Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation

Institute for Research in Biomedicine (IRB) and Department of Organic Chemistry, University of Barcelona, Baldiri Reixac, 10. 08028 Barcelona, Spain
*
Author to whom correspondence should be addressed.
Molecules 2010, 15(2), 1041-1073; https://doi.org/10.3390/molecules15021041
Submission received: 8 January 2010
/
Revised: 9 February 2010
/
Accepted: 20 February 2010
/
Published: 23 February 2010
(This article belongs to the Special Issue Asymmetric Synthesis)
Abstract
:An overview of the synthesis and applications of chiral 2,3-epoxy alcohols containing unsaturated chains is presented. One of the fundamental synthetic routes to these compounds is Sharpless asymmetric epoxidation, which is reliable, highly chemoselective and enables easy prediction of the product enantioselectivity. Thus, unsaturated epoxy alcohols are readily obtained by selective oxidation of the allylic double bond in the presence of other carbon-carbon double or triple bonds. The wide availability of epoxy alcohols with unsaturated chains, the versatility of the epoxy alcohol functionality (e.g. regio- and stereo-selective ring opening; oxidation; and reduction), and the arsenal of established alkene chemistries, make unsaturated epoxy alcohols powerful starting materials for the synthesis of complex targets such as biologically active molecules. The popularization of ring-closing metathesis has further increased their value, making them excellent precursors to cyclic compounds.

Abbreviations
| Ac | acetyl |
| ACN | acetonitrile |
| Ac2O | acetic anhydride |
| aq. | aqueous |
| 9-BBN | 9-borabicyclo[3.3.1]nonane |
| Bn | benzyl |
| Boc2O | di-tert-butyl dicarbonate |
| cat. | catalyst |
| CuTC | copper(I)-thiophene-2-carboxylate |
| Cy | cyclohexyl |
| CM | cross metathesis |
| DCC | dicyclohexylcarbodiimide |
| DDQ | 2,3-dichloro-5,6-dicyano-1,4-benzoquinone |
| de. | diastereomeric excess |
| DIPT | diisopropyl tartrate |
| DMAP | 4-dimethylaminopyridine |
| DME | dimethoxyethane |
| DMF | dimethylformamide |
| DMP | Dess-Martin periodinane: 1,1,1-triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one |
| DMPM | 3,4-dimethoxybenzyl |
| DMSO | dimethyl sulfoxide |
| dr | diastereomeric ratio |
| ee | enantiomeric excess |
| HMPA | hexamethylphosphoramide |
| Imid. | imidazole |
| Mes | 2,4,6-trimethylphenyl |
| MS | molecular sieves |
| MsCl | mesityl chloride |
| NMO | N-methyl-morpholine-N-oxide |
| nm | not measured |
| Pip. | piperidine |
| PMB | p-methoxybenzyl |
| PSA | p-TsOH.H2O: p-toluenesulfonic acid monohydrate |
| Pyr. | pyridine |
| RedAl | sodium bis(2-methoxyethoxy)aluminum hydride |
| RCM | ring-closing metathesis |
| SAE | Sharpless asymmetric epoxidation |
| TASF | tris(dimethylamino)sulfonium difluorotrimethylsilicate |
| TBAF | tetra-n-butylammonium fluoride |
| TBS | tert-butyldimethylsilyl |
| TBDPS | tert-butyldiphenylsilyl |
| TEMPO | 2,2,6,6-tetramethylpiperidine-1-oxyl |
| TES | triethylsilyl |
| Tf | triflate |
| THF | tetrahydrofuran |
| TIPS | triisopropylsilyl |
| TMS | trimethylsilyl |
| TPAP | tetrapropylammonium perruthenate |
| TsCl | m-toluenesulfonyl chloride |
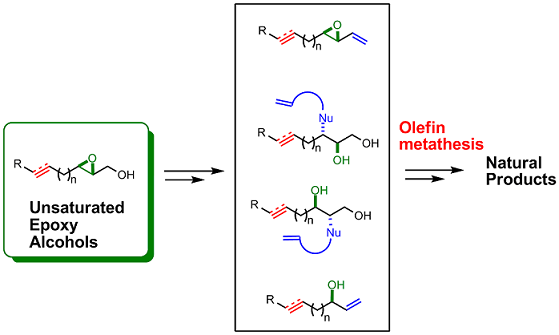
1. Introduction
The Sharpless asymmetric epoxidation (SAE) [1,2,3] is among the most powerful enantioselective catalytic reactions available. This well-known reaction comprises enantioselective epoxidation of an allyl alcohol using tert-butyl hydroperoxide as oxidant. The catalyst can be easily prepared in situ by reacting titanium isopropoxide and a chiral tartrate in dichloromethane. The absolute configuration of the resulting epoxide can be easily predicted using a rule developed by Sharpless: it correlates to the enantiomer of the tartrate used. Moreover, Sharpless asymmetric epoxidation is chemoselective. For example, unsaturated epoxy alcohols can be obtained by selective oxidation of an allylic double bond in compounds containing other carbon-carbon double or triple bonds.
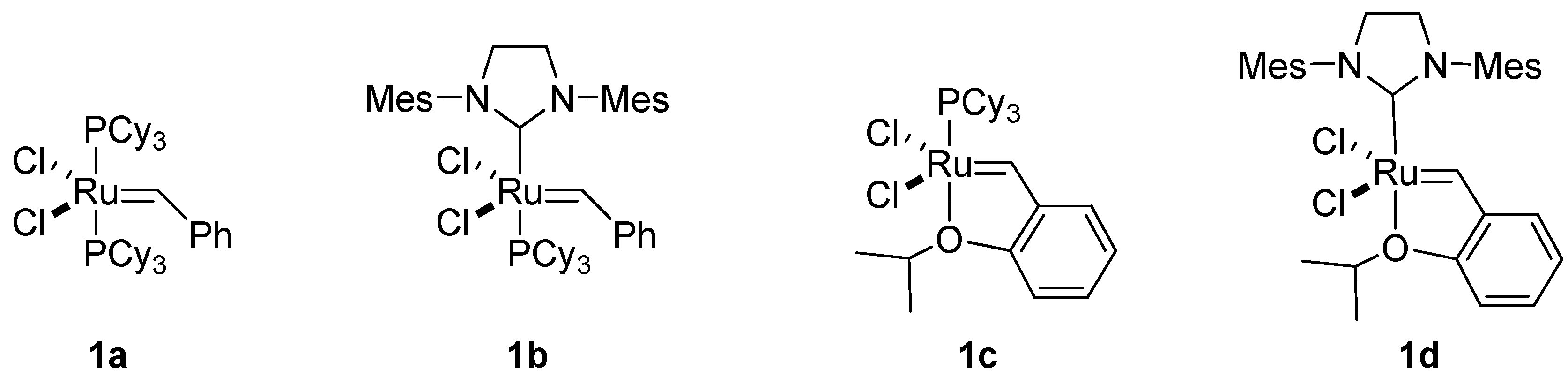
Reliable olefin metathesis reactions – chiefly, ring-closing metathesis (RCM) [4,5,6,7] and cross metathesis (CM) [8,9] – have had a major impact on organic synthesis. For example, alkene metatheses have proven invaluable for assembling complex small-molecule targets. Commercialization of the first- and second-generation Grubbs’ catalysts (1a and 1b, respectively; Figure 1), and of the first- and second-generation Hoveyda-Grubb’s catalysts [10,11] (1c and 1d, respectively; Figure 1), has greatly facilitated their use and has fostered synthetic exploitation of chiral unsaturated intermediates, which are excellent precursors for cyclic compounds. Likewise, olefin metatheses have increased the synthetic applications of chiral epoxy alcohols containing olefins in their side chain.
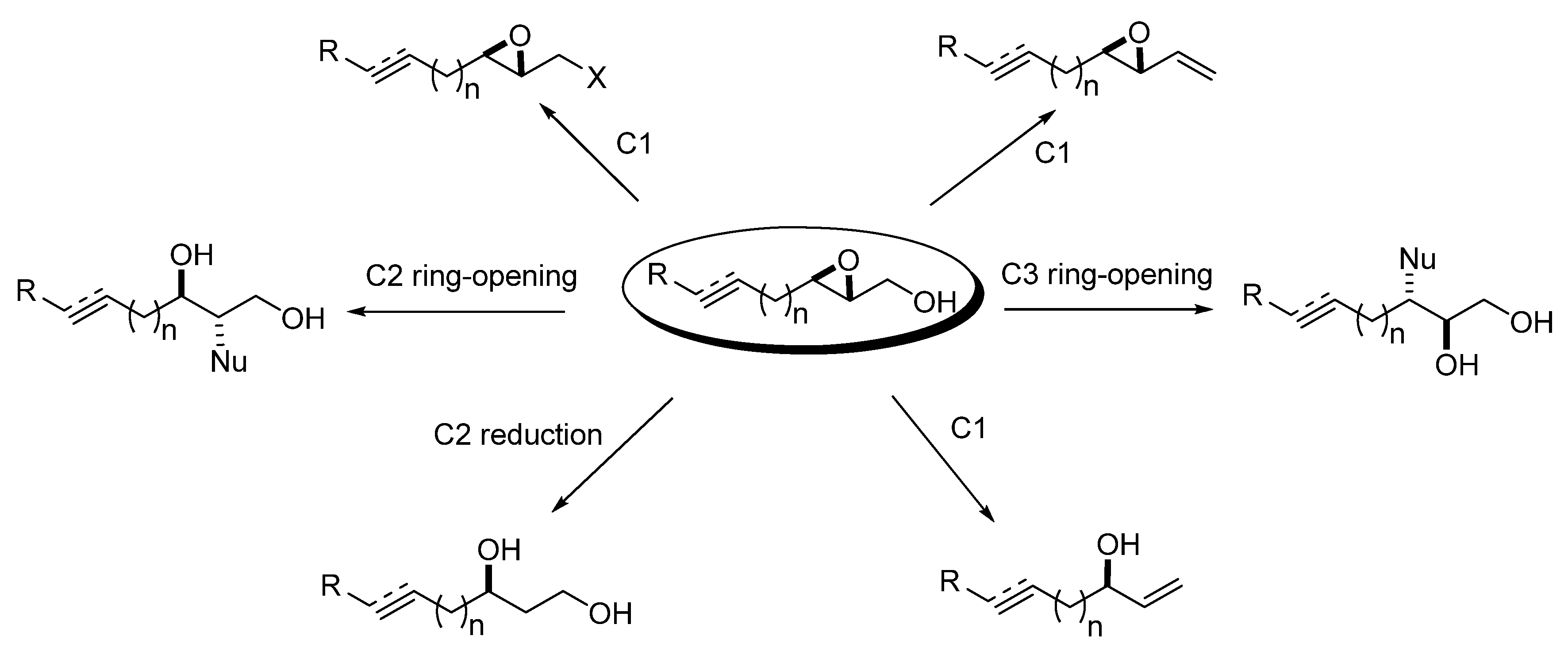
The chemistry of the epoxy alcohol fragment in unsaturated epoxy alcohols encompasses the following reactions: a) functional group transformations of the primary hydroxyl group (C1 substitution); b) olefination at C1; c) deoxygenation to an allyl alcohol; d) reduction at C2; and e) epoxide ring-opening at C2 or C3. All of these transformations generate highly functionalized products with excellent regio- and stereo-selectivities (Figure 2).
Figure 1.
Ruthenium catalysts for metatheses: first-generation Grubbs’ catalyst (1a); second- generation Grubbs’ catalyst (1b); first-generation Hoveyda-Grubbs’ catalyst (1c); and second-generation Hoveyda-Grubbs’ catalyst (1d).
Figure 1.
Ruthenium catalysts for metatheses: first-generation Grubbs’ catalyst (1a); second- generation Grubbs’ catalyst (1b); first-generation Hoveyda-Grubbs’ catalyst (1c); and second-generation Hoveyda-Grubbs’ catalyst (1d).

Figure 2.
Chemistry of unsaturated 2,3-epoxy alcohols.

2. Synthesis of Chiral Epoxy Alcohols
There are many examples in the literature of chiral unsaturated epoxides, most of which have been prepared in enantiomerically enriched form by SAE. Table 1 summarizes known epoxy alcohols that contain a double or triple bond less than four carbon atoms away from the epoxide and that were prepared by SAE. As observed, the yields are generally good (except for highly volatile or water-soluble epoxides) and the enantiomeric purities were high (90 to 99% ee).
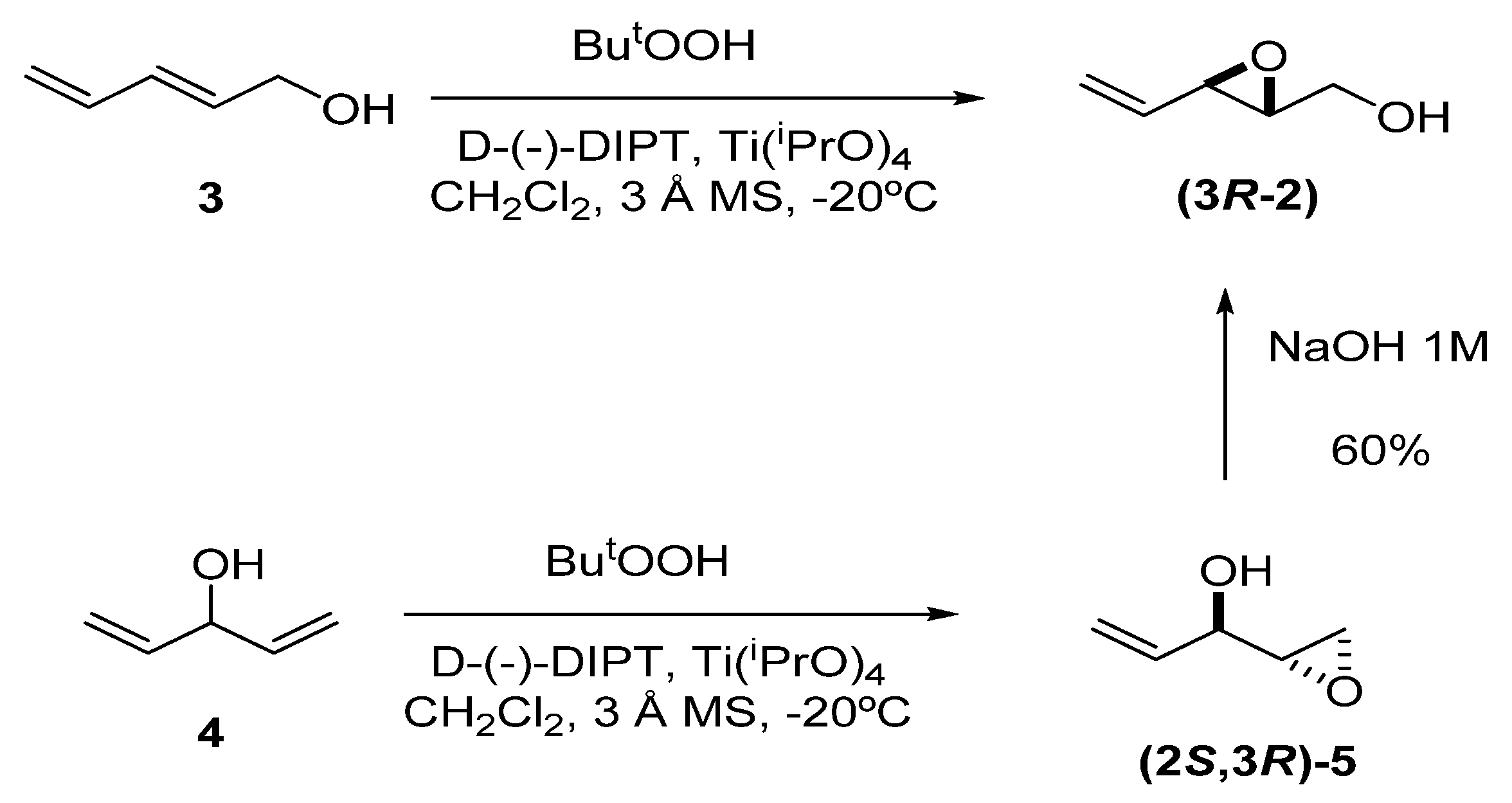
Although the simplest unsaturated epoxide 2 can be prepared by SAE of 2,4-pentadien-1-ol (3), a more convenient route is the kinetic resolution methodology developed by Jagger et al. [12,13,14]. This entails SAE of 1,4-pentadien-3-ol (4) to give epoxide 5 in excellent enantiomeric excess (>99%), followed by Payne rearrangement [15] of 5 with NaOH 1M to afford 2 (60%). The reason that Jagger’s method provides high enantiomeric excess is because the minor epoxy alcohol formed in the first reaction is removed by epoxidation to the double epoxide; thus, the stereoisomer which forms slower than its homolog, also reacts quicker, generating a double epoxide that can be removed by chromatography.
Scheme 1.
Preparation of epoxy alcohol 2 using either Sharpless asymmetric epoxidation (SAE) or the methodology developed by Jagger, which combines SAE and Payne rearrangement.
Scheme 1.
Preparation of epoxy alcohol 2 using either Sharpless asymmetric epoxidation (SAE) or the methodology developed by Jagger, which combines SAE and Payne rearrangement.


| Epoxy alcohol | Structure | Yield (%) | ee (%) | References |
|---|---|---|---|---|
| 2 |  | n.d.* | 96 | [13,57,58,85] |
| 6 |  | 50 | nd | [74] |
| 7 |  | 98 | 61 | [53] |
| 8 |  | 83 | 88 | [52] |
| 9 |  | 85 | 95 | [51] |
| 10 |  | 21** | 96 | [51] |
| 11 |  | 82 | 92 | [82],[47] |
| Z 11 |  | 82 | >99 | [28],[45] |
| 12 |  | 75 | 93.6 | [44] |
| 13 |  | 83 | 93 | [88] |
| 14 |  | 90 | nm | [41] |
| 15 |  | 72 | nm | [16] |
| 16 |  | 91 | 90 (de.) | [34] |
| 17 |  | 90 | nm | [30] |
| 18 |  | 92 | >90 | [17] |
| 19 |  | 92 | 94 | [82],[68] |
| 20 |  | 92 | 91 | [18] |
| 21 |  | nm | 63 | [42] |
| 22 |  | 99 | 92 | [19] |
| 23 |  | 72 | 98 (de) | [29] |
| 24 |  | 93 | 97 | [26] |
| 25 |  | 70 | >95 | [20] |
| 26 |  | 75 | 99 | [37] |
| 27 |  | 85 | 91 | [82],[25],[50],[49] |
| 28 |  | 91 | >94 | [21] |
| 29 |  | 92 | >99 | [33] |
* This compound was not isolated due to its high volatility and reactivity; it was used directly in the subsequent chemistry, without further purification; ** yield for three steps, starting from hexa-2,4-diyne-1,6-diol .
3. Transformations of C1
Many of the standard functional group modifications of the primary alcohol (C1) in 2,3-epoxy alcohols [22,23,24] have been used in compounds with unsaturated side chains. The most common of these are oxidation to the aldehyde, halogenation and esterification. The aldehyde offers rich synthetic potential, mainly for nucleophilic attack and olefination. The double bond of the side chain can later be exploited in various ways.
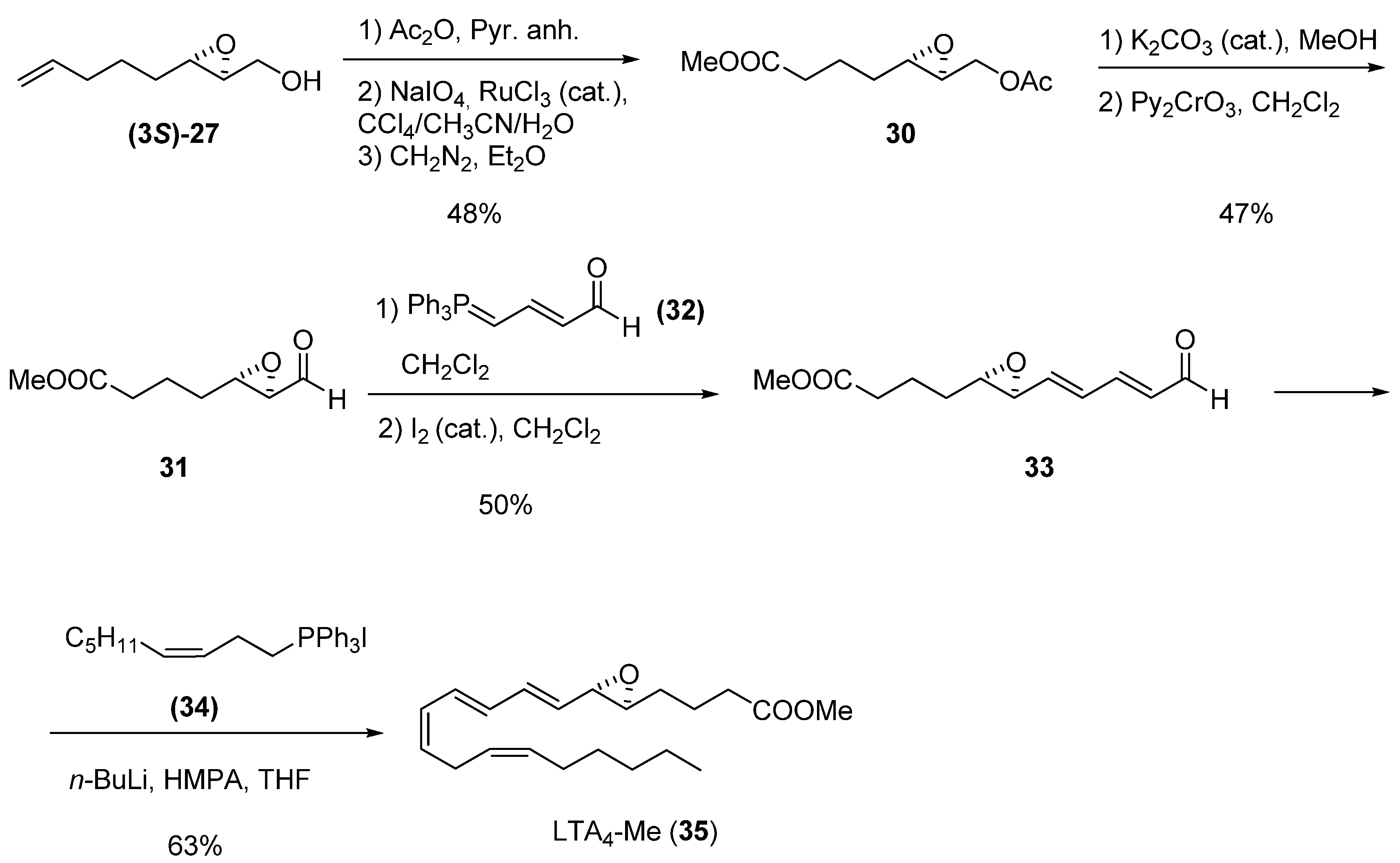
Before the RCM was developed, one of the classical uses of terminal double bonds was as latent acid derivatives. The synthesis of leukotrienes developed by Spur et al. [25] is a prime example: epoxide (3S)-27 (>94% ee) was converted into the epoxy acetate 30 by acetylation followed by oxidation to acid under Sharpless conditions (RuCl3, NaIO4), and esterification with CH2N2 in good overall yield. The acetate was readily hydrolyzed in MeOH using a catalytic amount of K2CO3 plus anhydrous Na2SO4 (no Payne rearrangement or methyl ester cleavage was observed), providing an epoxy alcohol that was oxidized with Py2CrO3 to the aldehyde 31. Crude 31 was converted into the trans- epoxydienal 33 in 50% yield. Finally, Wittig reaction with the appropriate phosphorane, 34, gave leukotriene LTA4 methyl ester (35).
Scheme 2.
Synthesis of leukotriene LTA4 methyl ester (Spur et al. [25]).
Scheme 2.
Synthesis of leukotriene LTA4 methyl ester (Spur et al. [25]).

