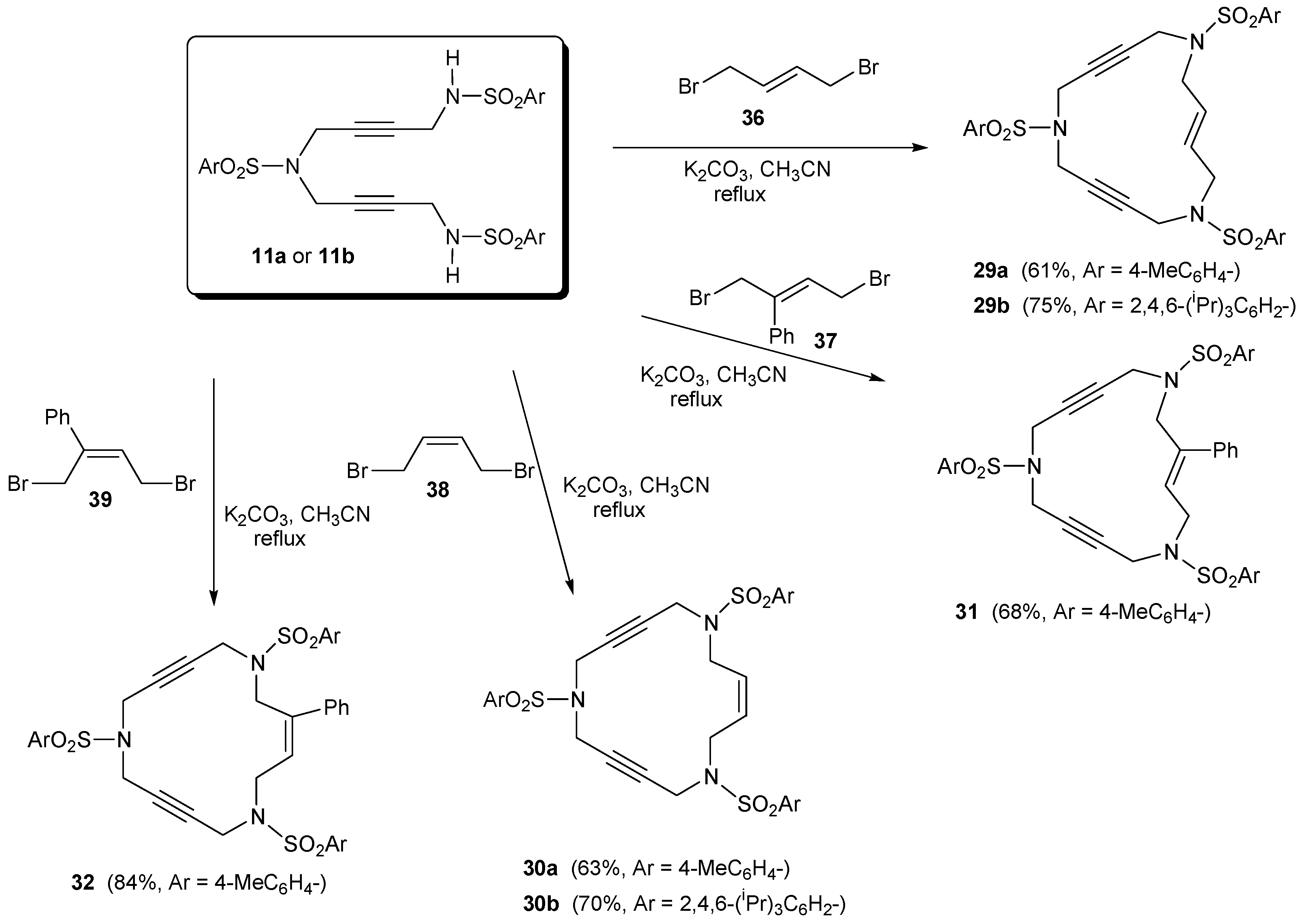
2.1. Preparation of tri-, tetra-, and pentaacetylenic azamacrocycles






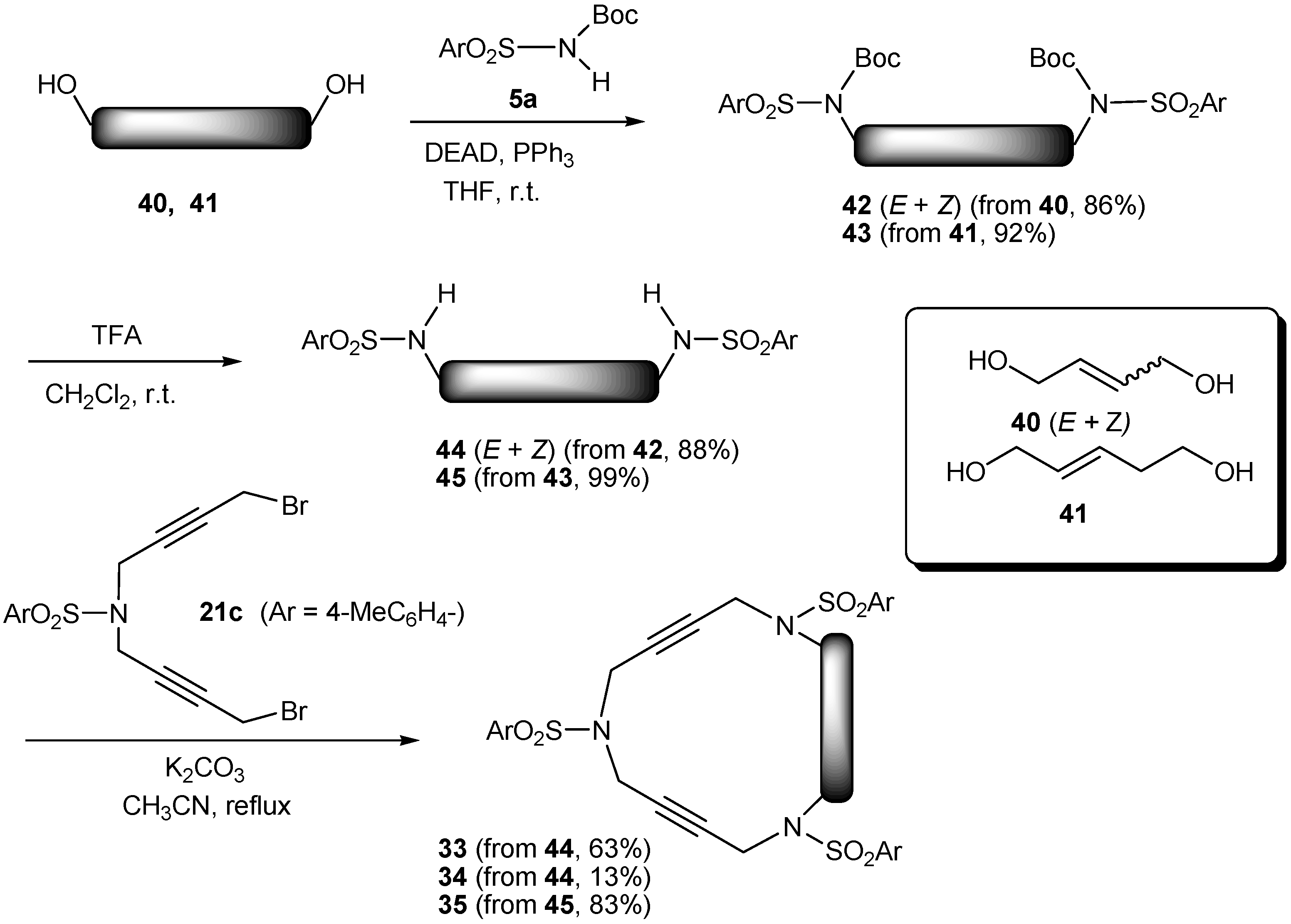
2.2. Preparation of enediyne azamacrocycles



3. [2+2+2] Cycloaddition Reactions
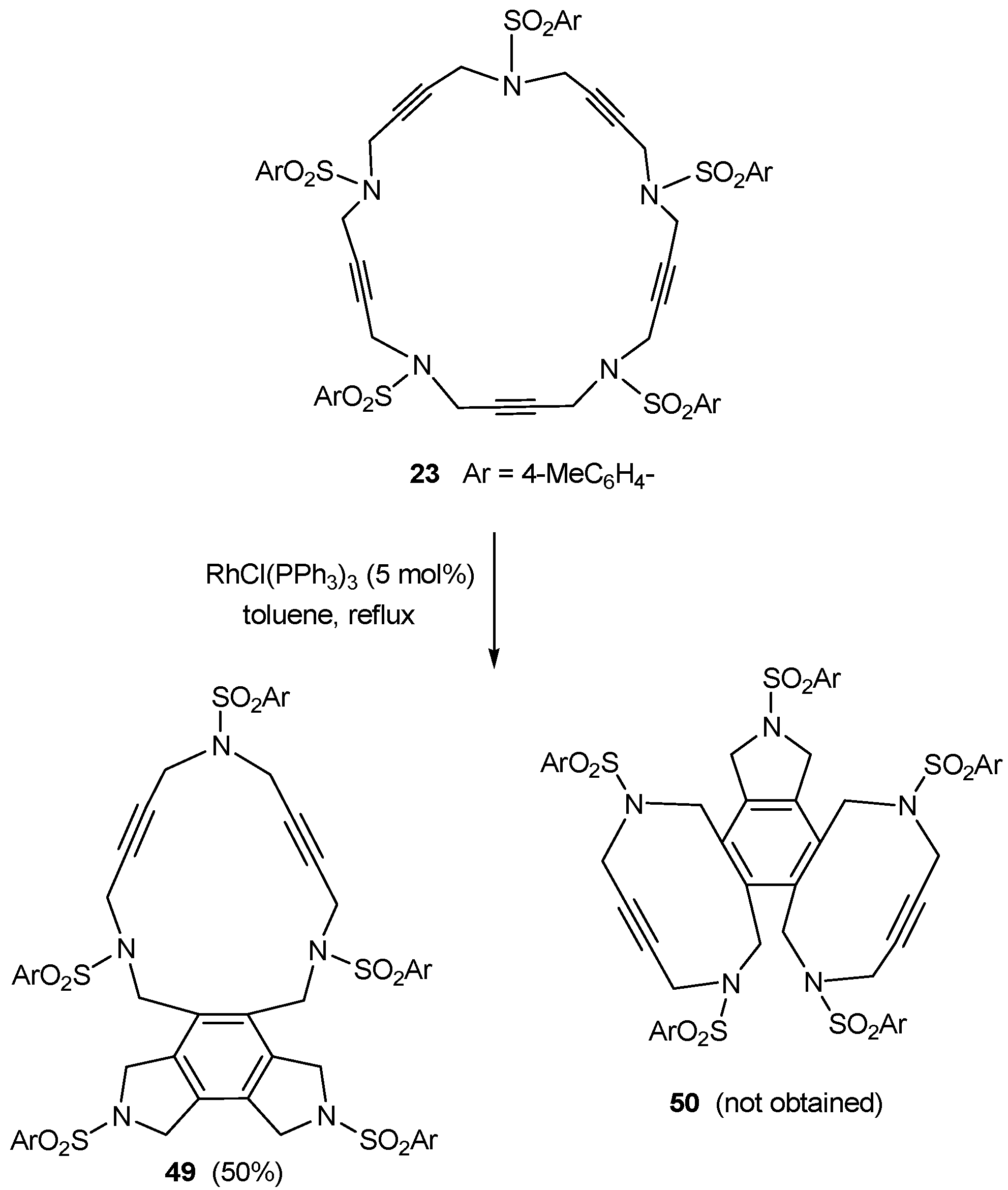
3.1. Cycloaddition reactions of tri-, tetra-, and pentaacetylenic azamacrocycles


| Entry | MCC | Catalyst(% molar) | Reaction conditions | Product | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | 1a | Pd(PPh3)4 (110) | toluene, reflux, 22 h | 46a | 54 | [38] |
| 2 | 1b | Pd(PPh3)4 (110) | toluene, reflux, 24 h | 46b | 54 | [39] |
| 3 | 1c | Pd(PPh3)4 (110) | toluene, reflux, 24 h | 46c | 65 | [39] |
| 4 | 1d | Pd(PPh3)4 (110) | toluene, reflux, 24 h | 46d | 54 | [39] |
| 5 | 1e | Pd(PPh3)4 (110) | toluene, reflux, 24 h | 46e | 45 | [39] |
| 6 | 1b | CpCo(CO)2 (5) | decane, 140 ºC, 3.5 h | 46b | 44 | [39] |
| 7 | 1b | CpCo(CO)2 (100) | decane, 140 ºC, 1 h | 46b | 88 | [39] |
| 8 | 1b | Grubbs’ cat.a (7) | toluene, reflux, 22 h | 46b | 36 | [39] |
| 9 | 1d | Grubbs’ cat.a (7) | toluene, reflux, 22 h | 46d | 42 | [39] |
| 10 | 1b | Grubbs’ cat.a (20) | toluene, reflux, 22 h | 46b | 36 | [39] |
| 11 | 1a | RhCl(CO)(PPh3)2 (5) | toluene, 65 ºC, 24 h | 46a | 88 | [39] |
| 12 | 1b | RhCl(CO)(PPh3)2 (5) | toluene, 65 ºC, 18 h | 46b | 96 | [39] |
| 13 | 1d | RhCl(CO)(PPh3)2 (5) | toluene, 65 ºC, 24 h | 46d | 89 | [39] |
| 14 | 1b | RhCl(CO)(PPh3)2 (1) | toluene, 65 ºC, 72 h | 46b | 80 | [39] |
| 15 | 1a | RhCl(PPh3)3 (10) | toluene, r.t., 1.5 h | 46a | 84 | b |
| 16 | 2 | RhCl(PPh3)3 (10) | toluene, r.t., 26 h | 47 | 91 | b |
| 17 | 2 | RhCl(PPh3)3 (10) | toluene, 60 ºC, 24 h | 47 | 99 | [41] |
| 18 | 3 | RhCl(PPh3)3 (5) | toluene, 90 ºC, 28 h | 48 | 81 | [41] |

| Entry | Catalyst (% molar) | Reaction conditions | Yield of 46b (%) | Ref. |
|---|---|---|---|---|
| 1 | RhCl(IiPr)(cod)a (5) | CH2Cl2, r.t., 7d | 90 | [42] |
| 2 | RhCl(IiPr)(cod)a (5) | toluene, 50ºC, 48h | 98 | [42] |
| 3 | RhCl(IMes)(cod)b (5) | toluene, 90ºC , 24h | 97 | [42] |
| 4 | RhCl(PPh3)3 (5) | n-Bu4NBr,130ºC, 7h | 80 | [40] |