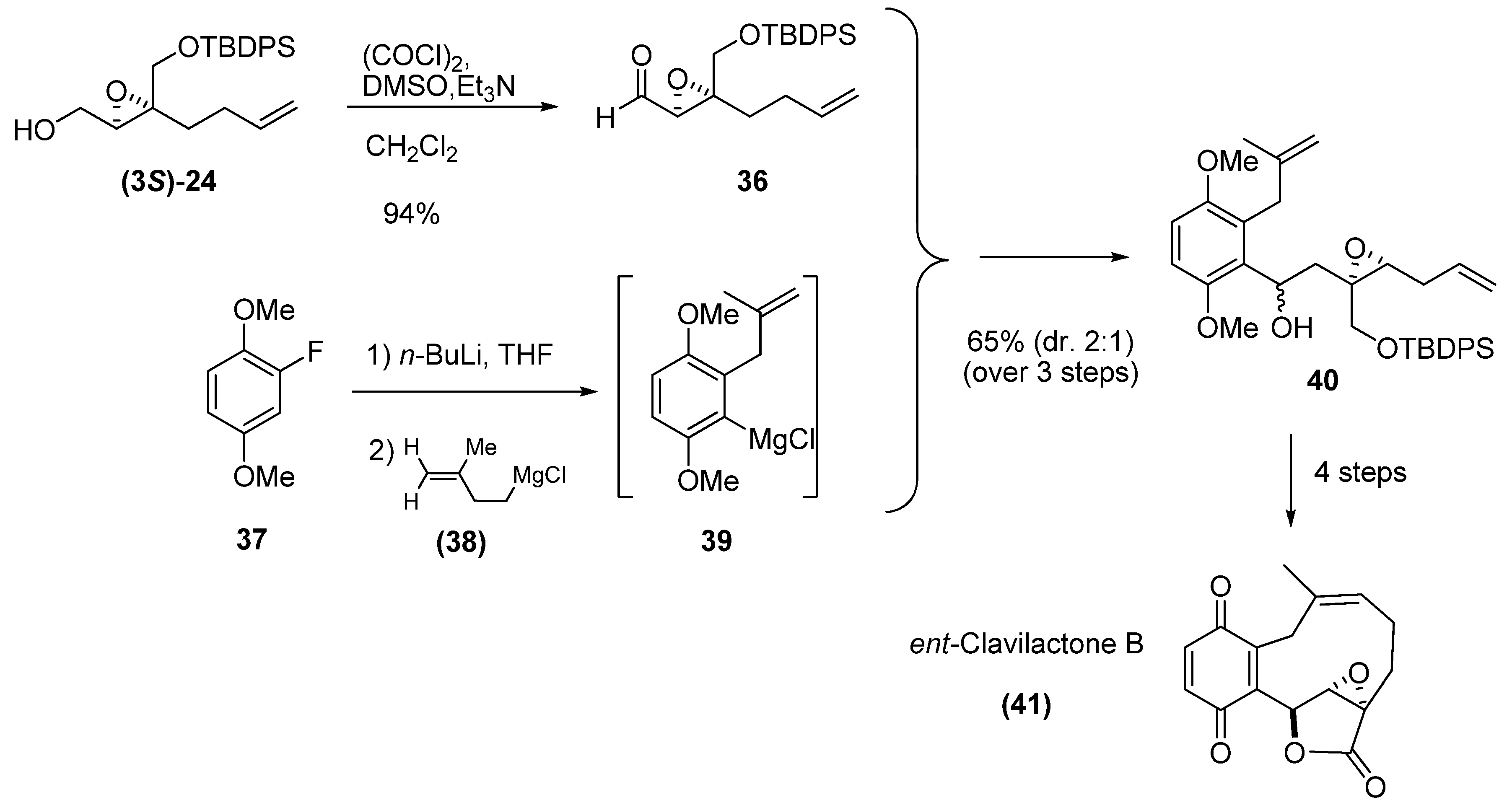
Among the synthetic advantages of unsaturated epoxides is the ease with which a second point of unsaturation can be introduced into their chain for their subsequent use in RCM. Barrett et al. [26] used the enantiomerically enriched (-)-epoxy alcohol (3S)-24 as starting material in their synthesis of ent-Clavilactone B. They oxidized the epoxide under Swern conditions to obtain the (+)-epoxy-aldehyde 36 with 97% ee. As benzyne precursor, they chose 2-fluoro-1,4-dimethoxybenzene (37), which upon reaction with n-BuLi gave the corresponding o-fluoroaryl lithium species, which in turn was allowed to fragment to a benzyne derivative in the presence of 38, to afford the aryl Grignard species 39. Finally, reaction of 39 with epoxy aldehyde 36 gave the two diastereomeric adducts 40. The crude 40 was converted into a lactone that was then subjected to RCM using the second-generation Grubbs’ catalyst (1b). Oxidative demethylation of the resulting adduct gave the target compound 41 with the opposite configuration to the natural clavilactone B.

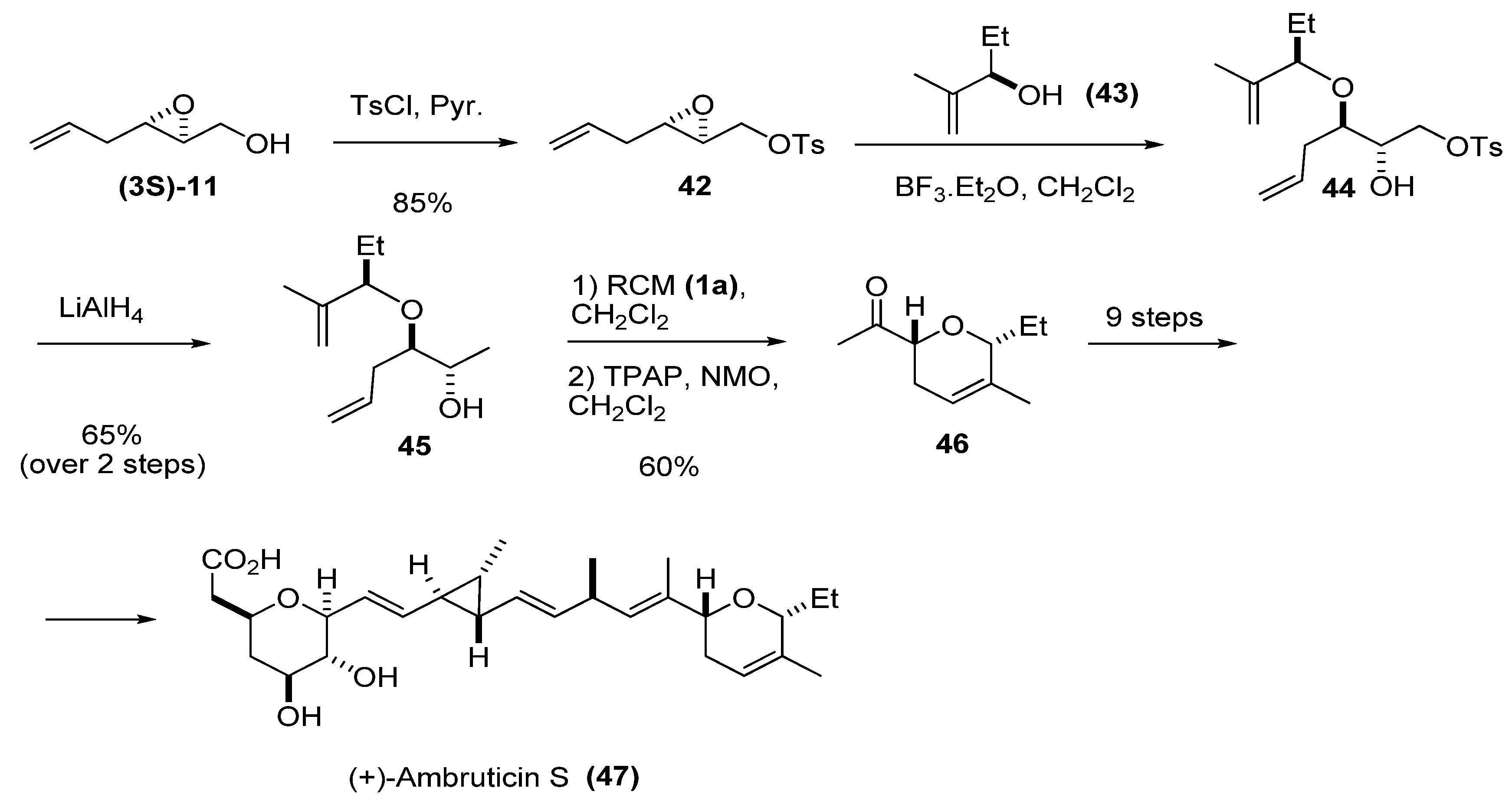
Another possible transformation at C1 is reduction of the tosylate of the primary alcohol to a methyl group. The tosylate favors the regioselective introduction of an unsaturated chain at C3 by nucleophilic ring-opening of the epoxide. This strategy was used by Martin et al. [27] in their synthesis of one of the fragments of the antifungal antibiotic ambruticin S. Tosylation of the epoxy alcohol (3S)-11 furnished epoxide 42, which was treated with the chiral allyl alcohol 43 in the presence of BF3·Et2O, to give diene 44. The tosylate was reduced to the diene 45, which was then subject to RCM using the first-generation Grubbs’ catalyst (1a). Oxidation of the secondary alcohol afforded 46, one of the pyranyl fragments of (+)-ambruticin S (47).
Scheme 4.
Synthesis of a chiral pyranyl fragment (46) of (+)-ambruticin S (47) (Martin et al. [27]).
Scheme 4.
Synthesis of a chiral pyranyl fragment (46) of (+)-ambruticin S (47) (Martin et al. [27]).

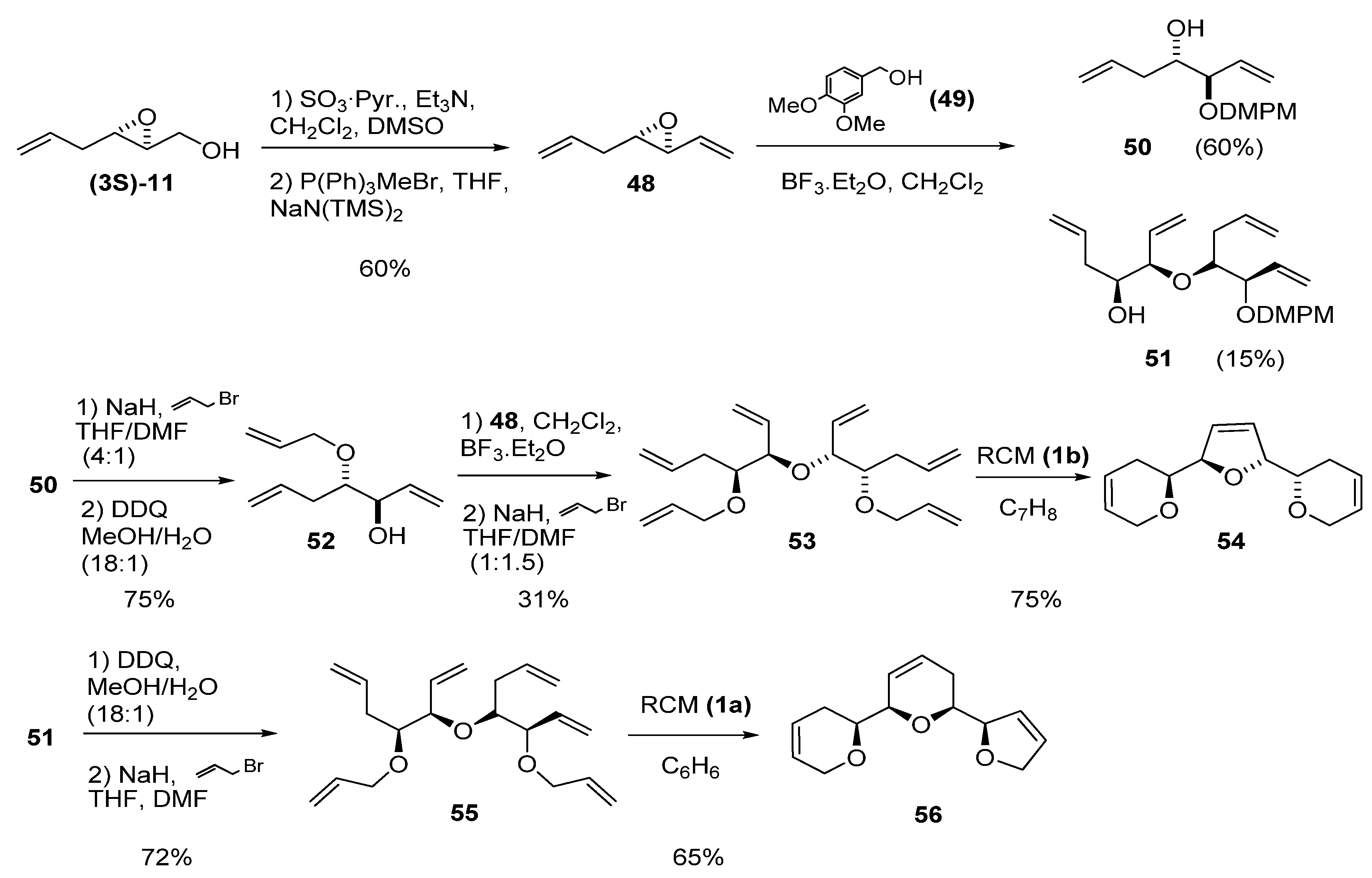
The two-step sequence of oxidation of the primary alcohol to aldehyde followed by Wittig olefination is a useful transformation for introducing a vinylic fragment adjacent to the epoxide. Mioskowski et al. [28] combined this methodology with the aforementioned ring-opening with an unsaturated alcohol to prepare polyenes as precursors of tricyclic ethers (found in various natural products) via RCM. Thus, epoxy alcohol (3S)-11 was oxidized to the corresponding aldehyde and submitted to Wittig olefination to yield the vinyl epoxide 48 (Scheme 5). Epoxide ring-opening of 48 with 3,4-dimethoxybenzyl alcohol (49) in CH2Cl2 gave the expected C2 product 50 plus the alcohol 51 as by-product (formed by ring-opening of 48 with 50). Alkylation of 50 with allyl bromide followed by deprotection with DDQ afforded the alcohol 52, which was then used as nucleophile in the ring-opening of 48. The resulting alcohol was alkylated again with allyl bromide to give the hexaene 53. A two-step sequence of deprotection followed by alkylation with allyl bromide afforded hexaene 55 from by-product 51. Both acyclic hexaenes 53 and 55 were finally submitted to RCM to obtain the triadjacent cyclic ethers 54 and 56, respectively, in good yields (75 and 65%, respectively).
Scheme 5.
Synthesis of adjacent polycyclic ethers by multiple RCM (Mioskovski et al. [28]).
Scheme 5.
Synthesis of adjacent polycyclic ethers by multiple RCM (Mioskovski et al. [28]).

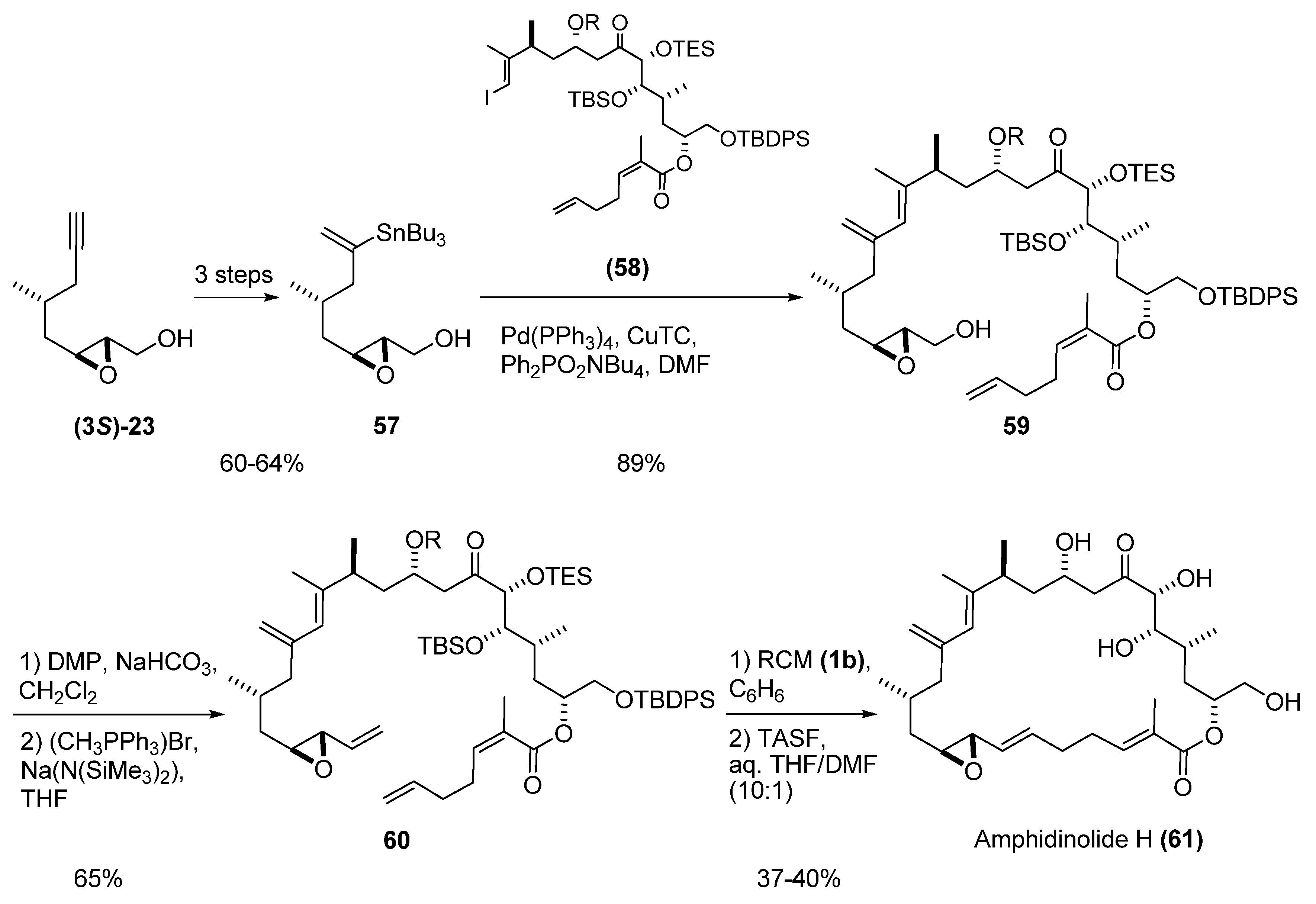
Fürstner et al. [29] used RCM macrocyclization of a vinyl epoxide as the basis of a total synthesis of amphidinolide H (61). Thus, the epoxy alcohol (3S)-23 (prepared by SAE) was transformed into the vinyl stannane 57 (Scheme 6). Cross-coupling reaction with the appropriate vinyl iodide (58) afforded compound 59, which was submitted to the aforementioned sequence of oxidation and Wittig olefination to give the vinyl epoxide 60. RCM of 60 with the second-generation Grubbs’ catalyst (1b) proceeded cleanly, and subsequent deprotection of the silyl protecting groups afforded the target, amphidinolide H (61).
Scheme 6.
RCM macrocyclization of a vinyl epoxide in the synthesis of amphidinolide H (Fürstner et al. [29]).
Scheme 6.
RCM macrocyclization of a vinyl epoxide in the synthesis of amphidinolide H (Fürstner et al. [29]).

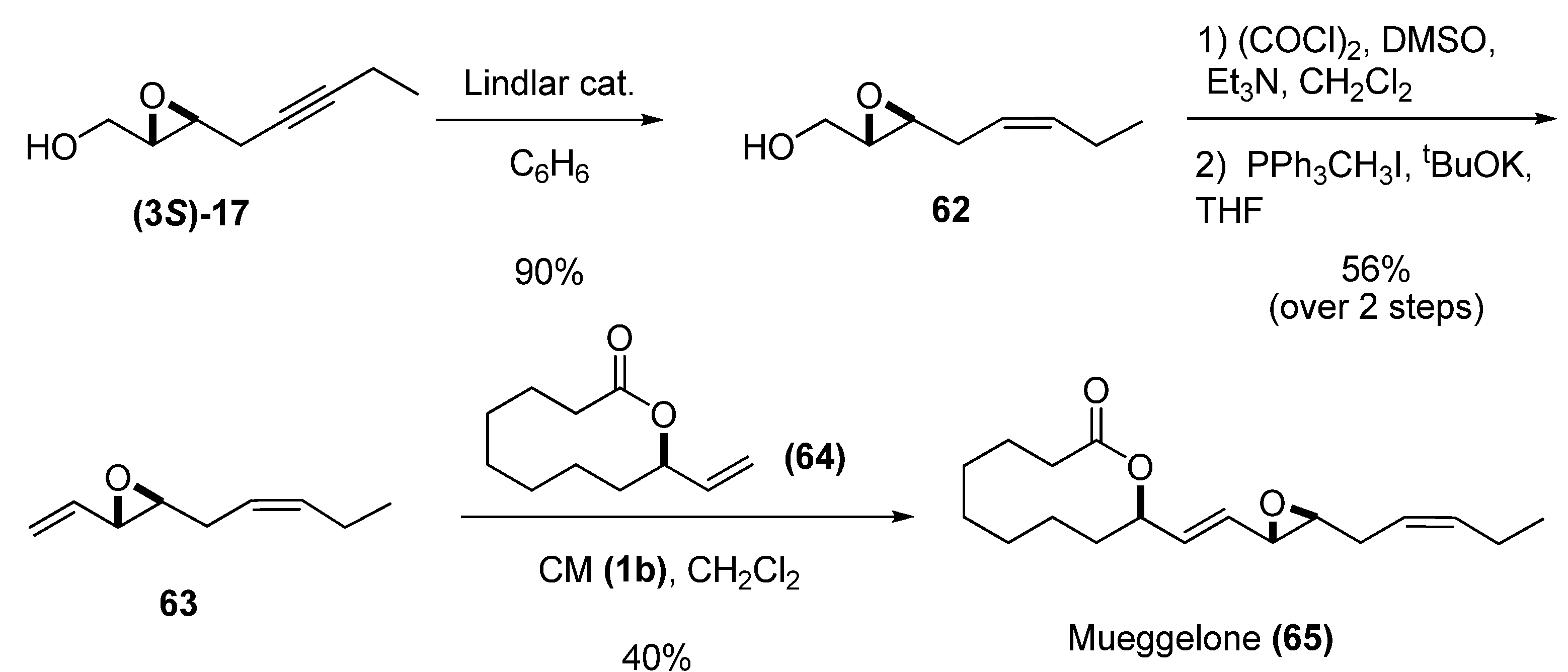
Vinyl epoxides can also be substrates for cross metathesis (CM), as demonstrated by Yadav et al. [30] in their synthesis of the fish-development inhibitor mueggelone from the chiral epoxide (3S)-17 via the sequence shown in Scheme 7. Reduction of (3S)-17 by Lindlar hydrogenation gave the cis-olefin 62, which was subjected to Swern oxidation followed by C1-Wittig homologation [31] to provide the vinyl epoxide 63 in good yield. CM between precursors 63 and 64, catalyzed by the second-generation Grubbs’ catalyst (1b), afforded the target, (+)-mueggelone (65), in 40% yield with complete E selectivity.
Scheme 7.
Synthesis of mueggelone based on cross-metathesis (Yadav et al. [30])
Scheme 7.
Synthesis of mueggelone based on cross-metathesis (Yadav et al. [30])

Reduction of epoxy alcohols to secondary allyl alcohols is a straightforward route to chiral alcohols with fully defined stereochemistry. This transformation can be done using (C5H5)2TiCl, prepared in situ from (C5H5)2TiCl2 and granulated zinc containing ZnCl2, as described by Yadav et al. [32]. Interestingly, the chirality of C3 in the epoxide (3R)-17 is totally conserved in the allylic alcohol 66.
Scheme 8.
Regioselective deoxygenation of 2,3-epoxy alcohols with (C5H5)2TiCl.

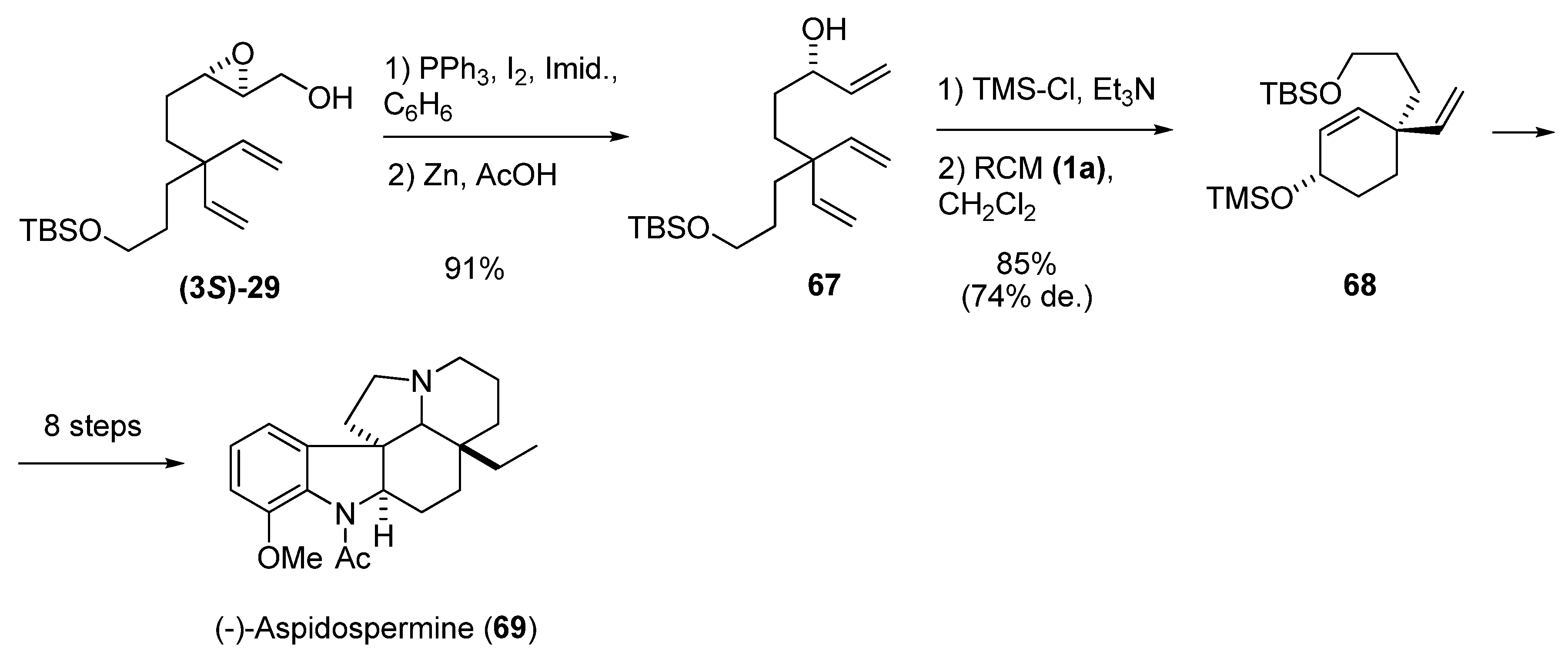
A similar transformation is reduction of the iodomethyl derivative of the epoxy alcohol. In the synthesis of aspidospermine by Shishido et al. [33], the optically pure (> 99% ee) hydroxytriene 67 was prepared by iodination of the epoxy alcohol (3S)-29 followed by treatment with Zn/AcOH (Scheme 9). After protection of the allyl alcohol, the triene was subjected to diastereoselective RCM to give the cyclohexene 68, a key intermediate in the synthesis of (-)-aspidospermine (69).
Scheme 9.
Synthesis of a key intermediate of (-)-Aspidospermine by diastereoselective RCM (Shishido et al. [33]).
Scheme 9.
Synthesis of a key intermediate of (-)-Aspidospermine by diastereoselective RCM (Shishido et al. [33]).

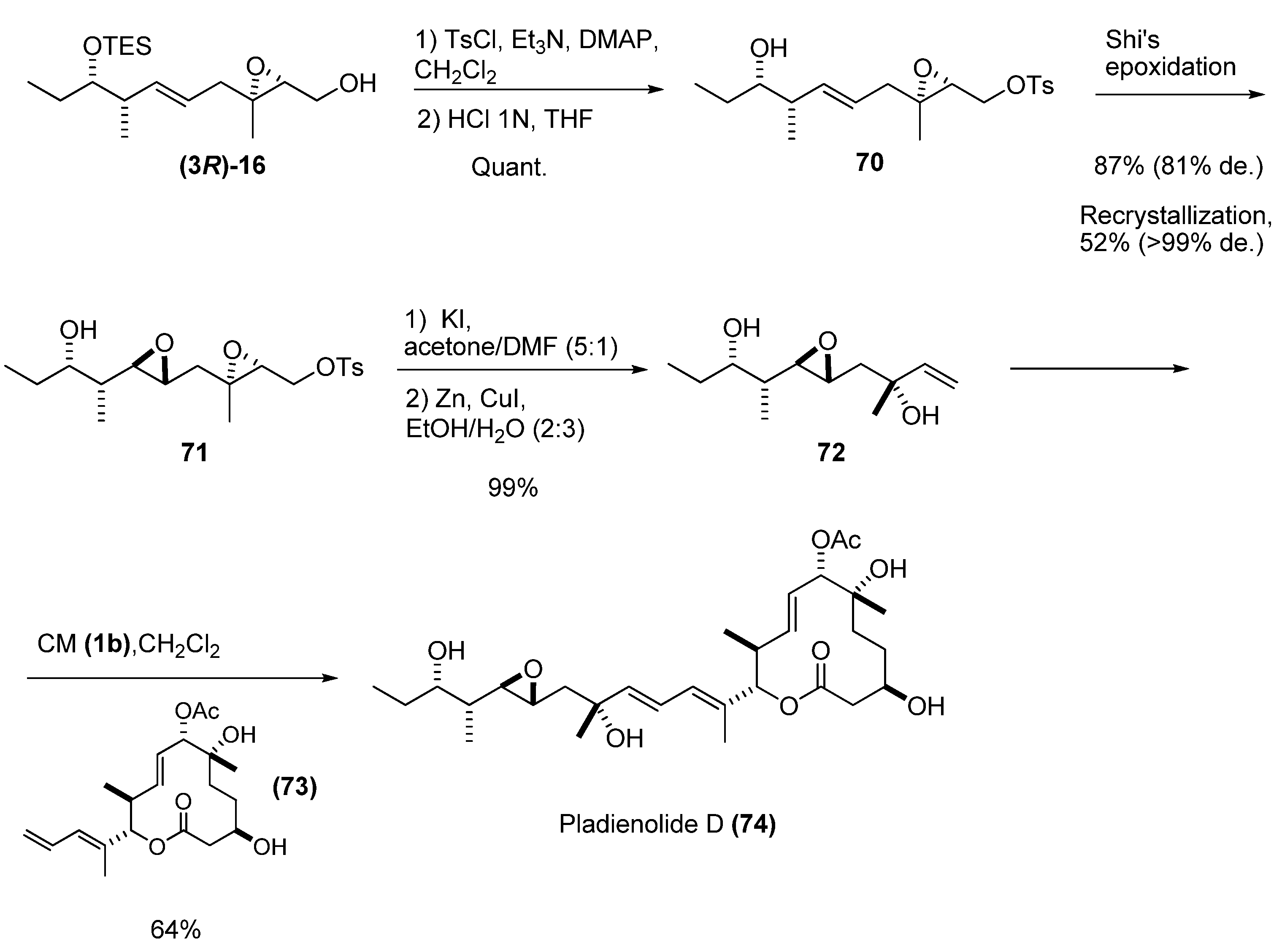
Kotake et al. [34] also used a chiral allylic alcohol prepared by deoxygenation of an epoxy alcohol to synthesize the potent antitumor compound pladienolide D. The allylic alcohol 72 was prepared from the epoxy alcohol (3R)-16 (Scheme 10). Tosylation of the primary hydroxyl group and removal of the TES group under weakly acidic conditions provided the alcohol 70, which was subjected to asymmetric Shi’s epoxidation [35] to the diepoxide 71. Nucleophilic substitution of the tosylate by iodide and subsequent cleavage of the epoxide with zinc-copper couple [36], gave the desired tertiary allylic alcohol 72 in excellent yield. Finally, CM with the appropriate olefin fragment (73) and the second-generation Grubbs’ catalyst 1b afforded pladienolide D (74) in 64% yield and with excellent stereoselectivity.
Scheme 10.
Synthesis of Pladienolide D via cross metathesis of a chiral allylic alcohol (Kotake et al. [34]).
Scheme 10.
Synthesis of Pladienolide D via cross metathesis of a chiral allylic alcohol (Kotake et al. [34]).

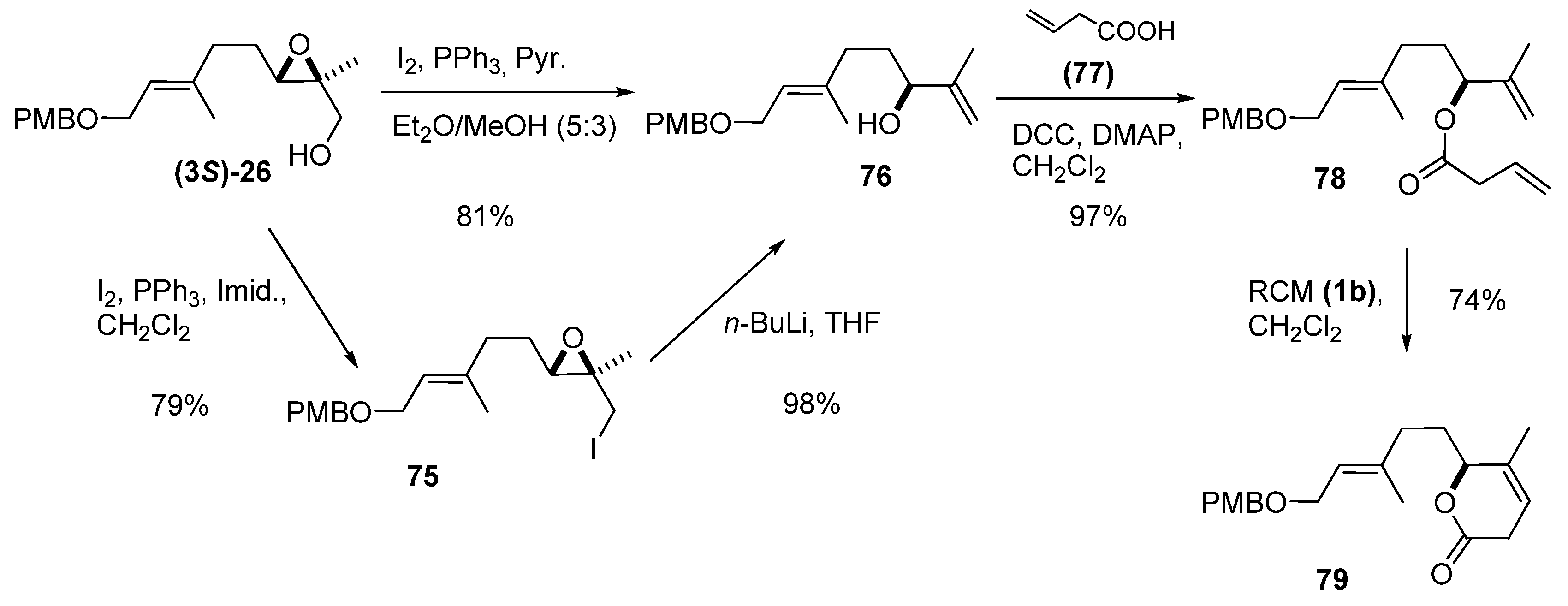
Nakata et al. [37] used the same transformation in their synthesis of a lactone fragment (79) (and synthetic precursor) of the marine biscembranoid methyl sarcophytoate (Scheme 11). The enantiomerically enriched epoxy alcohol (3S)-26 was treated with iodine, triphenylphosphine and imidazole to provide epoxy iodide 75, which was treated with n-BuLi to give the allylic alcohol 76. Alternatively, 76 was directly obtained from (3S)-26 in 81% yield by one-pot treatment of the intermediate iodide with water. To improve its enantiomeric purity, 76 was subjected to kinetic resolution [38], which provided an ee of > 98%. Condensation of 76 with vinyl acetic acid (77) using DCC and a catalytic amount of DMAP gave 78, the substrate for the RCM. Treatment with the second-generation Grubbs’ catalyst (1b) afforded the target, 79, in good yield.
Scheme 11.
Synthesis of an intermediate in the synthesis of methyl sarcophytoate (Nakata et al. [37]).
Scheme 11.
Synthesis of an intermediate in the synthesis of methyl sarcophytoate (Nakata et al. [37]).

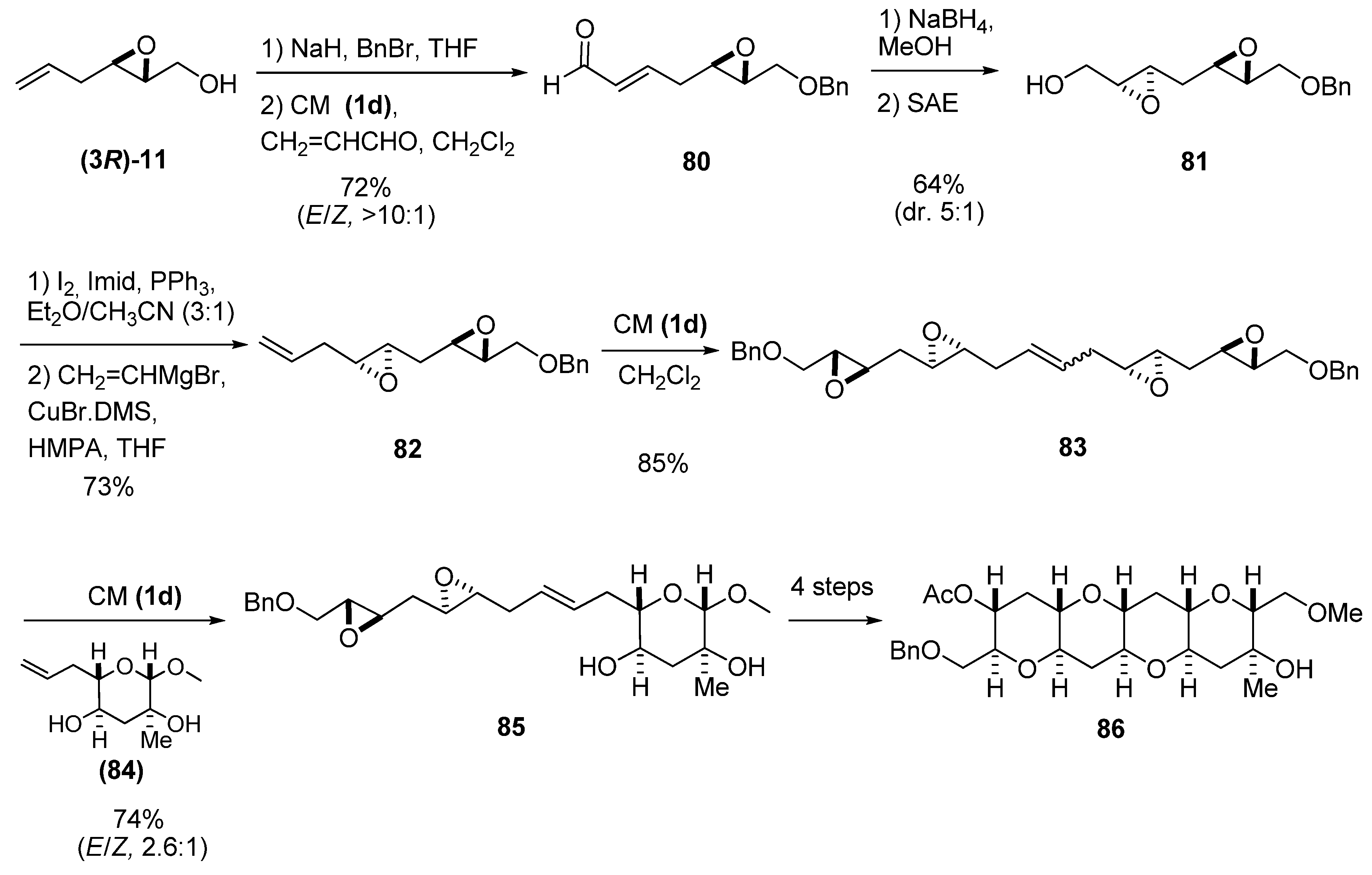
Recently, Jamison et al. [39] used epoxide (3R)-11 prepared by SAE as starting material to synthesize compound 86, a tetracyclic fragment of gymnocin A (Scheme 12). The key step was an epoxide-opening cascade reaction of a triepoxide prepared from 85. This compound was prepared by CM of alkene 84 and either the diepoxide 82 (synthesized via the sequence shown in Scheme 12) or the self-metathesis product 83. To this end, compound (3R)-11 was protected as the benzyl ether, and then subject to CM with acroleine using the second-generation Hoveyda-Grubbs’ catalyst 1d to give the aldehyde 80. Reduction of 80 to the corresponding alcohol, followed by a second SAE, afforded the diepoxide 81. Then, a nucleophilic substitution by a vinyl cuprate at C1, gave vinyl diepoxide 82, which reacted with the second-generation Hoveyda-Grubbs’ catalyst 1d to give the self-metathesis product 83. A second CM with the olefin 84 afforded the key intermediate, the diepoxide 85.
Scheme 12.
Route to the diepoxide 85, a synthetic template for the synthesis of a tetracyclic fragment (86) of gymnocin A (Jamison et al. [39]).
Scheme 12.
Route to the diepoxide 85, a synthetic template for the synthesis of a tetracyclic fragment (86) of gymnocin A (Jamison et al. [39]).

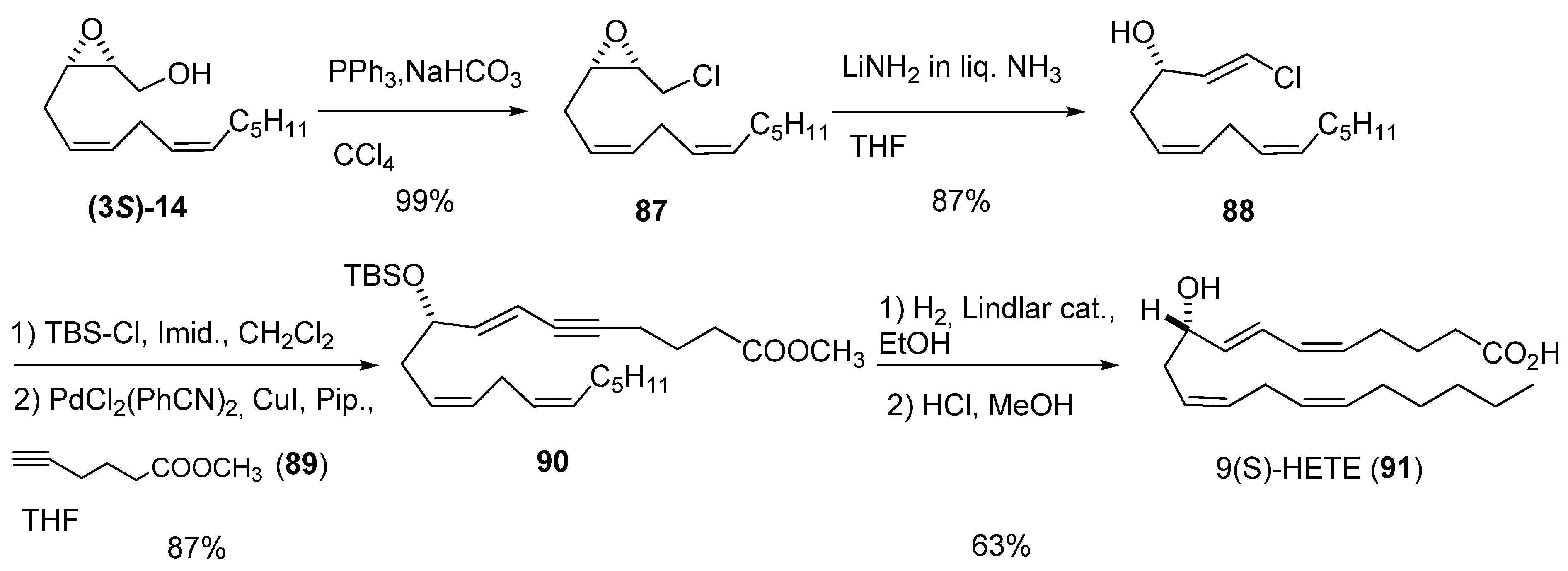
Yadav et al. [40] developed a methodology to form trans-3-hydroxy-vinyl chlorides—which are useful reagents for coupling reactions—by treatment of epoxy chlorides with LiNH2. They employed this strategy to prepare both enantiomers of 9-hydroxy eicosatetraenoic acid (HETE) [41]. The epoxy alcohol (3S)-14, prepared by SAE, was converted into the corresponding chloride 87 by treatment with PPh3 and NaHCO3 in refluxing CCl4 (Scheme 13). Addition of LiNH2 to 87 in liquid NH3 at -33 ºC afforded the trans vinyl chloride 88 exclusively. After protection of 88 as the tert-butyldimethyl silyl ether, Sonogashira reaction with methyl hex-5-inoate (89) afforded the intermediate ester 90. Finally, 90 was subjected to selective reduction of the triple bond using Lindlar catalyst, followed by deprotection with 1% HCl in MeOH, to obtain the target, 91. The 9(R)-HETE enantiomer was similarly prepared, starting from (3R)-14.
Scheme 13.
Synthesis of 9(S)-HETE based on preparation of a hydroxy vinyl chloride from an epoxy alcohol (Yadav et al. [40]).
Scheme 13.
Synthesis of 9(S)-HETE based on preparation of a hydroxy vinyl chloride from an epoxy alcohol (Yadav et al. [40]).