| 5 | Recycling entry 4 | 24h | 39 | [40] |
| 6 | Recycling entry 5 | 24h | 24 | [40] |
| 7 | RhCl(PPh3)3 (10) | n-Bu4NBr,130ºC, 3h | 82 | [40] |
| 8 | Recycing entry 7 | 24h | 61 | [40] |
| 9 | PdCl2 (10) | n-Bu4NBr,130ºC, 24h | 86 | [40] |
| 10 | Recycling entry 9 | 24h | 36 | [40] |

3.2. Cycloaddition reactions of enediyne azamacrocycles

| Entry | MCC | Reaction conditions | Product | Yield (%) | Ref. |
|---|---|---|---|---|---|
| 1 | 29a | cat. A (5% molar), 90 ºC, 24 h |  | 98 | [39,41] |
| 51a (Ar = 4-MeC6H4-) | |||||
| 2 | 29b | cat. A (5% molar), 90 ºC, 24 h | 51b (Ar = 2,4,6-(iPr)3C6H2-) | 80 | [39] |
| 3 | 30a | cat. A (5% molar), 90 ºC, 24 h |  | 79 | [39] |
| 52a (Ar = 4-MeC6H4-) | |||||
| 4 | 30b | cat. A (5% molar), 90 ºC, 24 h | 52b (Ar = 2,4,6-(iPr)3C6H2-) | 68 | [39] |
| 5 | 29b | cat. B (5% molar), 90 ºC, 24 h | 51b (Ar = 2,4,6-(iPr)3C6H2-) | 80 | [39] |
| 6 | 29b | cat. C (5% molar), 50 ºC, 3 d | 51b (Ar = 2,4,6-(iPr)3C6H2-) | 98 | [42] |
| 7 | 31 | cat. B (5% molar), reflux, 24 h |  | 95 | [41] |
| 53 | |||||
| 8 | 32 | cat. B (5% molar), reflux, 24 h |  | 71 | [41] |
| 54 | |||||
| 9 | 33 | cat. B (10% molar), 80 ºC, 5 h |  | 90 | [41] |
| 55 | |||||
| 10 | 34 | cat. B (10% molar), 80 ºC, 5 h |  | 87 | [41] |
| 56 | |||||
| 11 | 35 | cat. B (10% molar), 60 ºC, 4 h |  | 98 | [41] |
| 57 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | MCC | Catalyst (10% molar) | Reaction conditions | Product | Yield (%) | e.e (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | 29a |  | toluene, 65ºC, 24h | 51a | 95 | 44 | [41,44] |
| 2 | 33 |  | toluene, 65ºC, 24h | 55 | 46 | 41 | [41,44] |
| 3a | 29a |  | CH2Cl2, r.t., 28h | 51a | 77 | 48 | [44] |
| 4a | 29a |  | toluene, r.t., 5.5h | 51a | 79 | 50 | [44] |
| 5a | 33 |  | toluene, r.t., 5.5h | 55 | 94 | 7 | [44] |
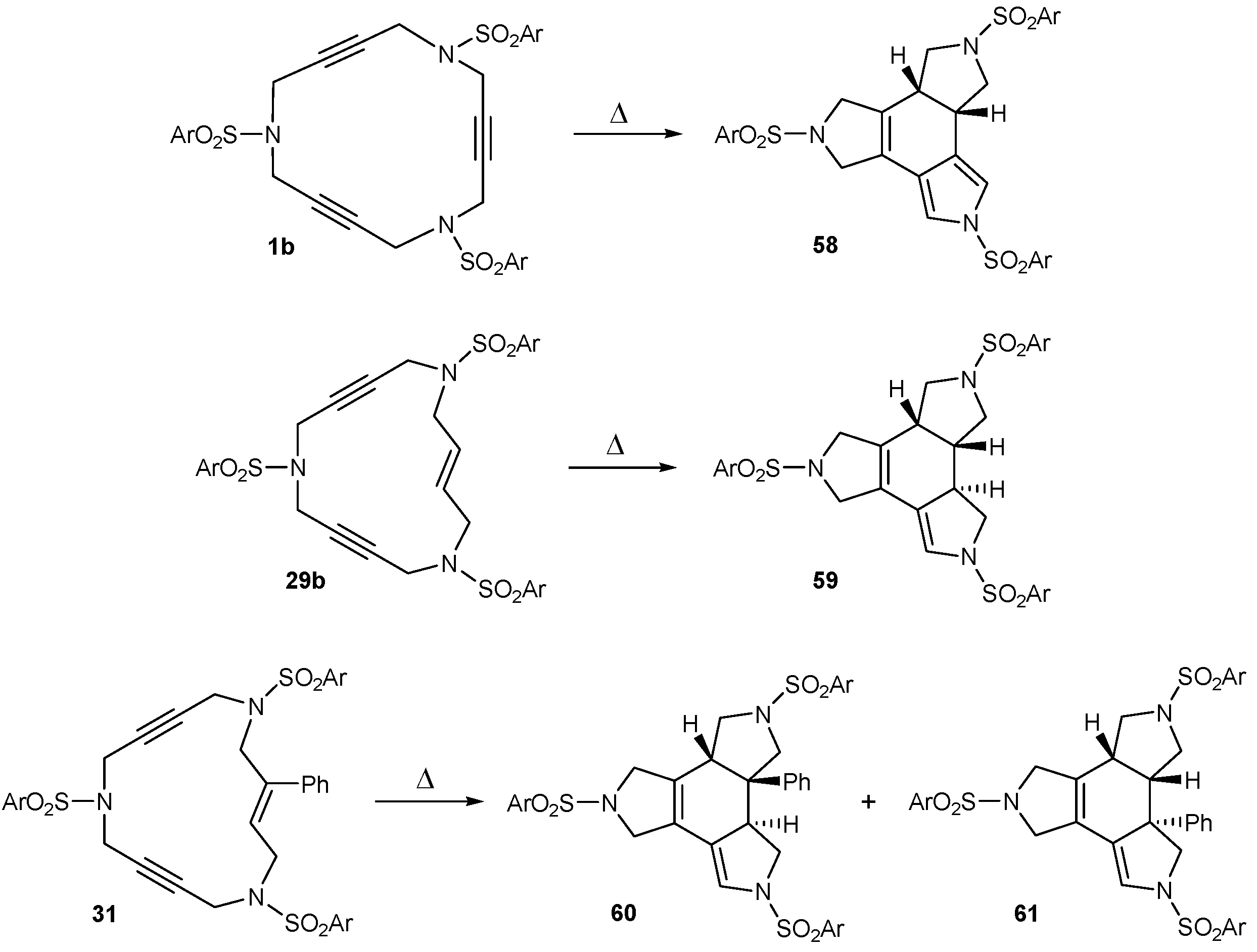
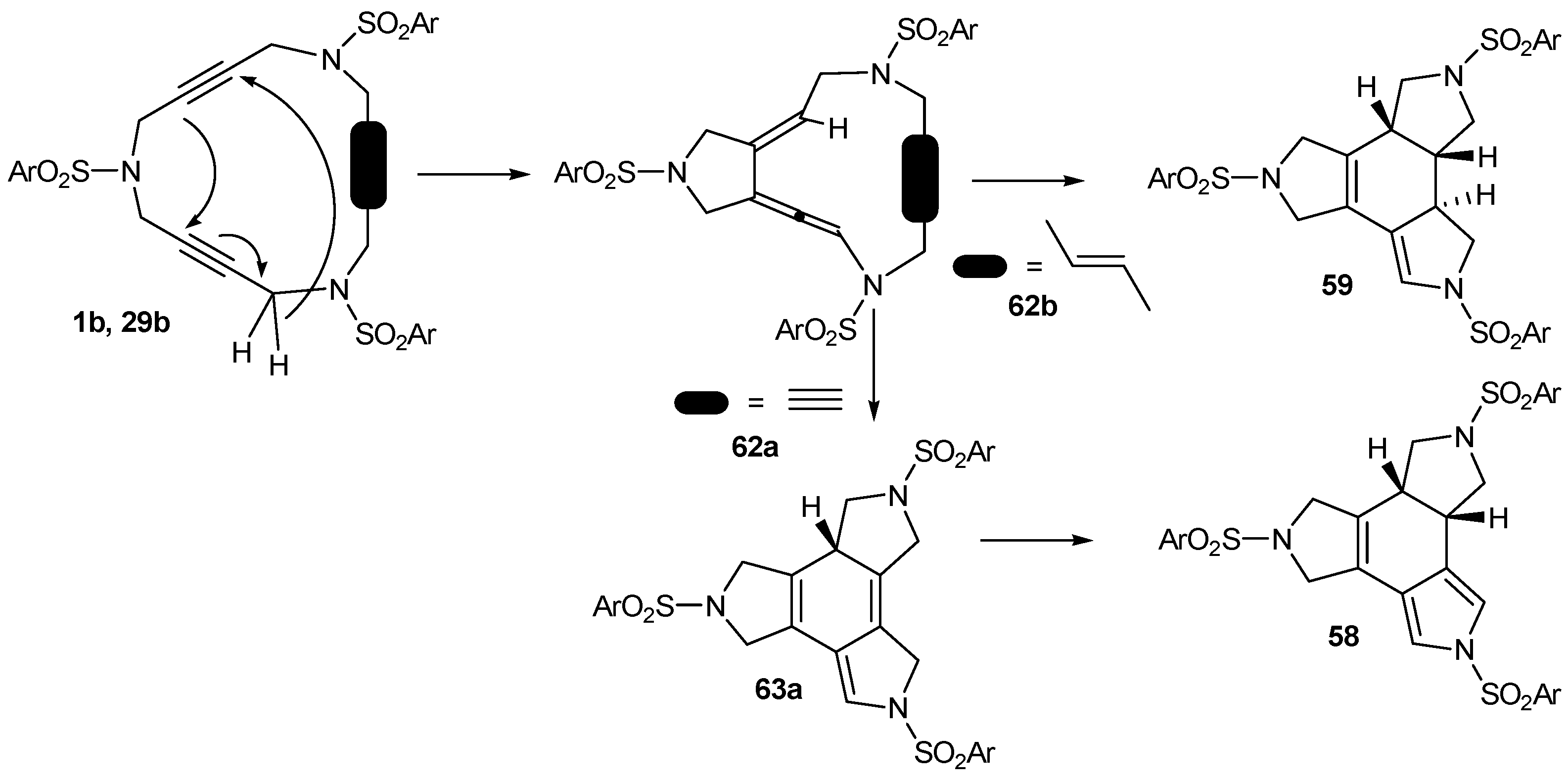
3.3. Cycloaddition reactions of triacetylene and enediyne azamacrocycles in the absence of transition metals

| Entry | Substrate | Additive | Reaction conditions | Product | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1b | --- | toluene, 110 ºC, 30 h | 58 | 32 |