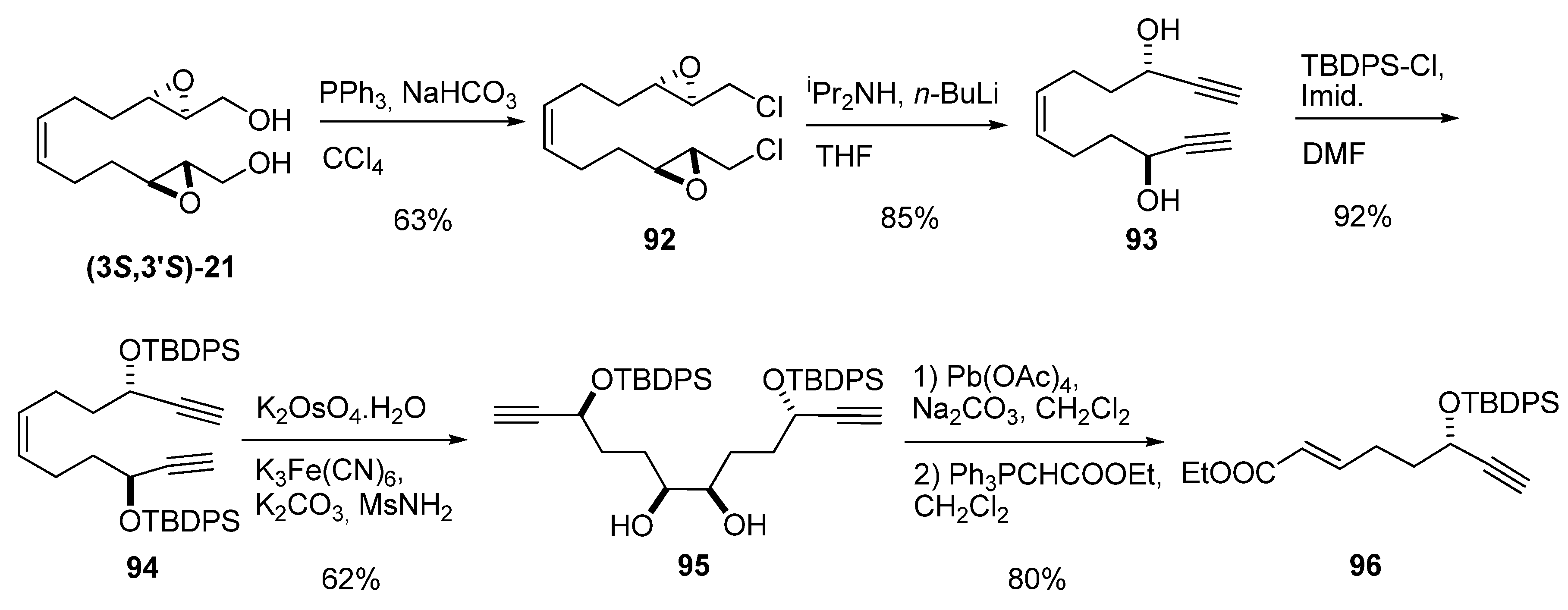
Marshall et al. [42] in a modular synthesis of the Annonaceous acetogenin Asimin, prepared the diepoxy diol (3S,3’S)-21, in four steps from 1,5-cyclooctadiene [43], to avoid volatility problems during synthesis of the key intermediate 96 (Scheme 14). Derivatization of (3S,3’S)-21 to the chloride 92 was followed by elimination to the dialkyne diol 93 using LDA. Both alcohols were protected as tert-butyldiphenyl silyl ethers, and the double bond was dihydroxylated to afford the diol 95. Oxidative cleavage with lead tetraacetate and subsequent Wittig homologation yielded the conjugate ester 96.
Scheme 14.
Route to the conjugate ester 96, a key intermediate in the synthesis of asimin (Marshall et al. [42]).
Scheme 14.
Route to the conjugate ester 96, a key intermediate in the synthesis of asimin (Marshall et al. [42]).

4. Epoxide Ring-Opening at C2
4.1. Nucleophilic attack at C2 with carbon nucleophiles
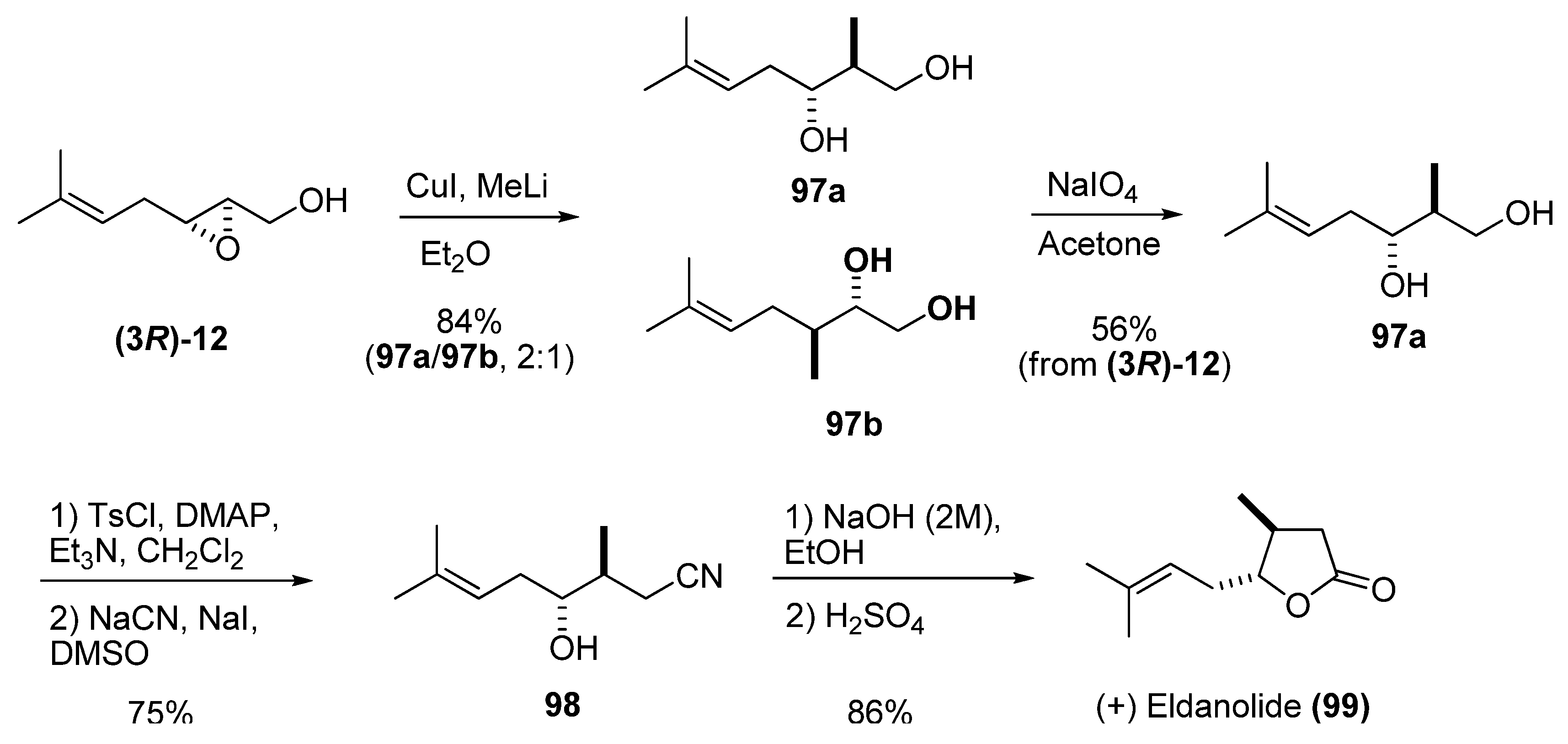
Reaction of epoxy alcohols with carbon nucleophiles such as cuprates leads to opening of the epoxide ring. The major isomer is usually the 1,3-diol resulting from the attack at C2. Although the regioselectivity is generally moderate, the minor isomer (the 1,2-diol) can easily be removed by oxidation with sodium periodate. In the synthesis of (+)-eldanolide (99), a monoterpenoid pheromone with an unsaturated chain, Zhai et al. [44] established the stereochemistry of both stereocenters by opening the unsaturated epoxy alcohol (3R)-12 with lithium dimethylcuprate, which provided moderate selectivity (Scheme 15). The resulting inseparable mixture of 1,2- and 1,3-diols was treated with NaIO4 in acetone to provide the diol 97a in 56% yield (starting from (3R)-12). Selective tosylation of 97a at the primary alcohol followed by SN2 displacement gave the nitrile 98 in 75% yield for the two steps. Basic hydrolysis followed by acid-catalyzed lactonization smoothly converted 98 into the desired (+)-eldanolide (99).
Scheme 15.
Enantioselective synthesis of (+)-eldanolide (Zhai et al. [44]).
Scheme 15.
Enantioselective synthesis of (+)-eldanolide (Zhai et al. [44]).

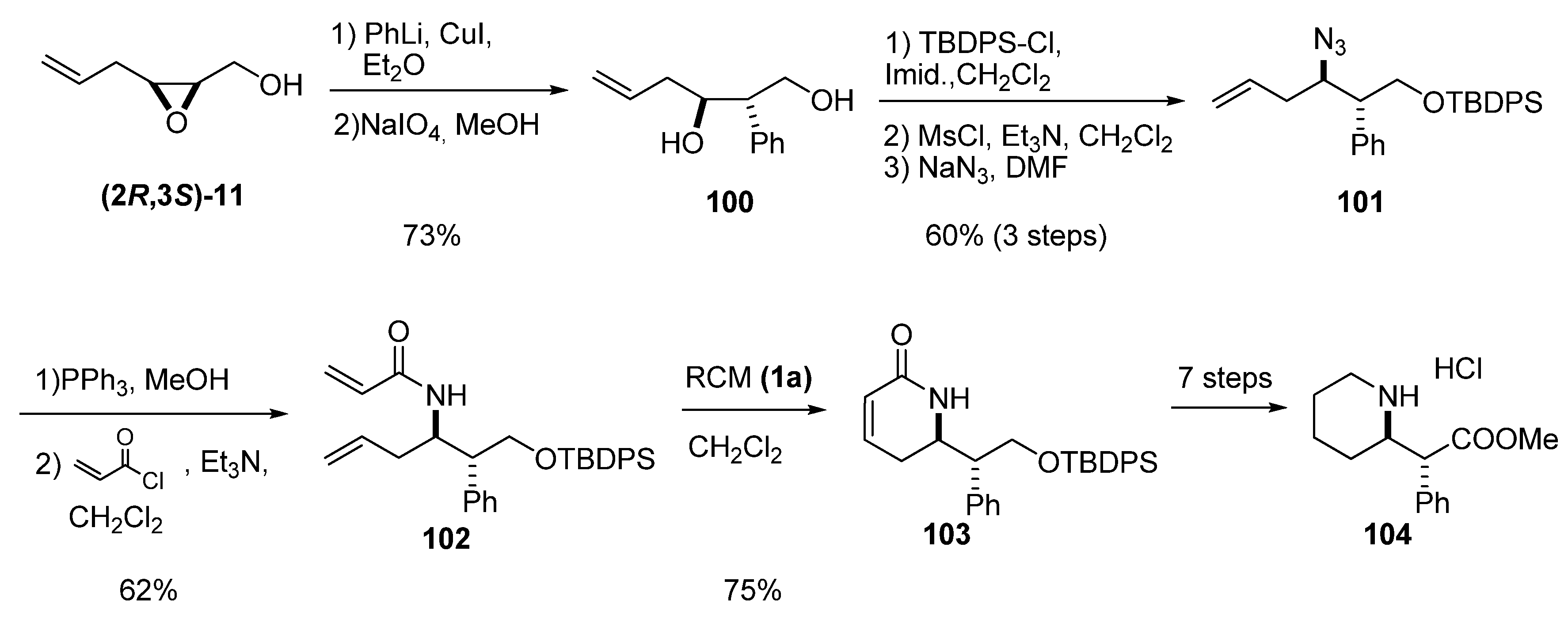
As mentioned above, the most useful reactivity of the terminal alkenes of the epoxy alcohol’s side chain is in olefinic metatheses. Krishna et al. [45], in their synthesis of threo-(+)-methylphenidate hydrochloride, treated the epoxy alcohol (2R,3S)-11 (from Z-hexa-2,5-dien-1-ol) with lithium diphenylcuprate to give a 9:1 mixture of C2/C3 regioisomers (Scheme 16). Once they obtained the desired diol 100, they protected the primary alcohol and substituted the secondary alcohol with azide to prepare the vinylic azide 101. Reduction of 101, followed by treatment of the resulting intermediate with acryloyl chloride, afforded the bisolefin 102. RCM using the first-generation Grubbs’ catalyst (1a) afforded the lactam 103, which was transformed into the desired (2R, 2’R)-threo-(+)-methylphenidate hydrochloride (104). This compound is currently used in racemic form (under the brand name Ritalin) for the treatment of attention deficit hyperactivity disorder (ADHD) in children.
Ma et al. [46] used ring-opening of epoxy alcohols with vinyl cuprate, electrophilic introduction of allyl groups, and double RCM to prepare various isomers of the quinolizidine alkaloid lupinine. Epoxide (3S)-11 was opened by vinyl cuprate to afford the diene 105, which was subjected to a four-step reaction sequence to generate the azido alcohol 106 (Scheme 17). Protection of the alcohol followed by reduction of the azide and double alkylation provided compound 107, the precursor for the double RCM. The RCM afforded three different products: 108a, 108b and 109. Reduction and deprotection of 108a/108b afforded (+)-epilupinine (110) in good yield. The other three isomers of lupinine were likewise synthesized via double RCM reactions of optically active propenamides analogous to 107.
Scheme 16.
Enantioselective synthesis of threo-(+)-methylphenidate hydrochloride (104) (Krishna et al. [45]).
Scheme 16.
Enantioselective synthesis of threo-(+)-methylphenidate hydrochloride (104) (Krishna et al. [45]).

Scheme 17.
Synthesis of (+)-epilupinine by double RCM (Ma et al. [46]).
Scheme 17.
Synthesis of (+)-epilupinine by double RCM (Ma et al. [46]).

4.2. Nucleophilic reduction at C2 with hydride
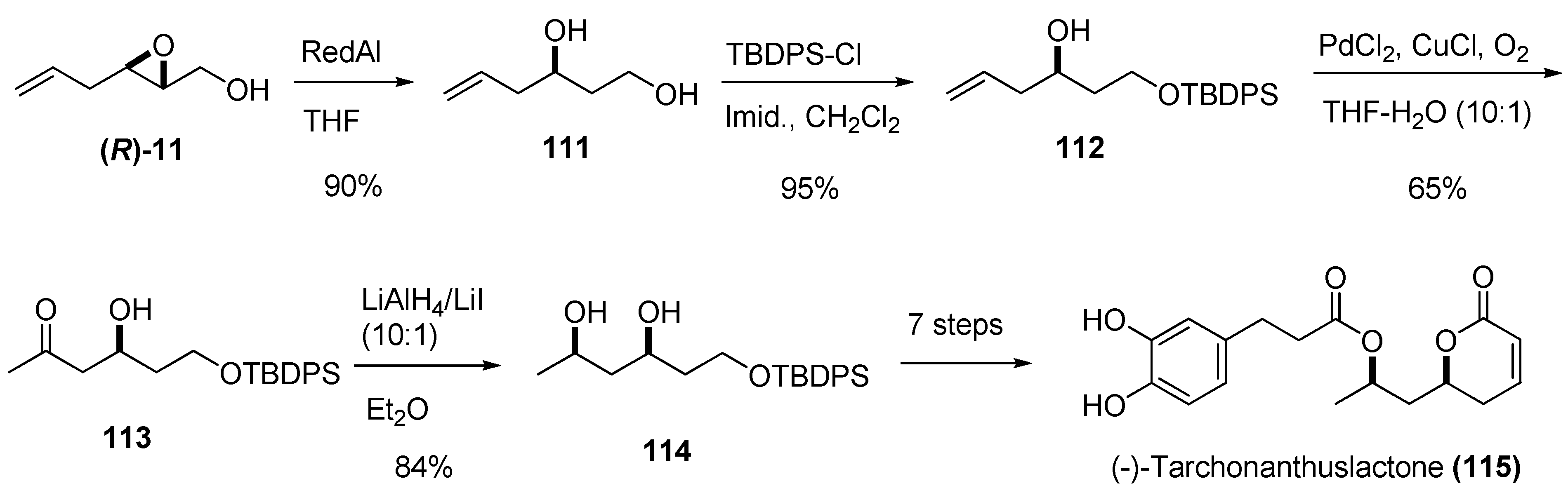
Treatment of 2,3-epoxy alcohols with a metal hydride such as Red-Al usually provides regioselective reduction at C2. The corresponding diol is a valuable synthetic intermediate: the two hydroxy groups can be selectively functionalized and the stereochemistry of the secondary alcohol is completely defined. Yadav et al. used this strategy to prepare (-)-tarchonanthuslactone (115) [47], a pyrone isolated from the leaves of the tree Tarchonanthus trilobus (Scheme 18). Reduction of the epoxide (R)-11 with Red-Al yielded the diol 111, which was selectively protected at the primary alcohol as the tert-butyldiphenyl silyl ether 112. The terminal double bond of the side chain was converted into the methyl ketone by Wacker oxidation, giving the ß-hydroxy ketone 113 (65%). The key intermediate, the syn-1,3-diol 114, was conveniently prepared with high diastereoselectivity (syn:anti selectivity of up to 99:1 or greater) by chelation-controlled reduction of 113 using LiI/LiAlH4 [48]. Conversion of 114 into (-)-tarchonanthuslactone (115) required seven steps.
Scheme 18.
Synthesis of (-)-tarchonanthuslactone (115) based on regioselective C2 reduction of the epoxy alcohol (R)-11 (Yadav et al. [47]).
Scheme 18.
Synthesis of (-)-tarchonanthuslactone (115) based on regioselective C2 reduction of the epoxy alcohol (R)-11 (Yadav et al. [47]).

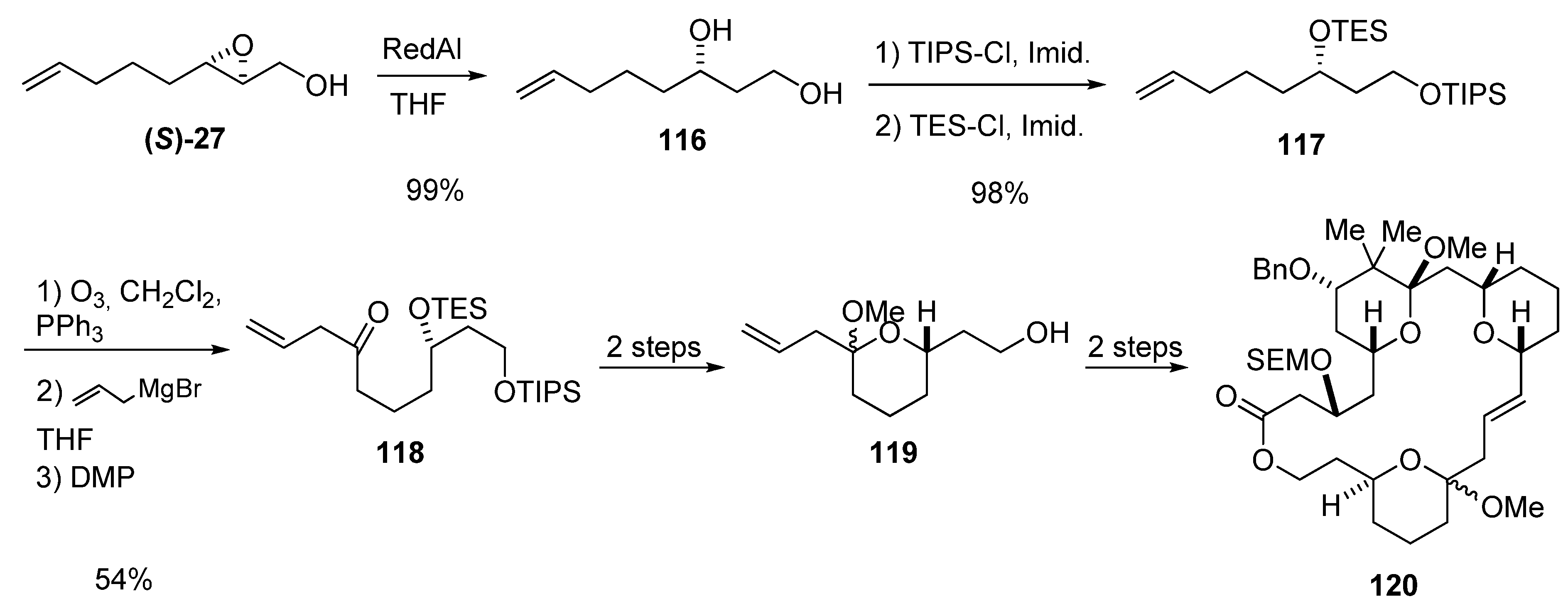
Thomas et al. [49], in their studies on bryostatin synthesis, constructed the hemiacetal fragment present in these important macrolides from the epoxy alcohol (S)-27 (Scheme 19). Regioselective reduction of (S)-27 gave the diol 116, which was selectively protected to give the bis-silyl ether 117. The terminal alkene was ozonized and the resulting aldehyde treated with allyl magnesium bromide. Oxidation of the resulting alcohol with the Dess-Martin periodinane afforded 118, which was easily converted into the hemiacetal 119, the precursor of 120, which is a macrocyclic bryostatin analog in which the C16-C17 double bond has the (E)-configuration.
Scheme 19.
Synthesis of the hemiacetal fragment 119 of the bryostatin analog 120 (Thomas et al. [49]).
Scheme 19.
Synthesis of the hemiacetal fragment 119 of the bryostatin analog 120 (Thomas et al. [49]).