| 2 | 29b | --- | toluene, 110 ºC, 6 d | 59 | 45 |
| 3 | 1b | 1,4-CHD | toluene, 110 ºC, 60 h | 58 | 77 |
| 4 | 29b | 1,4-CHD | toluene, 110 ºC, 6 d | 59 | 78 |
| 5 | 31 | 1,4-CHD | toluene, 110 ºC, 11 d | 60+61 | 81a |
| 6 | 1b | --- | chlorobenzene, 110 ºC, 30 h | 58 | 62 |
| 7 | 29b | --- | chlorobenzene, 110 ºC, 60 h | 59 | 60 |

4. Concluding Remarks
Acknowledgements
References and Notes
- Vollhardt, K.P.C. Cobalt-mediated [2+2+2]-cycloadditions: A maturing synthetic strategy [New Synthetic Methods (43)]. Angew. Chem. Int. Ed. Engl. 1984, 23, 539–556. [Google Scholar] [CrossRef]
- Schore, N.E. Transition metal-mediated cycloaddition reactions of alkynes in organic synthesis. Chem. Rev. 1988, 88, 1081–1119. [Google Scholar]
- Lautens, M.; Klute, W.; Tam, W. Transition metal-mediated cycloaddition reactions. Chem. Rev. 1996, 96, 49–92. [Google Scholar]
- Saito, S.; Yamamoto, Y. Recent advances in the transition-metal-catalyzed regioselective approaches to polysubstituted benzene derivatives. Chem. Rev. 2000, 100, 2901–2915. [Google Scholar] [CrossRef]
- Yamamoto, Y. Recent advances in intramolecular alkyne cyclotrimerization and its applications. Curr. Org. Chem. 2005, 9, 503–519. [Google Scholar] [CrossRef]
- Kotha, S.; Brahmachary, E.; Lahiri, K. Transition metal catalyzed [2+2+2] ccloaddition and application in organic synthesis. Eur. J. Org. Chem. 2005, 4741–4767. [Google Scholar]
- Chopade, P.R.; Louie, J. [2+2+2] Cycloaddition reactions catalyzed by transition metal complexes. Adv. Synth. Catal. 2006, 348, 2307–2327. [Google Scholar] [CrossRef]
- Gandon, V.; Aubert, C.; Malacria, M. Recent progress in cobalt-mediated [2+2+2] cycloaddition reactions. Chem. Commun. 2006, 2209–2217. [Google Scholar]
- Tanaka, K. Cationic rhodium(I)/BINAP-type bisphosphine complexes: versatile new catalysts for highly chemo-, regio-, and enantioselective [2+2+2] cycloadditions. Synlett 2007, 1977–1993. [Google Scholar] [CrossRef]
- Agenet, N.; Buisine, O.; Slowinski, F.; Gandon, V.; Aubert, C.; Malacria, M. Cotrimerizations of acetylenic compounds. Org. React. 2007, 68, 1–302. [Google Scholar]