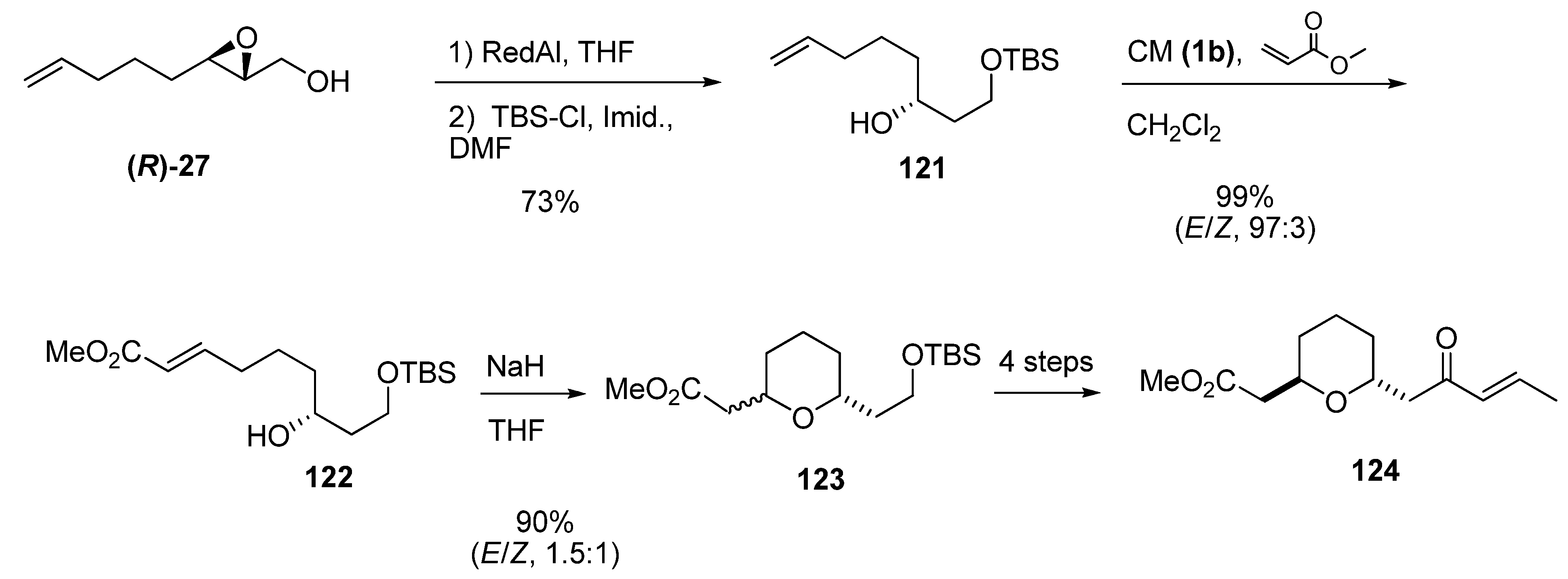
Piva et al. [50] used a similar reduction to synthesize the nor-methyl tetrahydropyran subunit of bistramide A, starting with the enantiomeric epoxide (R)-27 (Scheme 20). This epoxide was opened by regioselective hydride reduction at C2 with Red-Al, and the primary alcohol of the resulting diol was selectively protected as the tert-butyldimethylsilyl ether 121. CM between 121 and methyl acrylate using the second-generation Grubbs’ catalyst (1b) gave the ester 122 (96%). Intramolecular oxa-Michael cyclization of 122 afforded a 1.5:1 mixture of the trans and cis isomers of the tetrahydropyran 123. After chromatographic separation and functional group manipulation, the tetrahydropyran trans-124 was obtained in 40% yield (over three steps and a final isomerization) and with 96% ee (as determined by chiral HPLC).
Scheme 20.
Synthesis of the nor-methyl tetrahydropyran subunit (124) of bistramide A (Piva et al. [50]).
Scheme 20.
Synthesis of the nor-methyl tetrahydropyran subunit (124) of bistramide A (Piva et al. [50]).

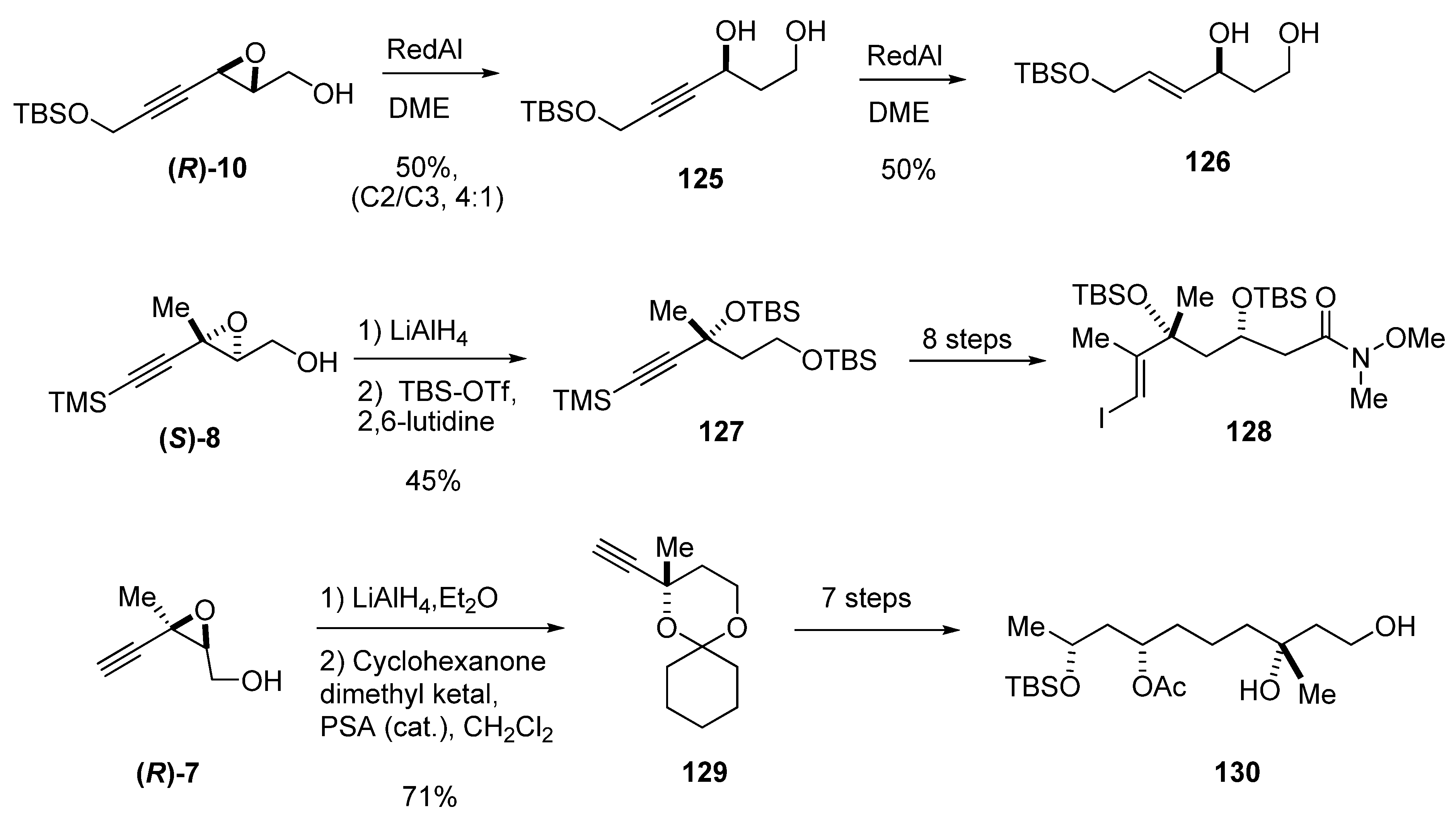
The regioselective hydride reduction at C2 has also been used in epoxy alcohols with triple bonds in the chain. In 3-alkynyl epoxy alcohols the regioselectivity at C2 is moderate due to the competition from the intramolecular reduction at the activated propargylic position (C3-attack) to give the 1,2-diol. For instance, reduction of (R)-10 gave the 1,3-diol 125 with a ring-opening selectivity C2/C3 of 4:1 [51]. Diol 125 was further reduced to the E allylic alcohol 126 by slow addition of Red-Al, and then purified by chromatography (Scheme 21).
Substitution at C3 further increases the selectivity at C2. Thus, LiAlH4 reduction of alkynyl epoxides with a fully substituted C3 center, such as (S)-8 and (R)-7, is completely regioselective and has proven invaluable for the preparation of chiral tertiary alcohols (Scheme 21). In an asymmetric synthesis of the C7-C20 synthon of the anti-cancer agent amphidinolide B (128), Nelson et al. [52] used this methodology to fix the chirality of the tertiary alcohol at C16. Regioselective hydride-mediated oxirane opening of (S)-8 afforded a diol that was then protected with tert-butyldimethylsilyl trifluoromethanesulfonate and 2,6-lutidine to give 127, which was converted into the iodide 128 (subsequently used for coupling) in eight steps (Scheme 21).
Similarly, Ghosh et al. [53] efficiently established the chiral center at C3 in Taurospongin A, a non-nucleoside reverse transcriptase inhibitor. The epoxide (R)-7 was reduced with LiAlH4 in ether at 0 ºC to the corresponding 1,3-diol which was subsequently treated with cyclohexanone dimethyl ketal using a catalytic amount of p-TsOH·H2O to provide the ketal 129 (Scheme 21). Alcohol 130 which had been converted previously to taurospongin A by Lebel and Jacobsen [54] could be prepared from 129 in seven steps.
Scheme 21.
Representative examples of enantioselective syntheses of chiral alcohols by regioselective C2 reduction of alkynyl epoxides.
Scheme 21.
Representative examples of enantioselective syntheses of chiral alcohols by regioselective C2 reduction of alkynyl epoxides.

4.3. Nucleophilic attack at C2 with nitrogen nucleophiles
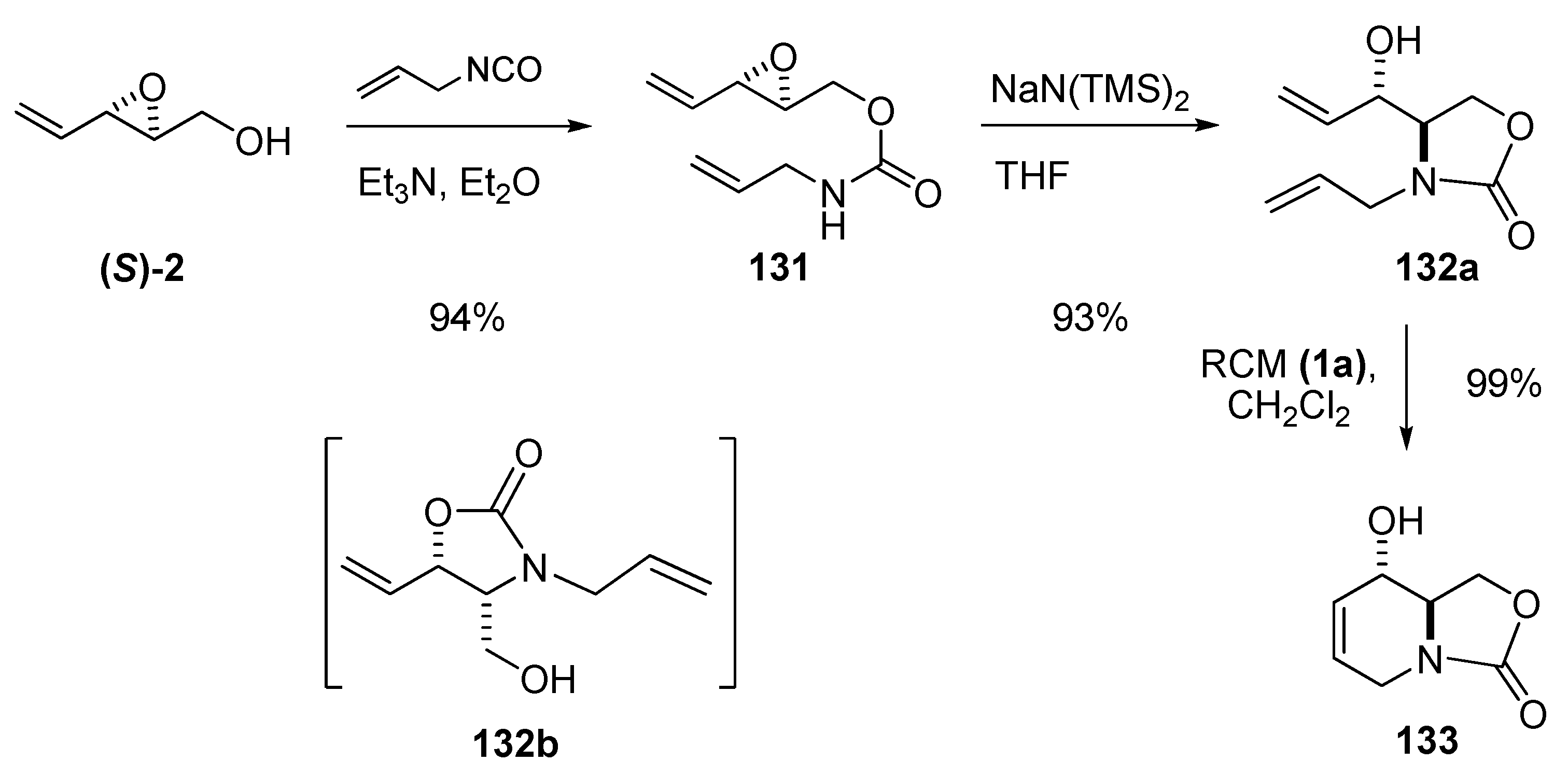
Nitrogen nucleophiles attack epoxy alcohols with low selectivity or with C3 selectivity in the presence of Ti(iOPr)4 or LiClO4 as Lewis acids. The pioneering work of Roush et al. [55,56] enabled directed attack at C2 via intramolecular attack by a carbamic nitrogen. A nitrogen nucleophile can be introduced at C2 with excellent regioselectivity by treating the epoxy alcohol with an isocyanate to form a carbamate that is subsequently cyclized under basic conditions. The selectivity probably stems from kinetic preferential formation of a five-membered ring as compared to a six-membered ring. However, this methodology suffers from a major drawback: certain substrates are prone to isomerize during the basic treatment because of easy acyl transfer from the primary to the secondary alcohol.
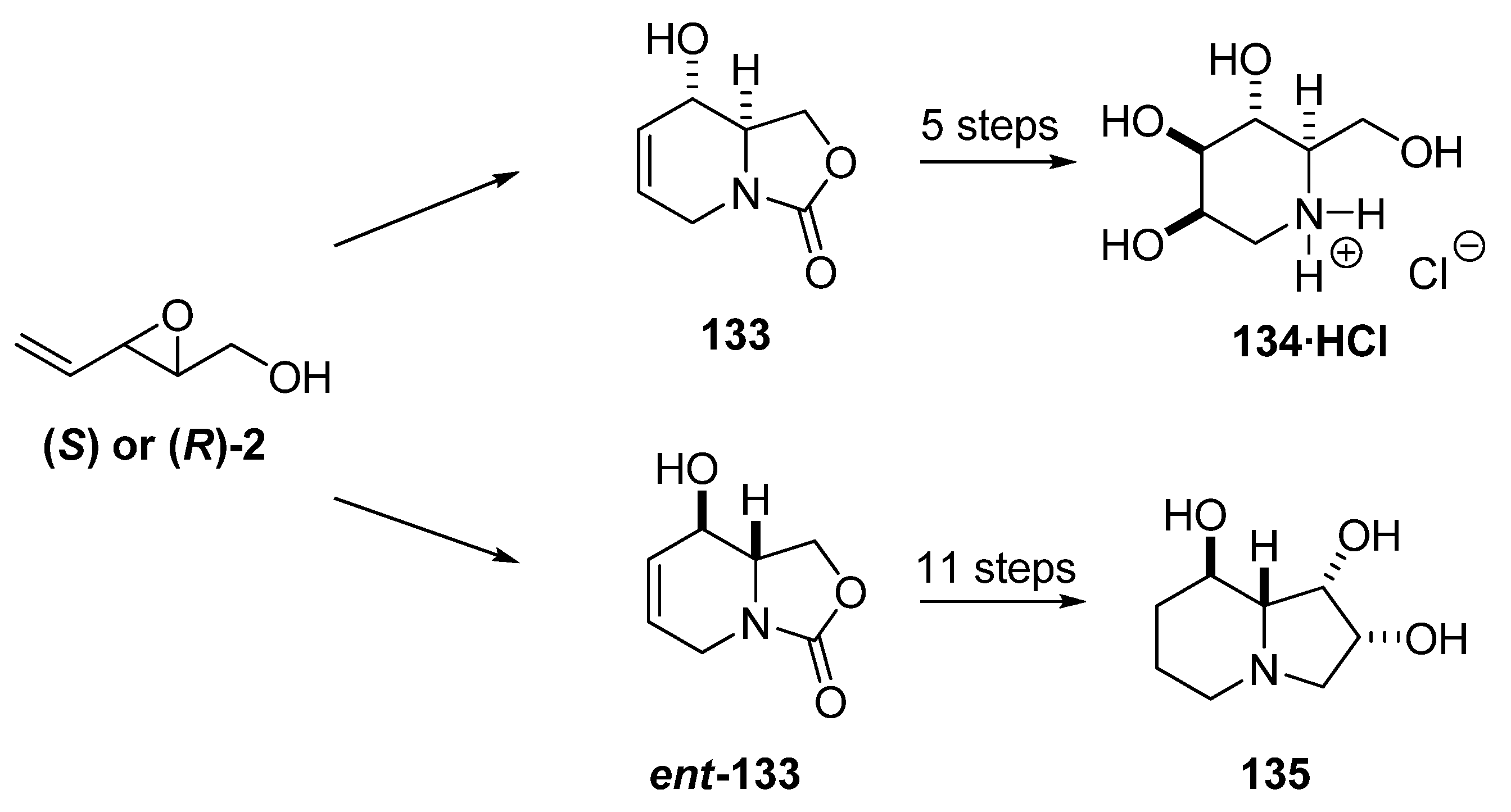
In seeking an efficient enantioselective synthesis of azasugars, Riera et al. [57] envisaged preparation of the key intermediate 133 by RCM of the carbamate 132a, which was prepared from chiral epoxide (S)-2. The crude epoxide (S)-2 was treated with allyl isocyanate/Et3N in refluxing ether to provide allyl carbamate 131 in 94% yield (Scheme 22). However, the subsequent intramolecular ring-opening of 131 under standard conditions (NaH/THF) gave a mixture of the desired oxazolidine 132a and the trans-acetylated isomer 132b[57]. Other bases (e.g. tert-BuOK) gave only slightly better yields, whereas treatment with Lewis acid catalysts such as LiClO4 and Ti(iOPr)4, led to decomposition of the starting material. Ultimately, sodium bis(trimethylsilyl)amide in THF provided 132a in 88% yield with no sign of 132b. Subsequent RCM of 132a using 5% first-generation Grubbs’ catalyst (1a) proceeded smoothly to afford the intermediate 133.
Scheme 22.
Synthesis of 133, a key synthetic precursor of several glycosidase inhibitors (Riera et al. [57]).
Scheme 22.
Synthesis of 133, a key synthetic precursor of several glycosidase inhibitors (Riera et al. [57]).

The bicyclic carbamate 133 has been converted by Riera et al. and others into several glycosidase inhibitors (e.g. 1-deoxymannojirimycin (134) [58,59,60] and 1-deoxygalactostatin [61]) and other biologically active compounds [62]. Moreover, Riera et al. [58] transformed the enantiomeric intermediate ent-133, prepared from the epoxide (R)-2, into enantiomerically pure swainsonine (135), a glycosidase inhibitor with antitumor and antiviral properties (Scheme 23).
Scheme 23.
Epoxide 2 as starting material for the synthesis of 1-deoxymannojirimycin (134) and swainsonine (135) (Riera et al. [58]).
Scheme 23.
Epoxide 2 as starting material for the synthesis of 1-deoxymannojirimycin (134) and swainsonine (135) (Riera et al. [58]).

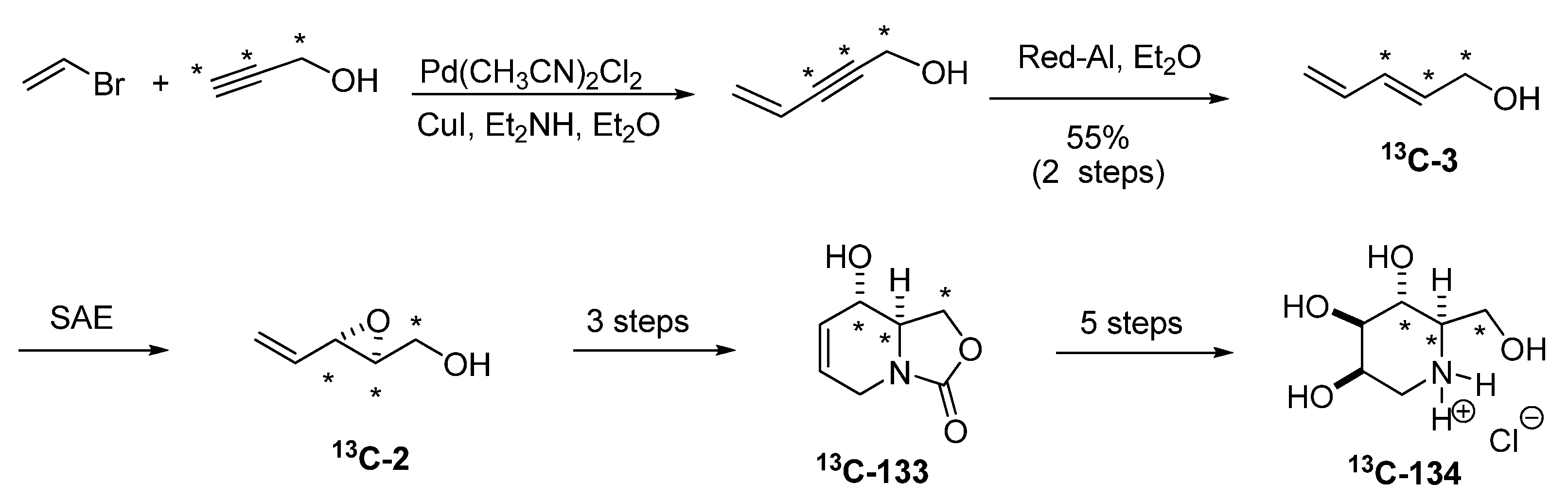
Riera et al. exploited the aforementioned methodology to gain access to a host of 13C labeled glycosidase inhibitors for NMR based protein-binding experiments, such as (4,5,6-13C)-deoxymannojirimycin [63]. 13C labeled (E)-2,4-pentadien-1-ol (13C-3), was prepared from 13C-propargyl alcohol by a Sonogashira coupling with allyl bromide followed by reduction (Scheme 24). SAE afforded the isotopically labeled (S)-13C-2, which was run through the sequence shown in Scheme 22, to obtain the isotopically labeled 133, which was converted into (4,5,6-13C)-deoxymannojirimycin (13C-134).
Scheme 24.
Synthesis of (4,5,6-13C)-deoxymannojirimycin (Riera et al. [63]).
Scheme 24.
Synthesis of (4,5,6-13C)-deoxymannojirimycin (Riera et al. [63]).