- Shibata, T.; Tsuchikama, K. Recent advances in enantioselective [2+2+2] cycloaddition. Org. Biomol. Chem. 2008, 6, 1317–1323. [Google Scholar] [CrossRef]
- Galan, B.R.; Rovis, T. Beyond Reppe: building substituted arenes by [2+2+2] cycloadditions of alkynes. Angew. Chem. Int. Ed. 2009, 48, 2830–2834. [Google Scholar] [CrossRef]
- Tanaka, K. Transition-metal-catalyzed enantioselective [2+2+2] cycloadditions for the synthesis of axially chiral biaryls. Chem. Asian J. 2009, 4, 508–518. [Google Scholar] [CrossRef]
- Ojima, I.; Vu, A.T.; McCullagh, J.V.; Kinoshita, A. Rhodium-catalyzed intramolecular silylcarbotricyclization (SiCaT) of triynes. J. Am. Chem. Soc. 1999, 121, 3230–3231. [Google Scholar] [CrossRef]
- Hoven, G.B.; Efskind, J.; Rømming, C.; Undheim, K. Ru(II)-catalyzed cascade reactions in stereocontrolled construction of rigid as-indacene-bridged bis(α-amino acid) derivatives. J. Org. Chem. 2002, 67, 2459–2463. [Google Scholar] [CrossRef]
- Kinoshita, H.; Shinokubo, H.; Oshima, K. Rhodium-catalyzed [2+2+2] cyclotrimerization in an aqueous-organic biphasic system. J. Am. Chem. Soc. 2003, 125, 7784–7785. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Arakawa, T.; Ogawa, R.; Itoh, K. Ruthenium(II)-catalyzed selective intramolecular [2+2+2] alkyne cyclotrimerizations. J. Am. Chem. Soc. 2003, 125, 12143–12160. [Google Scholar]
- Bong, D.T.-Y.; Chan, E.W.L.; Diercks, R.; Dosa, P.I.; Haley, M.M.; Matzger, A.J.; Miljanic, O.Š.; Vollhardt, K.P.C.; Bond, A.D.; Teat, S.J.; Stanger, A. Syntheses of syn and anti doublebent [5]phenylene. Org. Lett. 2004, 6, 2249–2252. [Google Scholar]
- Saino, N.; Kogure, D.; Okamoto, S. Intramolecular cyclotrimerization of triynes catalyzed by N-heterocyclic carbene-CoCl2/Zn or -FeCl3/Zn. Org. Lett. 2005, 7, 3065–3067. [Google Scholar] [CrossRef]
- Shibata, T.; Tsuchikama, K.; Otsuka, M. Enantioselective intramolecular [2+2+2] cycloaddition of triynes for the synthesis of atropisomeric chiral ortho-diarylbenzene derivatives. Tetrahedron Asymmetry 2006, 17, 614–619. [Google Scholar] [CrossRef]
- Chouraqui, G.; Petit, M.; Phansavath, P.; Aubert, C.; Malacria, M. From an acyclic, polyunsaturated precursor to the polycyclic taxane ring system: the [4+2]/[2+2+2] and [2+2+2]/[4+2] cyclization strategies. Eur. J. Org. Chem. 2006, 1413–1421. [Google Scholar]
- Tanaka, K.; Sagae, H.; Toyoda, K.; Noguchi, K.; Hirano, M. Enantioselective synthesis of planar-chiral metacyclophanes through rhodium-catalyzed alkyne cyclotrimerization. J. Am. Chem. Soc. 2007, 129, 1522–1523. [Google Scholar] [CrossRef]
- Shibata, T.; Uchiyama, T.; Endo, K. Enantioselective synthesis of chiral tripodal cage compounds by [2+2+2] cycloaddition of branched triynes. Org. Lett. 2009, 11, 3906–3908. [Google Scholar]
- Nicolaus, N.; Strauss, S.; Neudörfl, J.-M.; Prokop, A.; Schmalz, H.-G.A. [2+2+2]-Cycloaddition approach toward 6-oxa-allocolchicinoids with apoptosis-inducing activity. Org. Lett. 2009, 11, 341–344. [Google Scholar] [CrossRef]
- Songis, O.; Mísek, J.; Schmid, M.B.; Kollárovic, A.; Stará, I.G.; Saman, D.; Císarová, I.; Starý, I. A Versatile synthesis of functionalized pentahelicenes. J. Org. Chem. 2010, 75, 6889–6899. [Google Scholar]
- Severa, L.; Adriaenssens, L.; Vávra, J.; Saman, D.; Císarová, I.; Fiedler, P.; Teplý, F. Highly modular assembly of cationic helical scaffolds: rapid synthesis of diverse helquats via differential quaternization. Tetrahedron 2010, 66, 3537–3552. [Google Scholar] [CrossRef]
- Sternberg, E.D.; Vollhardt, K.P.C. Cobalt-mediated [2+2+2] cycloadditions en route to natural products: A novel total synthesis of steroids via the one-step construction of the B,C,D framework from an A-ring precursor. J. Org. Chem. 1982, 47, 3447–3453. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kuwabara, S.; Ando, Y.; Nagata, H.; Nishiyama, H.; Itoh, K. Palladium(0)-catalyzed cyclization of electron-deficient enynes and enediynes. J. Org. Chem. 2004, 69, 6697–6705. [Google Scholar] [CrossRef]
- Schelper, M.; Buisine, O.; Kozhushkov, S.; Aubert, C.; de Meijere, A.; Malacria, M. Cobalt(I)-mediated intramolecular [2+2+2] cocyclizations of (methylenecyclopropyl)diynes as an easy access to cyclopropanated oligocycles. Eur. J. Org. Chem. 2005, 3000–3007. [Google Scholar]