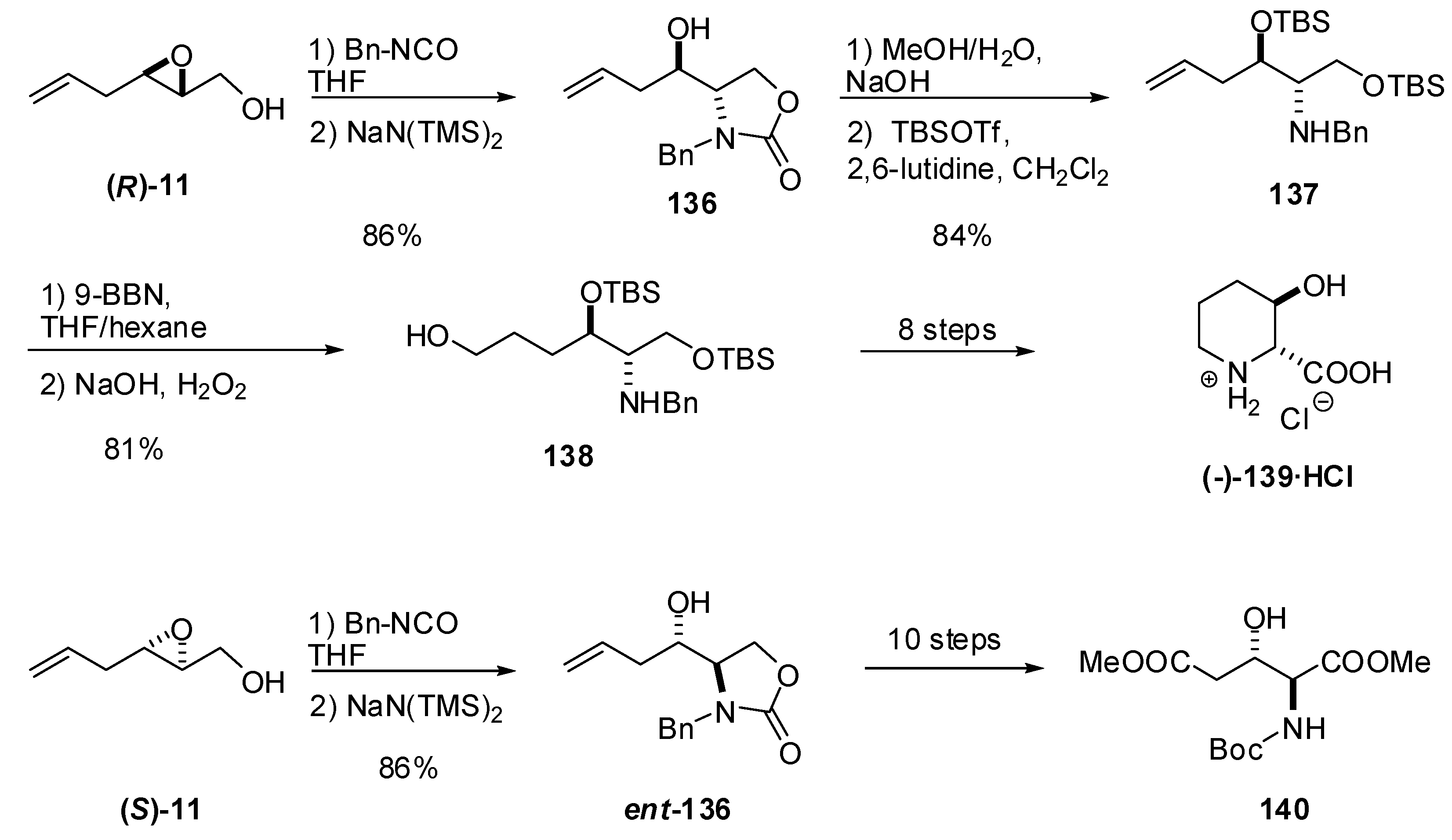
Both enantiomers of benzyl carbamate 136 can be readily prepared by regioselective C2 ring opening of epoxy alcohols 11 with benzyl isocyanate (Scheme 25). Riera et al. transformed epoxy alcohol (R)-11 into 3-hydroxypipecolic acid (139) [64]. Hydroboration of the terminal double bond was essential for preparing the alcohol 138, which, after protecting group manipulation, was cyclized and oxidized to 139.Analogously, Riera et al. used the enantiomer (S)-8 as starting material to synthesize N-Boc erythro-β-hydroxyglutamic acid methyl ester (140) [65]. Oxidation of the terminal double bond to an ethyl ester in an adequately protected derivative of 136 enabled preparation of 140.
Scheme 25.
Synthesis of 3-hydroxypipecolic acid hydrochloride (139) from the epoxide (R)-11 and of erythro-β-hydroxyglutamic acid methyl ester (140) from the epoxide (S)-11 (Riera et al. [64]).
Scheme 25.
Synthesis of 3-hydroxypipecolic acid hydrochloride (139) from the epoxide (R)-11 and of erythro-β-hydroxyglutamic acid methyl ester (140) from the epoxide (S)-11 (Riera et al. [64]).

5. Epoxide Ring-Opening at C3
5.1. Nucleophilic attack at C3 with carbon nucleophiles
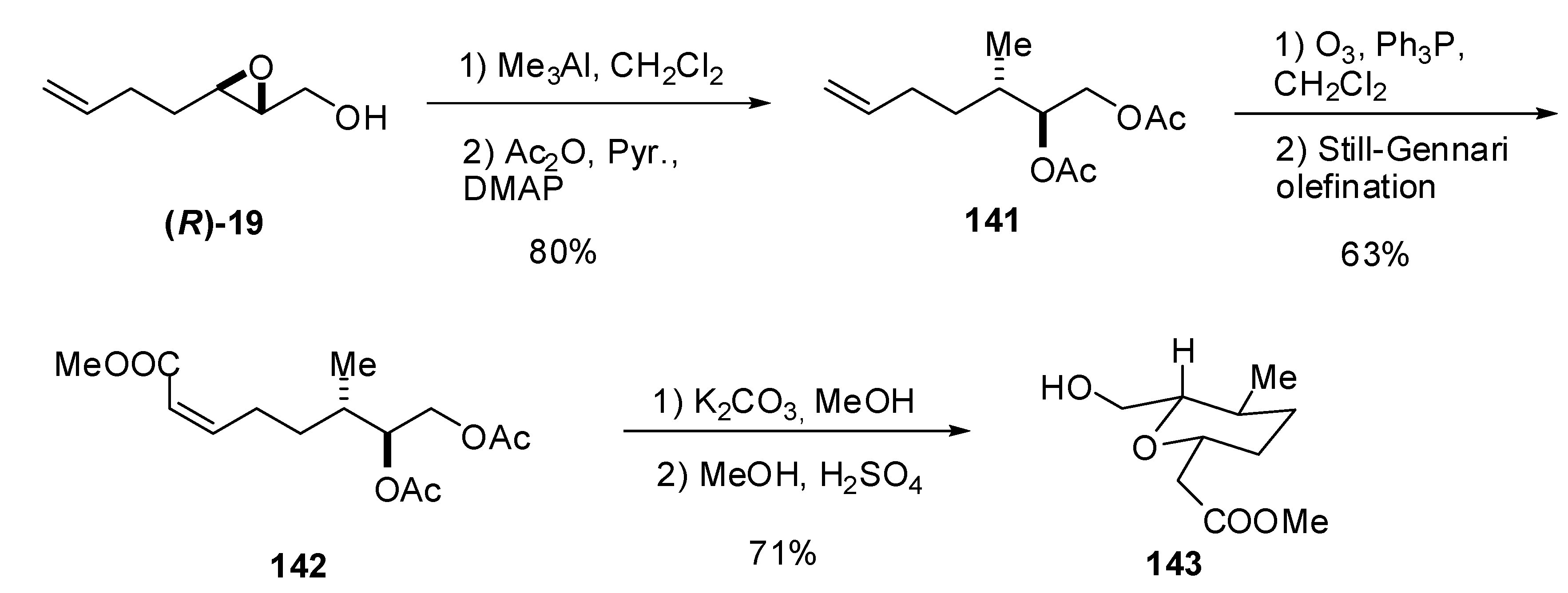
Whereas reaction of epoxy alcohols with cuprates leads to epoxide opening at C2, reaction with trimethylaluminum generally leads to methylation of C3, and consequently, affords a 1,2-diol as the major product. [66,67] Banwell et al. [68] exploited this transformation with the unsaturated epoxide (R)-19 to prepare the tetrahydropyranylic core 143 of the phytotoxic polyketide herboxidiene (Scheme 26). Thus, treatment of (R)-19 with trimethylaluminum provided smooth conversion (opening occurred exclusively on C3) into a diol that was acetylated under standard conditions to obtain 141. The terminal olefinic bond in 141 was subjected to ozonolytic cleavage, followed by reductive work up with triphenylphosphine, to give an aldehyde that was subjected to a Still-Gennari modification of the Wadsworth-Emmons olefination to afford the unsaturated ester 142. Finally, hydrolysis of the acetates, followed by acidic treatment, afforded the target 143.
Scheme 26.
Synthesis of the tetrahydropyranylic core (142) of the phytotoxic compound herboxidiene (Banwell et al. [68]).
Scheme 26.
Synthesis of the tetrahydropyranylic core (142) of the phytotoxic compound herboxidiene (Banwell et al. [68]).

In their studies towards the synthesis of briaran diterpenes, Nantz et al. [69] reported trimethylaluminum-promoted C3-ring opening of substrates having a fully substituted C3 center.
5.2. Nucleophilic attack at C3 with oxygen or sulfur nucleophiles
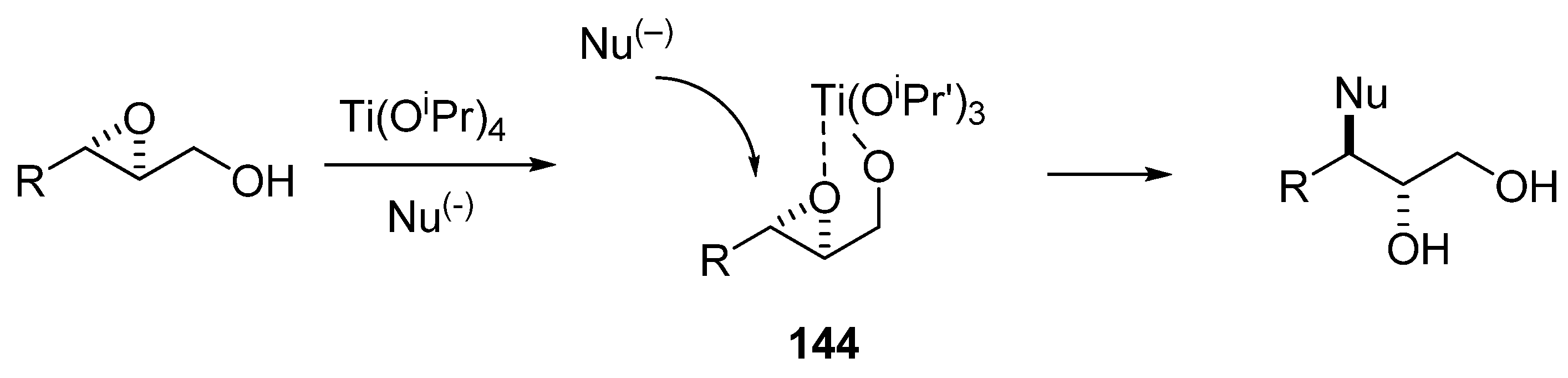
Sharpless [70] developed the use of Ti(iOPr)4 to direct the nucleophilic ring-opening of an epoxy alcohol to C3. Computational studies have shown that attack of the intermediate 144 at C3 is kinetically favored (Scheme 27) [71]. The Sharpless conditions are popular because they provide very high regioselectivity. However, nearly the same level of selectivity can be attained using lithium perchlorate in acetonitrile as Lewis acid, as first described by Crotti et al. [72]. Although these conditions offer slightly lower regioselectivity, they entail a very simple reaction and work-up.
Scheme 27.
Regioselective C-3 ring-opening of epoxy alcohols under Sharpless conditions.

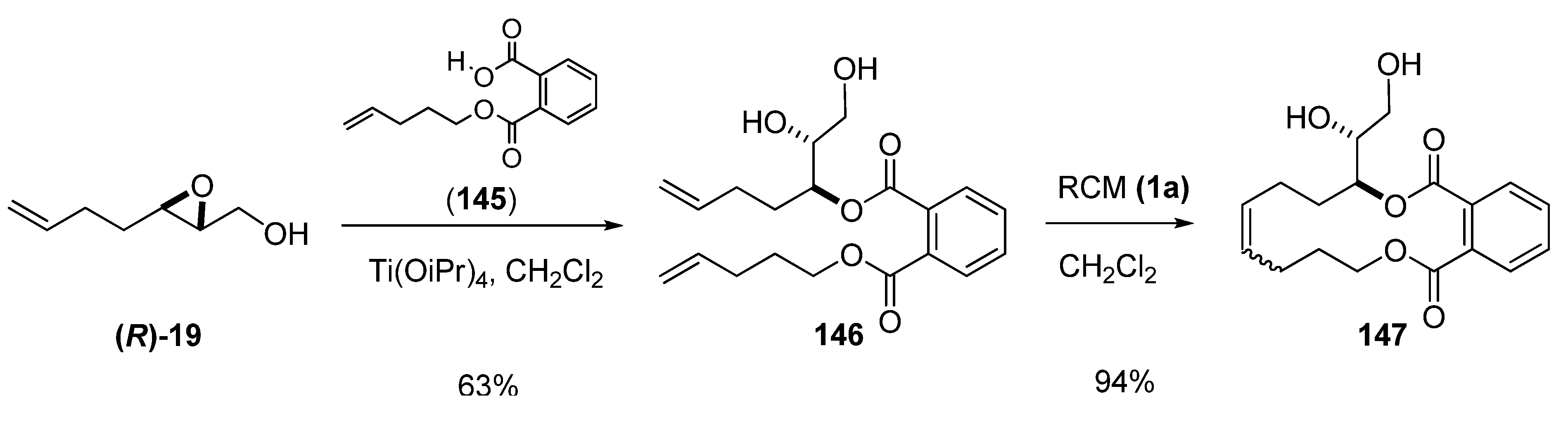
In the preparation of remotely functionalized compounds, Nakata [73] employed RCM of phthalate-tethered dienes. As a representative example, RCM of diol 146 using the first-generation Grubbs’ catalyst (1a) gave the cyclized diol 147 in 94% yield (Scheme 28). The chiral diene 146 had been prepared from the epoxy alcohol 19 by regioselective epoxide-opening at C3 using a phthalic acid monoester as a nucleophile under Sharpless conditions.
Scheme 28.
Ring-closing metathesis of a phthalate-tethered diene (Nakata et al. [73]).
Scheme 28.
Ring-closing metathesis of a phthalate-tethered diene (Nakata et al. [73]).

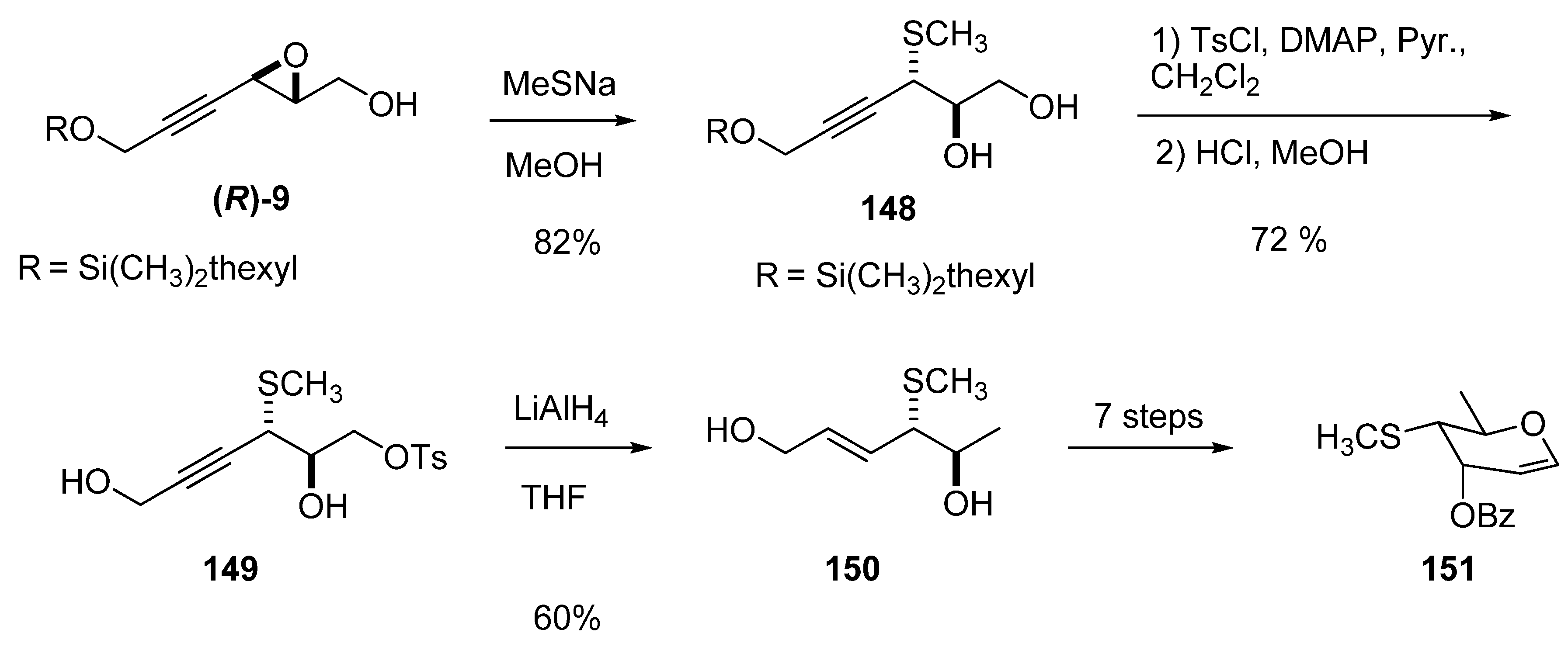
Ring-opening of 3-alkynylepoxy alcohols such as 9 at the propargylic position (C3) by a good nucleophile does not usually require Lewis acid promotion. For example, Beau et al. [51] prepared the glycal 151 (a synthetic precursor of the trisaccharide present in calicheamycins and esperamycins) by opening the epoxide (R)-9 with sodium methanethiolate at 0 ºC in dry MeOH (Scheme 29). The reaction was completely regioselective at C3 to give the diol 148 in 82% yield. The primary hydroxyl group was protected as the tosylate, and then the ether in the resulting adduct was deprotected with acidic MeOH to form the diol 149. With the correct stereochemistry and substitution at C2 and C3, the C1 position of diol 149 was deoxygenated with concomitant triple bond reduction using LiAlH4 in THF at 0 ºC. The stereochemistry of the new double bound was E. Finally, the crude diol was recrystallized from toluene to enantiomerically pure 150 and then transformed into the target glycal 151 in seven steps.
Scheme 29.
Synthesis of a glycal precursor (151) of the trisaccharide found in calicheamycins and esperamycins (Beau et al. [51]).
Scheme 29.
Synthesis of a glycal precursor (151) of the trisaccharide found in calicheamycins and esperamycins (Beau et al. [51]).

5.3. Nucleophilic attack at C3 with nitrogen nucleophiles
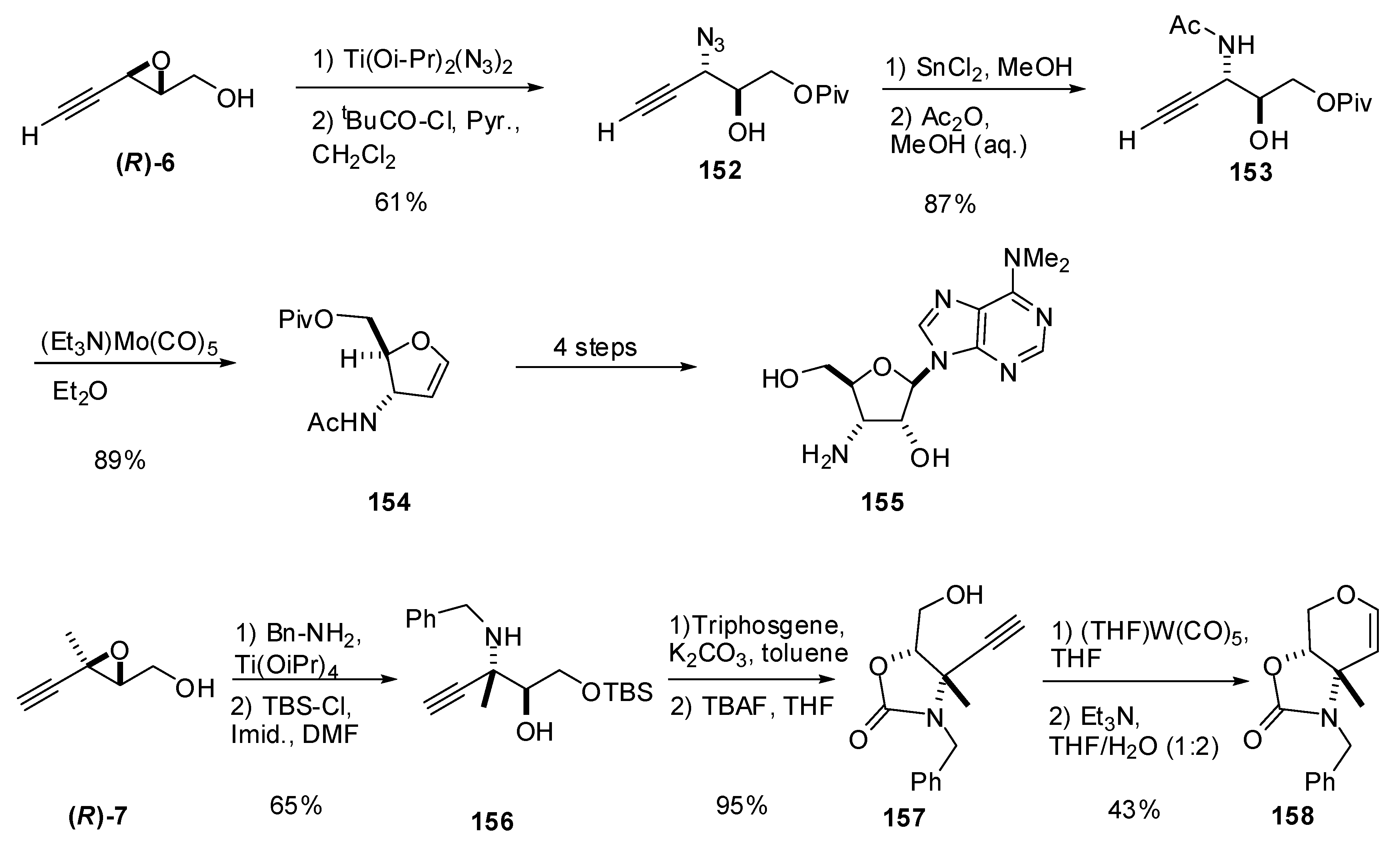
In a program devoted to carbohydrate synthesis, McDonald et al. [74] studied the preparation of glycals by metal-catalyzed alkynol cyclizations. The starting chiral alkynols were prepared from alkynyl epoxides via C3 ring-opening of alkynyl epoxy alcohols with nitrogen nucleophiles. The epoxide (R)-6 was subjected to titanium-mediated regioselective addition of azide at C3, giving a diol that was then selectively protected at the primary alcohol to give 152 (Scheme 30). Mosher analysis of the secondary alcohol of 152 indicated an ee of 92%. Reduction of the azide followed by acetylation provided the 3-amidoalkynol 153, which was submitted to the molybdenum-catalyzed alkynol cyclization to give the 3-amidoglycal 154 (a powerful synthetic precursor to myriad biologically active nucleosides, including puromycin aminonucleoside (155)).
The same research group used the acetylenic epoxide (R)-7 as starting material to prepare the enantiomerically enriched carbamate 157 [75]. This compound was used as a benchmark to study a novel tungsten-carbonyl induced cyclization used to assemble pyranose glycals. Reaction of benzylamine with (R)-7 under Sharpless conditions cleanly opened the epoxide at C3 (Scheme 30). The primary alcohol was then protected as the silyl ether to give 156. The amine and secondary alcohol were protected as the cyclic carbamate, and the silyl ether was cleaved to afford the alkynyl alcohol 157, the alkynol substrate for the cyclization. The tungsten carbonyl-induced cyclization of 157 gave a tungsten oxacarbene in good yield, which was converted into the organic pyranose glycal 158 under mildly basic conditions.
Scheme 30.
Synthesis of glycals via tungsten or molybdenum carbonyl-induced cyclizations of chiral alkynols (McDonald et al. [74]).
Scheme 30.
Synthesis of glycals via tungsten or molybdenum carbonyl-induced cyclizations of chiral alkynols (McDonald et al. [74]).

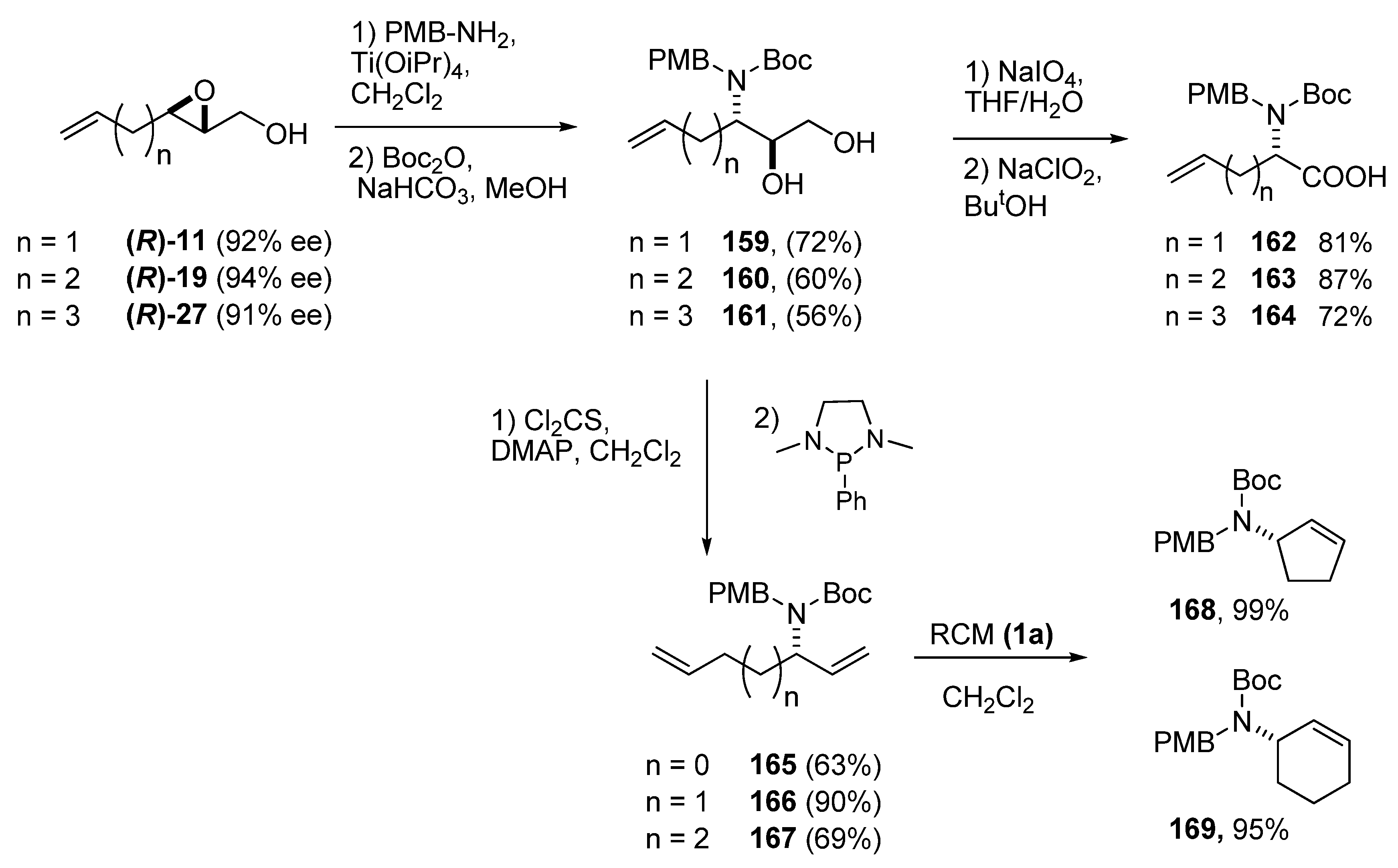
Riera et al. has extensively employed C3 ring-opening of epoxy alcohols with nitrogen nucleophiles [76,77,78,79,80,81] to prepare biologically-active amino acids. Simple, unsaturated amino acids were prepared by using p-methoxybenzylamine as ammonia equivalent [82]. The epoxy alcohols 11, 19 and 27 were treated with p-methoxybenzylamine and Ti(iPrO)4, and subsequently protected with Boc2O give the N-Boc-N-(4’-methoxybenzyl)-3-(amino)-1,2-diols 159-161 (Scheme 31). The 1,2-diol fragments were first cleaved with sodium periodate to the corresponding aldehyde, which was immediately oxidized by sodium chlorite to give the corresponding PMB/Boc protected amino acids 162-164 in good yields. The two protecting groups could be selectively deprotected in any order. Corey-Hopkins deoxygenation of the diol fragment to the terminal alkene [83] enabled preparation of the chiral allylamines 165-167, which were then submitted to RCM [84]. The cyclopentenyl and cyclohexenyl amines 168 and 169, respectively, were obtained in excellent yields. However, attempts at cyclizing 165 failed completely (the starting material was completely recovered), most likely due to the strain of the cyclobutane ring.
Scheme 31.
Enantioselective synthesis of unsaturated amino acids and cyclic allyl amines via nucleophilic ring-opening of epoxy alcohols at C3 (Riera et al. [76,77,78,79,80,81]).

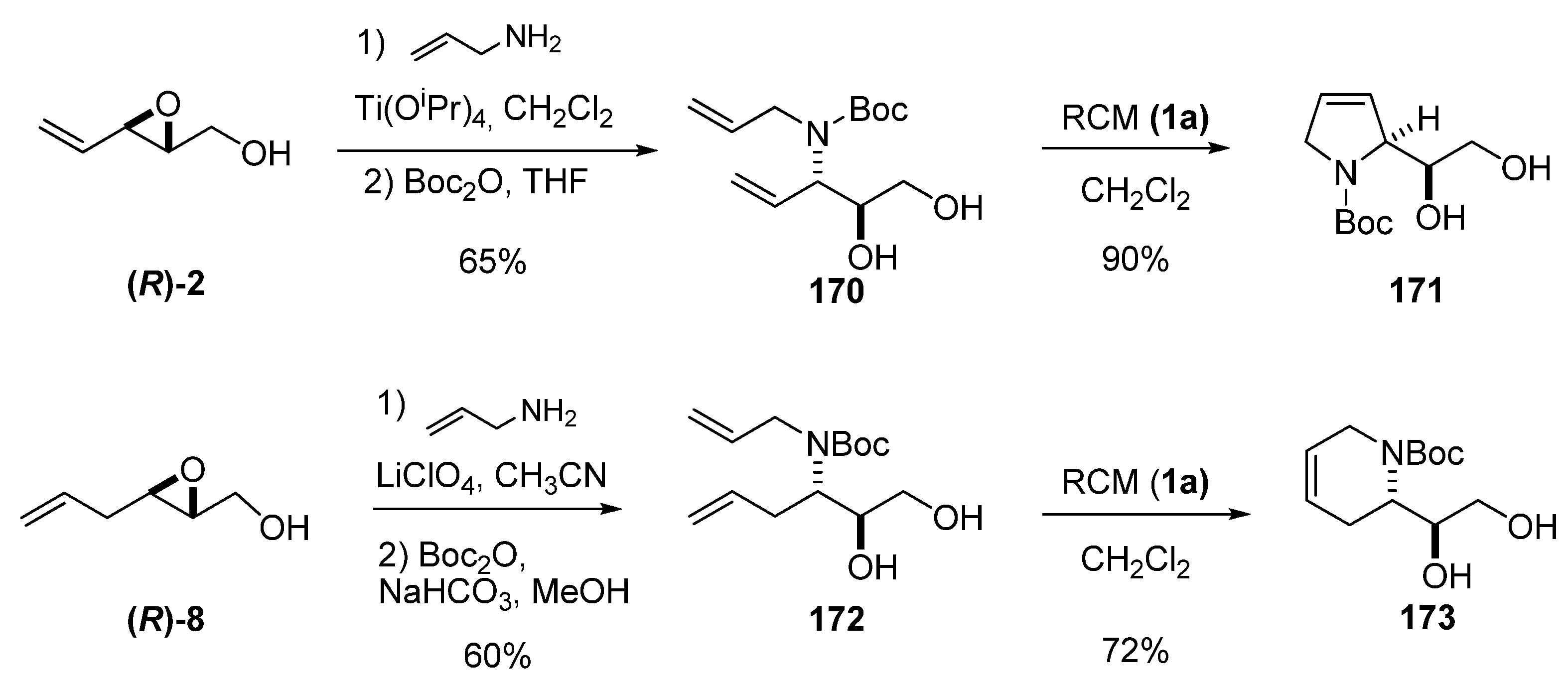
Regioselective and regiospecific ring-opening of unsaturated epoxy alcohols at C3 with allylamine is the most straightforward procedure for introducing the second unsaturation needed to prepare cyclic compounds by RCM. Riera et al. [85,86] developed several enantioselective syntheses of biologically interesting compounds based on this methodology. The aminodiol 170 was prepared via ring-opening of the epoxide (R)-2 by allyl amine under Sharpless conditions followed by N-Boc-protection in 65% overall yield (Scheme 32). RCM of 170 using the first-generation Grubbs’ catalyst 1a gave 171 (a key intermediate in the preparation of polyhydroxylated pyrrolidines) in excellent yield. An analogous sequence with the epoxide (R)-8 afforded aminodiol 172, which was submitted to RCM to give the dehydropiperidine 173 in excellent yield. The ring-opening was performed under Crottis’s conditions using lithium perchlorate. It is worth noting that the key intermediate 173 was enantiomerically enriched by crystallization.

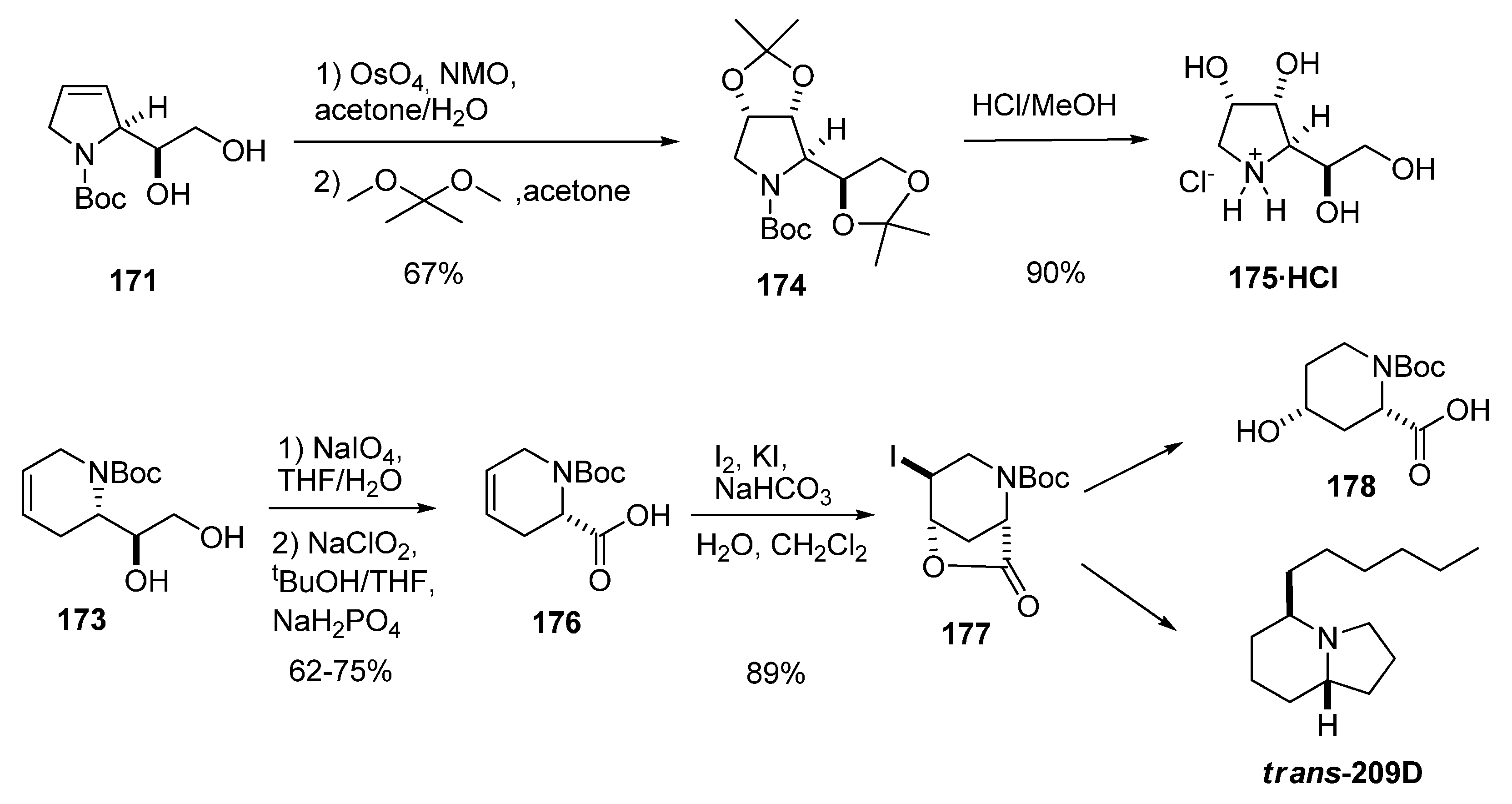
Both intermediates were transformed into biologically active compounds (Scheme 33): 171 was converted into several polyhydroxylated pyrrolidines such as 1,4-dideoxy-1,4-imino-D-allitol (175), whereas enantiomerically pure 173 was transformed, via oxidation to baikianine 176, into 4-hydroxy N-Boc-cis-4-hydroxypipecolic acid (178) [64] and the indolizidine alkaloid trans-209 (isolated from the skin of certain neotropical frogs) [87].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
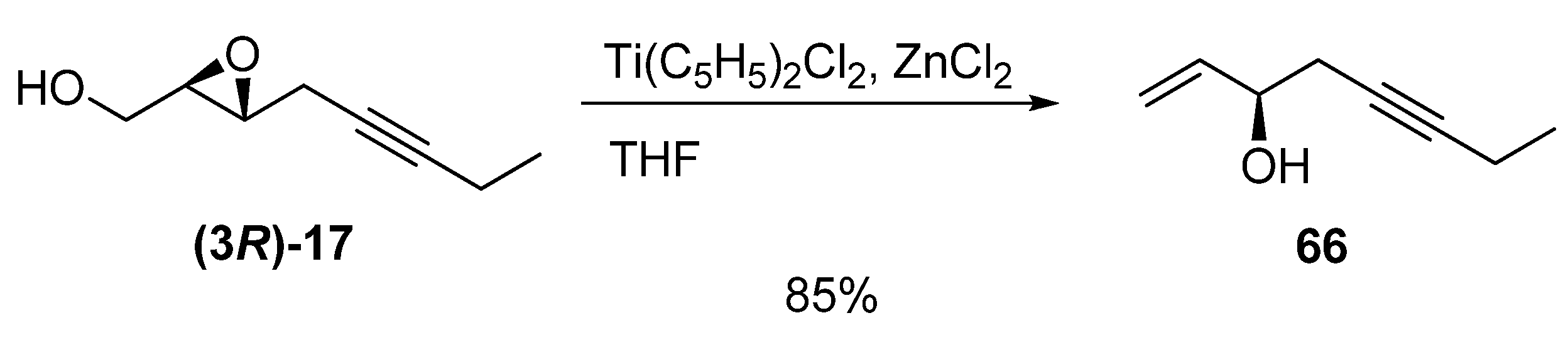{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
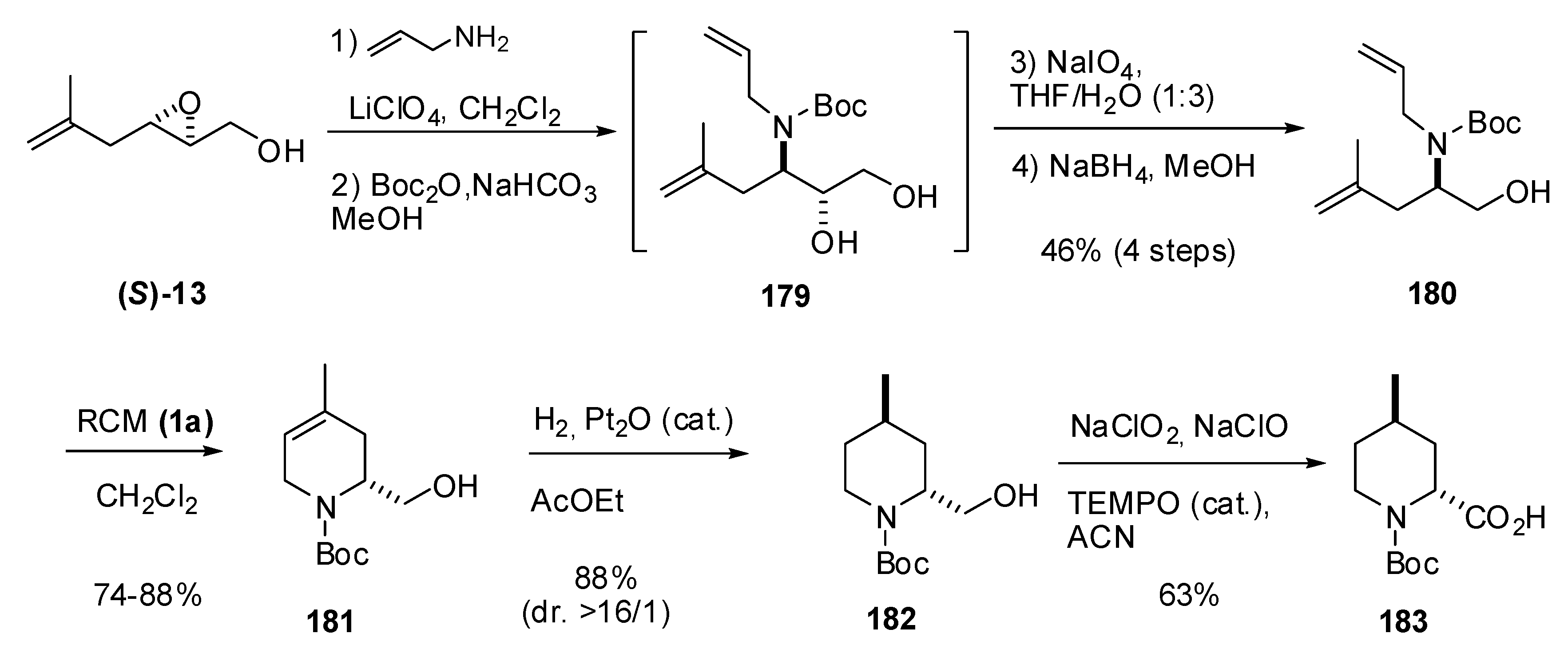
The same methodology was used in the synthesis of trans-4-methylpipecolic acid (183), a fragment of the thrombin inhibitor argatroban [88]. In this case, the regioselectivity of the C3 ring-opening in the epoxide (S)-13 was somewhat lower than in (R)-8 due to the increased steric hindrance of the side chain in the former (Scheme 34). However, oxidation with sodium perchlorate, followed by reduction of the aldehyde to the alcohol, enabled preparation of the diene 180 in good yield [46% from (S)-13)]. Subsequent RCM afforded dehydropiperidine 181 that was diastereoselectively hydrogenated to the trans isomer 182. The primary alcohol was required for obtaining good diastereoselectivities in the hydrogenation. Oxidation of the alcohol to the acid enabled completion of the first asymmetric synthesis of trans-4-methylpipecolic acid (183).