- Bennacer, B.; Fujiwara, M.; Lee, S.-Y.; Ojima, I. Silicon-initiated carbonylative carbotricyclization and [2+2+2+1] cycloaddition of enediynes catalyzed by rhodium complexes. J. Am. Chem. Soc. 2005, 127, 17756–17767. [Google Scholar] [CrossRef]
- Kaloko, J.J.; Teng, Y.-H.G.; Ojima, I. One-step formation of fused tetracyclic skeletons from cyclohexene-diynes and carbon monoxide through Rh(I)-catalyzed [2+2+2+1] cycloaddition reaction. Chem. Commun. 2009, 4569–4571. [Google Scholar]
- Geny, A.; Gaudrel, S.; Slowinski, F.; Amatore, M.; Chouraqui, G.; Malacria, M.; Aubert, C.; Gandon, V. A straightforward procedure for the [2+2+2] cycloaddition of enediynes. Adv. Synth. Catal. 2009, 351, 271–275. [Google Scholar] [CrossRef]
- Jones, A.L.; Snyder, J.K. Intramolecular rhodium-catalyzed [2+2+2] cyclizations of diynes with enones. J. Org. Chem. 2009, 74, 2907–2910. [Google Scholar]
- Barkovich, A.J.; Vollhardt, K.P.C. 1,5,9-Cyclododecatriyne. Synthesis and conversion to intermediate 1,2:3,4:5,6-tricyclobutabenzene. J. Am. Chem. Soc. 1976, 98, 2667–2668. [Google Scholar] [CrossRef]
- Sakurai, H.; Nakadaira, Y.; Hosomi, A.; Eriyama, Y.; Hirama, K.; Kabuto, C. Intramolecular cyclotrimerization of macrocylic and acyclic triynes with group 6 metal carbonyls. The formation of fulvene and benzene. J. Am. Chem. Soc. 1984, 106, 8315–8316. [Google Scholar] [CrossRef]
- Sakurai, H. Novel π-electron systems derived from silicon-containing macrocyclic polyacetylenes. Pure Appl. Chem. 1996, 68, 327–333. [Google Scholar] [CrossRef]
- Ebata, K.; Matsuo, T.; Inoue, T.; Otsuka, Y.; Kabuto, C.; Sekiguchi, A.; Sakurai, H. Intramolecular oligomerization of disialkylene {-Me(2)Si(CH2)(n)SiMe(2)} bridged cyclic triacetylenes. Chem. Lett. 1996, 1053–1054. [Google Scholar]
- Pla-Quintana, A.; Roglans, A.; Torrent, A.; Moreno-Mañas, M.; Benet-Buchholz, J. Synthesis of nitrogen-containing 15-membered triacetylenic macrocycles. Stable complex with Palladium(0). Organometallics 2004, 23, 2762–2767. [Google Scholar]
- Torrent, A.; González, I.; Pla-Quintana, A.; Roglans, A.; Moreno-Mañas, M.; Parella, T.; Benet-Buchholz, J. Transition metal-mediated intramolecular [2+2+2] cycloisomerizations of cyclic triynes and enediynes. J. Org. Chem. 2005, 70, 2033–2041. [Google Scholar] [CrossRef]
- González, I.; Bouquillon, S.; Roglans, A.; Muzart, J. Palladium and rhodium-catalyzed intramolecular [2+2+2] cycloisomerizations in molten tetrabutylammonium bromide. Tetrahedron Lett. 2007, 48, 6425–6428. [Google Scholar] [CrossRef]
- Brun, S.; Garcia, L.; González, I.; Torrent, A.; Dachs, A.; Pla-Quintana, A.; Parella, T.; Roglans, A. Fused tetracycles with a benzene or cyclohexadiene core: [2+2+2] cycloadditions on macrocyclic systems. Chem. Commun. 2008, 4339–4341. [Google Scholar]
- González, I.; Pla-Quintana, A.; Roglans, A. Rhodium N-heterocyclic carbene complexes as effective catalysts for [2+2+2]-cycloaddition reactions. Synlett 2009, 2844–2848. [Google Scholar]
- Dachs, A.; Torrent, A.; Roglans, A.; Parella, T.; Osuna, S.; Solà, M. Rhodium(I)-catalysed intramolecular [2+2+2] cyclotrimerisations of 15-, 20- and 25-membered azamacrocycles: experimental and theoretical mechanistic studies. Chem. Eur. J. 2009, 15, 5289–5300. [Google Scholar]
- Brun, S.; Parera, M.; Pla-Quintana, A.; Roglans, A.; León, T.; Achard, T.; Solà, J.; Verdaguer, X.; Riera, A. Chiral N-phosphino sulfinamide ligands in rhodium(I)-catalyzed [2+2+2] cycloaddition reactions. Tetrahedron 2010, 66, 9032–9040. [Google Scholar]
- Moreno-Mañas, M.; Pleixats, R.; Roglans, A.; Sebastián, R.M.; Vallribera, A. 15-Membered triolefinic macrocycles, their coordination chemistry with transition metals, and the catalytic properties of their palladium metal complexes. A review. ARKIVOC 2004, (iv), 109–129. [Google Scholar]
- Moreno-Mañas, M.; Pleixats, R.; Sebastián, R.M.; Vallribera, A.; Roglans, A. Organometallic chemistry of 15-membered tri-olefinic macrocycles: catalysis by palladium(0) complexes in carbon-carbon bond-forming reactions. J. Organomet. Chem. 2004, 689, 3669–3684. [Google Scholar] [CrossRef]
- Compounds of type 21 were prepared by alkylation of the corresponding sulfonamide with an excess of dihalo-2-butyne (4 equiv.).
- Negishi, E.-I.; Harring, L.S.; Owczarczyk, Z.; Mohamud, M.M.; Ay, M. Cyclic cascade carbopalladation reactions as a route to benzene and fulvene derivatives. Tetrahedron Lett. 1992, 33, 3253–3256. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nagata, A.; Nagata, H.; Ando, Y.; Arikawa, Y.; Tatsumi, K.; Itoh, K. Palladium(0)-catalyzed intramolecular [2+2+2] alkyne cyclotrimerizations with electron-deficient diynes and triynes. Chem. Eur. J. 2003, 9, 2469–2483. [Google Scholar] [CrossRef]
- Peters, J.-U.; Blechert, S. Ruthenium-catalysed conversion of triynes to benzene derivatives—a novel metathesis cascade. Chem. Commun. 1997, 1983–1984. [Google Scholar]
- Das, S.K.; Roy, R. Mild ruthenium-catalyzed intermolecular alkyne cyclotrimerization. Tetrahedron Lett. 1999, 40, 4015–4018. [Google Scholar] [CrossRef]
- Witulski, B.; Stengel, T.; Fernández-Hernández, J.M. Chemo- and regioselective crossed alkyne cyclotrimerisation of 1,6-diynes with terminal monoalkynes mediated by Grubbs’ catalyst or Wilkinson’s catalyst. Chem. Commun. 2000, 1965–1966. [Google Scholar]
- Grigg, R.; Scott, R.; Stevenson, P. Rhodium catalysed [2+2+2] cycloadditions of acetylenes. Tetrahedron Lett. 1982, 23, 2691–2692. [Google Scholar] [CrossRef]
- Grigg, R.; Scott, R.; Stevenson, P. Rhodium-catalysed [2+2+2]-cycloadditions of acetylenes. J. Chem. Soc. Perkin Trans 1 1988, 1357–1364. [Google Scholar]
- Müller, E. The diyne reaction of 1,4-, 1,5-, 1,6-, and 1,7-diynes via transition metal complexes to new compounds. Synthesis 1974, 761–774. [Google Scholar] [CrossRef]
- Ma, S.; Ni, B. Intramolecular triple Heck reaction. An efficient entry to fused tetracycles with a benzene core. J. Org. Chem. 2002, 67, 8280–8283. [Google Scholar] [CrossRef]
- Shibata, T.; Kurokawa, H.; Kanda, K. Enantioselective intramolecular [2+2+2] cycloaddition of enediynes for the synthesis of chiral cyclohexa-1,3-dienes. J. Org. Chem. 2007, 72, 6521–6525. [Google Scholar] [CrossRef]
- Tanaka, K.; Nishida, G.; Sagae, H.; Hirano, M. Enantioselective synthesis of C2-symmetric dimethyl cyclohexadienedicarboxylates through cationic rhodium(I)/modified-BINAP-catalyzed [2+2+2] cycloadditions. Synlett 2007, 1426–1430. [Google Scholar]
- González, I.; Pla-Quintana, A.; Roglans, A.; Dachs, A.; Solà, M.; Parella, T.; Farjas, J.; Roura, P.; Lloveras, V.; Vidal-Gancedo, J. Ene reactions between two alkynes? Doors open to thermally induced cycloisomerization of macrocyclic triynes and enediynes. Chem. Commun. 2010, 46, 2944–2946. [Google Scholar]
- Robinson, J.M.; Sakai, T.; Okano, K.; Kitawaki, T.; Danheiser, R.L. Formal [2+2+2] cycloaddition strategy based on an intramolecular propargylic ene reaction/Diels−Alder cycloaddition cascade. J. Am. Chem. Soc. 2010, 132, 11039–11041. [Google Scholar]
- Sakai, T.; Danheiser, R.L. Cyano Diels−Alder and cyano ene reactions. Applications in a formal [2+2+2] cycloaddition strategy for the synthesis of pyridines. J. Am. Chem. Soc. 2010, 132, 13203–13205. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pla-Quintana, A.; Roglans, A. [2+2+2] Cycloaddition Reactions of Macrocyclic Systems Catalyzed by Transition Metals. A Review. Molecules 2010, 15, 9230-9251. https://doi.org/10.3390/molecules15129230
Pla-Quintana A, Roglans A. [2+2+2] Cycloaddition Reactions of Macrocyclic Systems Catalyzed by Transition Metals. A Review. Molecules. 2010; 15(12):9230-9251. https://doi.org/10.3390/molecules15129230
Chicago/Turabian StylePla-Quintana, Anna, and Anna Roglans. 2010. "[2+2+2] Cycloaddition Reactions of Macrocyclic Systems Catalyzed by Transition Metals. A Review" Molecules 15, no. 12: 9230-9251. https://doi.org/10.3390/molecules15129230