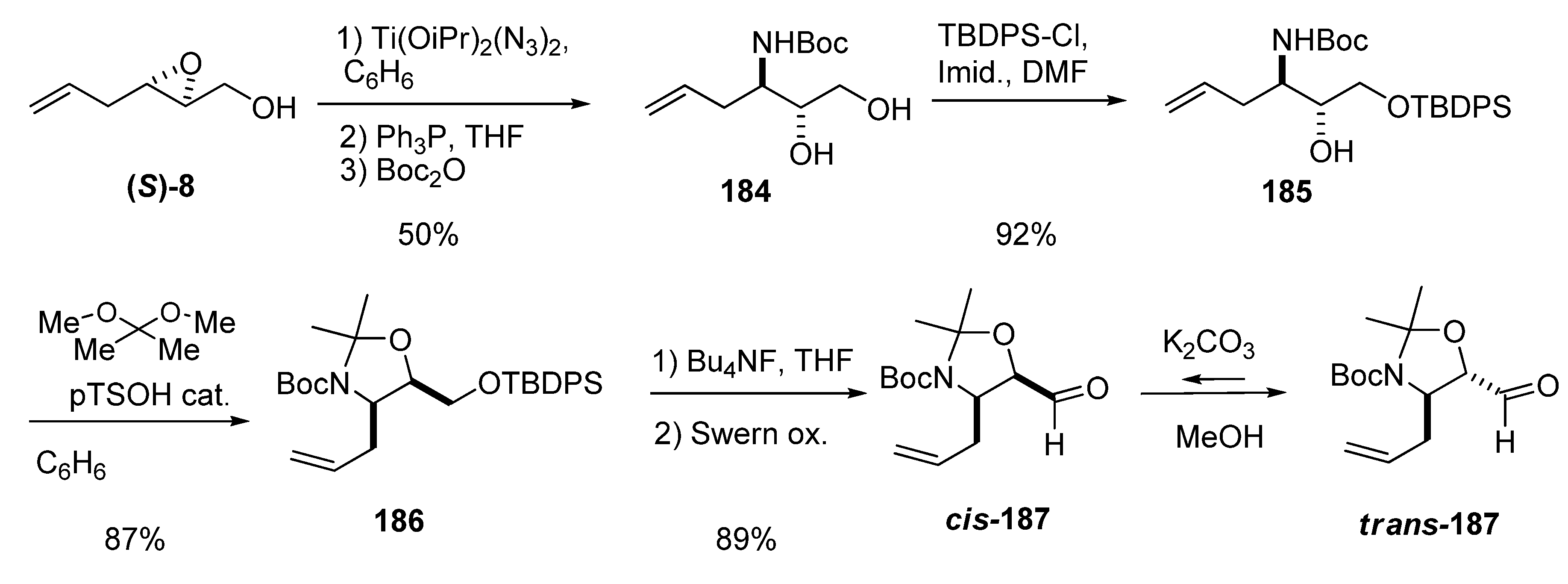
Ring-opening of epoxy alcohols at C3 with azide can provide excellent regioselectivity if performed with titanium diazidodiisopropoxide, whether isolated or prepared in situ. Riera et al. [89] used this reagent in a multigram synthesis of the N-Boc-3-amino-1,2-diol 184 that did not require purification of the intermediates. The epoxide (S)-8 was treated with titanium diazidodiisopropoxide at 75 ºC in benzene, and the resulting crude azidodiol was reduced with triphenylphosphine and then protected with Boc2O, affording the diol 184 in 50% overall yield (Scheme 35). Diol 184 was converted into carbaldehydes 187 through a high yielding protocol of protecting group manipulation and Swern oxidation. These carbaldehydes were later used in the enantioselective synthesis of 3-amino-2,3,6-trideoxysugars, conduramines and aminocyclitols.
Scheme 35.
Preparation of carbaldehydes cis and trans-187, key intermediates in the synthesis of 3-amino-2,3,6-trideoxysugars, conduramines and aminocyclitols.
Scheme 35.
Preparation of carbaldehydes cis and trans-187, key intermediates in the synthesis of 3-amino-2,3,6-trideoxysugars, conduramines and aminocyclitols.

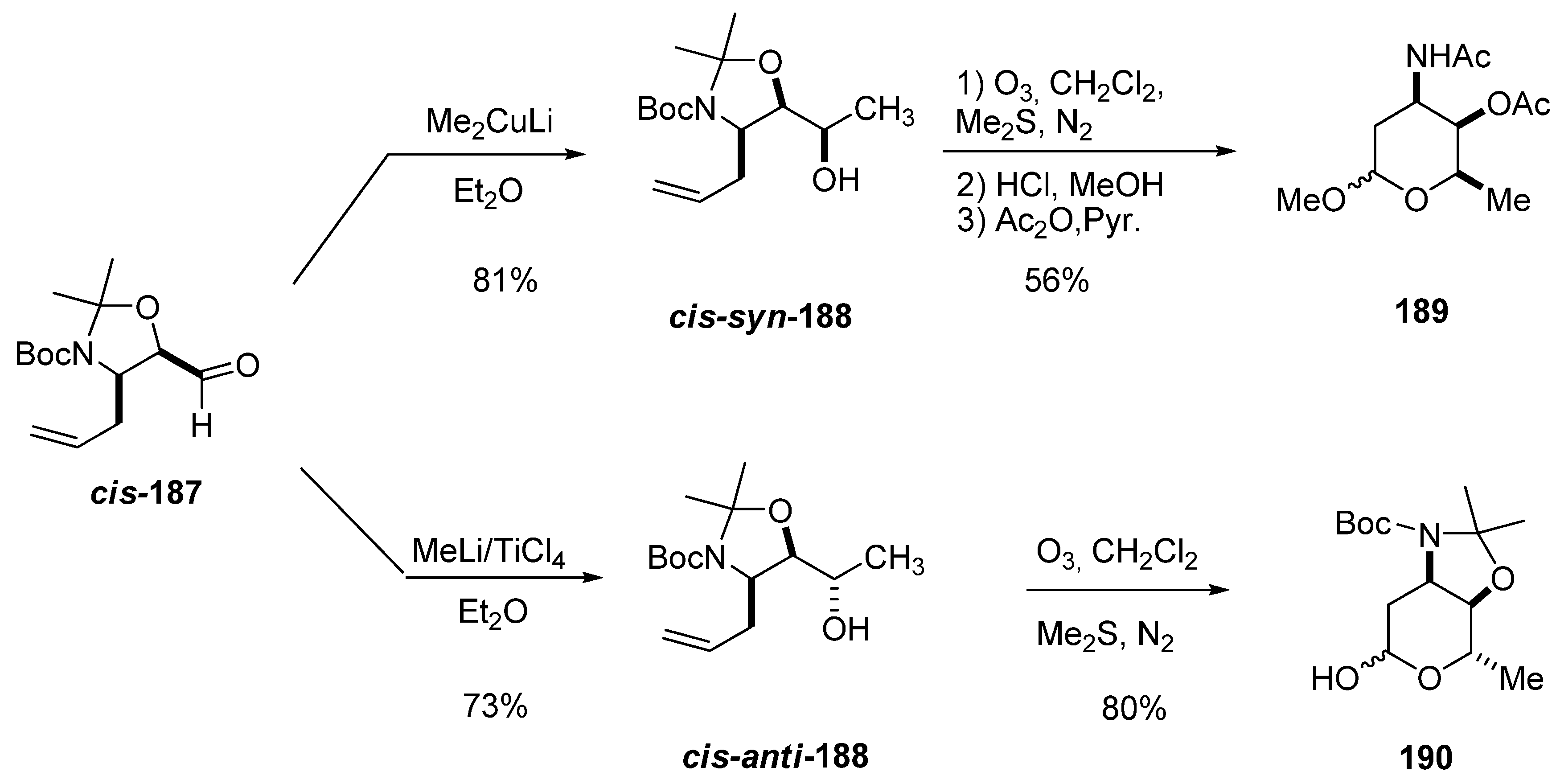
The 3-amino-2,3,6-trideoxysugars were prepared from carbaldehydes cis-187 and trans-187 via diastereoselective addition of a methyl metal reagent followed by ozonolysis of the double bond [89]. Interestingly, in the reactions of cis-187, lithium dimethyl cuprate mainly afforded the syn alcohol (93:7) cis-syn-188, whereas MeLi/TiCl4 led primarily to the anti alcohol (96:4) cis-anti-188. Reductive ozonolysis followed by acetylation afforded protected D-daunosamine (189) and L-ristosamine (190). A similar trend was observed for trans-187, which was converted into the protected L-epi-daunosamine and D-acosamine using the same reaction sequence. Thus, this approach constitutes a general synthesis of 3-amino-2,3,6-trideoxysugars with complete control of the stereochemistry of the three contiguous stereogenic centers.
Scheme 36.
Synthesis of protected D-daunosamine (189) and L-ristosamine (190) from aldehyde cis-187 (Riera et al. [89]).
Scheme 36.
Synthesis of protected D-daunosamine (189) and L-ristosamine (190) from aldehyde cis-187 (Riera et al. [89]).

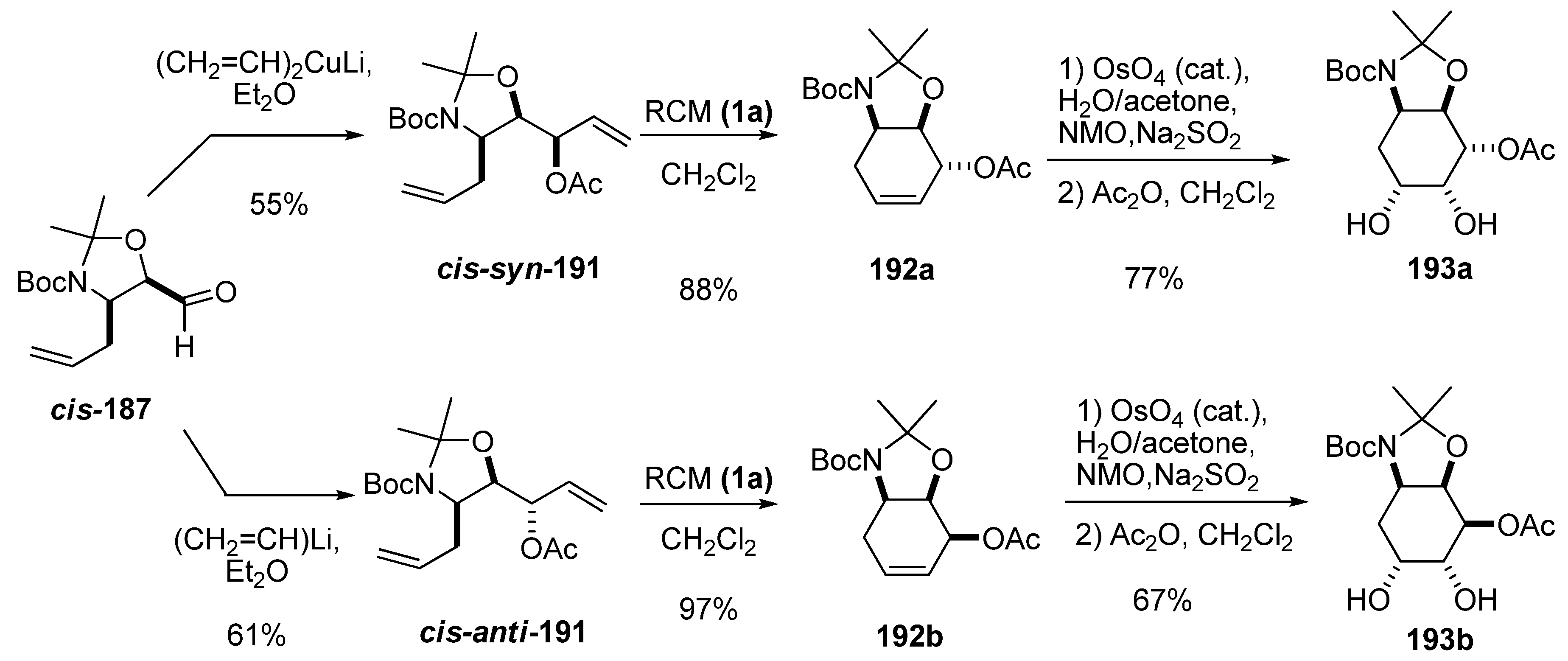
Diastereoselective addition of a vinyl group either to aldehydes cis and trans-187 enabled preparation of dienes suitable for RCM. This strategy was used to prepare 4-deoxy-3-conduramines with full stereochemical control of three chiral centers [90]. As in the addition of the methyl group, the addition of lithium divinyl cuprate to cis-187 gave excellent stereoselectivity (95:1) towards the syn isomer cis-syn-191. Vinyl lithium afforded the anti isomer cis-anti-191 with less selectivity (4:1). RCM with first-generation Grubbs’ catalyst (1a) of dienes 191 afforded deoxyconduramines 192 in excellent yields. All isomers of the deoxyconduramines could be selectively prepared by choosing the enantiomer of the epoxide, the cis/trans stereochemistry of the aldehyde, and the organometallic vinyl reagent. Moreover, diastereoselective dihydroxylation provided a new family of aminocyclitols 193 with full stereochemical control.
Scheme 37.
Syntheses of some isomers of deoxyconduramines and aminocyclitols from carbaldehyde cis-187 (Riera et al. [90]).
Scheme 37.
Syntheses of some isomers of deoxyconduramines and aminocyclitols from carbaldehyde cis-187 (Riera et al. [90]).

6. Conclusions
Epoxy alcohols with unsaturated side chains have been extensively applied to syntheses of biologically active compounds. They can be prepared in any absolute configuration by Sharpless asymmetric epoxidation. These epoxides have a rich chemistry of completely stereospecific transformations that yield products with predictable stereochemistry. Furthermore, the unsaturated side chain can be efficiently transformed via oxidation or olefin metatheses.
Acknowledgements
We thank MICINN (CTQ2008-000763/BQU) for financial support. MM thanks MICINN for a fellowship.
References and Notes
- Gao, Y.; Klunder, J.M.; Hanson, R.M.; Masamune, H.; Ko, S.Y.; Sharpless, K.B. Catalytic asymmetric epoxidation and kinetic resolution: Modified procedures including in situ derivatization. J. Am. Chem. Soc. 1987, 109, 5765–5780. [Google Scholar]
- Schinzer, D. Asymmetric synthesis.The Sharpless epoxidation. In Organic Synthesis Highlights II; VCH: Weinheim, Germany, 1995; pp. 3–8. [Google Scholar]
- Katsuki, T.; Martin, V. Asymmetric epoxidation of allylic alcohols: The Katsuki-Sharpless epoxidation reaction. Org. React. 1996, 48, 1–299. [Google Scholar]
- Grubbs, R.H.; Chang, S. Recent advances in olefin metathesis and its application in organic synthesis. Tetrahedron 1998, 54, 4413–4450. [Google Scholar] [CrossRef]
- Yet, L. Metal-mediated synthesis of medium-sized rings. Chem. Rev. 2000, 100, 2963–3007. [Google Scholar] [CrossRef]
- Van Otterlo, W.A.L.; de Koning, C.B. Metathesis in the synthesis of aromatic compounds. Chem. Rev. 2009, 109, 3743–3782. [Google Scholar]
- Grubbs, R.H.; Miller, S.J.; Fu, G.C. Ring-closing metathesis and related processes in organic synthesis. Acc. Chem. Res. 1995, 28, 446–452. [Google Scholar] [CrossRef]
- Gibson, S.E.; Keen, S.P. Cross-metathesis. In Topics in Organometallic Chemistry; Springer-Verlag: Berlin, Germany, 1998; pp. 155–181. [Google Scholar]
- Connon, S. Recent developments in olefin cross-metathesis. Angew. Chem. Int. Ed. 2003, 42, 1900. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar]
- Kingsbury, J.S.; Harrity, J.P.A.; Bonitatebus, P.J.; Hoveyda, A.H. A recyclable Ru-based metathesis catalyst. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar]
- Jaeger, V.; Huemmer, W.; Stahl, U.; Gracza, T. Controlled synthesis of enantio-, regio-, and diastereomers of amino-4-pentenediols from 1,4-pentadien-3-ol via epoxy-4-pentenols I. Erythro-1-amino-4-pentene-2,3-diols. Synthesis 1991, 769–776. [Google Scholar]
- Jaeger, V.; Schroeter, D.; Koppenhoefer, B. Asymmetric sharpless epoxidation of divinylcarbinol. erythro-D and -L-4-pentenitols by hydrolysis of regioisomeric epoxy-4-pentenols. Tetrahedron 1991, 47, 2195–2210. [Google Scholar] [CrossRef]
- Jaeger, V.; Stahl, U.; Huemmer, W. Controlled synthesis of regio-, enantio-, and diastereomers of amino-4-pentenediols from 1,4-pentadien-3-ol via epoxy-4-pentenols. II. erythro- and threo-3-amino-4-pentene-1,2-diols and erythro-2-benzylamino-4-pentene-1,3-diol. Synthesis 1991, 776–782. [Google Scholar]
- Payne, G.B. Epoxide migrations with alpha, beta-epoxy alcohols. J. Org. Chem. 1962, 27, 3819–3822. [Google Scholar] [CrossRef]
- Taber, D.F.; Zhang, Z. Synthesis of the enediol isofurans, endogenous oxidation products of arachidonic acid. J. Org. Chem. 2006, 71, 926–933. [Google Scholar]
- Parker, K.A.; Lim, Y. The total synthesis of (-)-SNF4435 C and (+)-SNF4435 D. J. Am. Chem. Soc. 2004, 126, 15968–15969. [Google Scholar] [CrossRef]
- Riou, M.; Barriault, L. De novo synthesis of (+)-isofregenedol. J. Org. Chem. 2008, 73, 7436–7439. [Google Scholar]
- Yuasa, H.; Makado, G.; Fukuyama, Y. Determination of the absolute configuration of vibsanin F by asymmetric synthesis via pi-allylpalladium complex. Tetrahedron Lett. 2003, 44, 6235–6239. [Google Scholar] [CrossRef]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar]
- Takahashi, S.; Kubota, A.; Nakata, T. Stereoselective total synthesis of muconin. Tetrahedron 2003, 59, 1627–1638. [Google Scholar] [CrossRef]
- Pena, P.C.A.; Roberts, S.M. The chemistry of epoxy alcohols. Curr. Org. Chem. 2003, 7, 555–571. [Google Scholar]
- Hanson, R.M. The synthetic methodology of nonracemic glycidol and related 2,3-epoxy alcohols. Chem. Rev. 1991, 91, 437–476. [Google Scholar] [CrossRef]
- Behrens, C.H.; Sharpless, K.B. New transformations of 2,3-epoxy alcohols and related derivatives. Easy routes to homochiral substances. Aldrichim. Acta 1983, 16, 67–80. [Google Scholar]
- Rodriguez, A.; Nomen, M.; Spur, B.W.; Godfroid, J.; Lee, T.H. Total synthesis of leukotrienes from butadiene. Eur. J. Org. Chem. 2000, 2991–3000. [Google Scholar]
- Larrosa, I.; Da Silva, M.I.; Gomez, P.M.; Hannen, P.; Ko, E.; Lenger, S.R.; Linke, S.R.; White, A.J.P.; Wilton, D.; Barrett, A.G.M. Highly convergent three component benzyne coupling: The total synthesis of ent-clavilactone B. J. Am. Chem. Soc. 2006, 128, 14042–14043. [Google Scholar]
- Berberich, S.M.; Cherney, R.J.; Colucci, J.; Courillon, C.; Geraci, L.S.; Kirkland, T.A.; Marx, M.A.; Schneider, M.F.; Martin, S.F. Total synthesis of (+)-ambruticin S. Tetrahedron 2003, 59, 6819–6832. [Google Scholar]
- Heck, M.; Baylon, C.; Nolan, S.P.; Mioskowski, C. Triple ring closing metathesis reaction: Synthesis of adjacent cyclic ethers. Org. Lett. 2001, 3, 1989–1991. [Google Scholar]
- Furstner, A.; Bouchez, L.C.; Funel, J.; Liepins, V.; Poree, F.; Gilmour, R.; Beaufils, F.; Laurich, D.; Tamiya, M. Total syntheses of amphidinolide H and G. Angew. Chem., Int. Ed. 2007, 46, 9265–9270. [Google Scholar]
- Yadav, J.S.; Somaiah, R.; Ravindar, K.; Chandraiah, L. Stereoselective total synthesis of (+)-mueggelone, a novel inhibitor of fish development. Tetrahedron Lett. 2008, 49, 2848–2850. [Google Scholar]
- Corey, E.J.; Marfat, A.; Laguzza, B.C. Total synthesis of 5S,12S-dihydroxy-6,10-E,8,14-Z-eicosatetraenoic acid, a new human metabolite of arachidonic acid. Tetrahedron Lett. 1981, 22, 3339–3342. [Google Scholar] [CrossRef]
- Yadav, J.S.; Shekharam, T.; Gadgil, V.R. Titanocene induced regioselective deoxygenation of 2,3-epoxy alcohols: A new reaction for the synthesis of allylic alcohols. J. Chem. Soc. Chem. Commun. 1990, 843–844. [Google Scholar]
- Fukuda, Y.; Shindo, M.; Shishido, K. Total synthesis of (-)-aspidospermine via Diastereoselective ring-closing olefin metathesis. Org. Lett. 2003, 5, 749–751. [Google Scholar] [CrossRef]
- Kanada, R.M.; Itoh, D.; Nagai, M.; Niijima, J.; Asai, N.; Mizui, Y.; Abe, S.; Kotake, Y. Total synthesis of the potent antitumor macrolides pladienolide B and D. Angew. Chem. Int. Ed. 2007, 46, 4350–4355. [Google Scholar]
- Wang, Z.; Tu, Y.; Frohn, M.; Zhang, J.; Shi, Y. An efficient catalytic asymmetric epoxidation method. J. Am. Chem. Soc. 1997, 119, 11224–11235. [Google Scholar] [CrossRef]
- Sarandeses, L.A.; Mourino, A.; Luche, J.L. Cleavage of 2,3-epoxyalkyl halides by the sonochemical zinc-copper couple. J. Chem. Soc. Chem. Commun. 1991, 818–820. [Google Scholar]
- Ichige, T.; Okano, Y.; Kanoh, N.; Nakata, M. Total synthesis of methyl sarcophytoate, a marine natural biscembranoid. J. Org. Chem. 2009, 74, 230–243. [Google Scholar] [CrossRef]
- Gao, Y.; Klunder, J.M.; Hanson, R.M.; Masamune, H.; Ko, S.Y.; Sharpless, K.B. Catalytic asymmetric epoxidation and kinetic resolution: Modified procedures including in situ derivatization. J. Am. Chem. Soc. 1987, 109, 5765–5780. [Google Scholar]
- Van Dyke, A.R.; Jamison, T.F. Functionalized templates for the convergent assembly of polyethers: Synthesis of the HIJK rings of gymnocin A. Angew. Chem. Int. Ed. 2009, 48, 4430–4432. [Google Scholar] [CrossRef] [Green Version]
- Yadav, J.S.; Deshpande, P.K.; Sharma, G.V.M. An effective, practical method for the synthesis of chiral propargyl alcohols. Tetrahedron 1990, 46, 7033–7046. [Google Scholar]
- Yadav, J.S.; Bhanu, L.R.M.; Dutta, D. Stereoselective total syntheses of (9S)- and (9R)-HETE. Tetrahedron 1998, 54, 3929–3934. [Google Scholar] [CrossRef]
- Marshall, J.A.; Piettre, A.; Paige, M.A.; Valeriote, F. A modular synthesis of annonaceous acetogenins. J. Org. Chem. 2003, 68, 1771–1779. [Google Scholar]
- Hoye, T.R.; Suhadolnik, J.C. Stereocontrolled synthesis of 2,5-linked bistetrahydrofurans via the triepoxide cascade reaction. Tetrahedron 1986, 42, 2855–2862. [Google Scholar] [CrossRef]
- Kong, L.; Zhuang, Z.; Chen, Q.; Deng, H.; Tang, Z.; Jia, X.; Li, Y.; Zhai, H. A facile asymmetric synthesis of (+)-eldanolide. Tetrahedron Asymmetry 2007, 18, 451–454. [Google Scholar] [CrossRef]
- Krishna, P.R.; Lopinti, K. A concise stereoselective total synthesis of (2R,2'R)-threo-(+)-methylphenidate via a ring-closing metathesis protocol. Synlett 2007, 1742–1744. [Google Scholar]
- Ma, S.; Ni, B. Double ring-closing metathesis reaction of nitrogen-containing tetraenes: Efficient construction of bicyclic alkaloid skeletons and synthetic application to four stereoisomers of lupinine and their derivatives. Chem. Eur. J. 2004, 10, 3286–3300. [Google Scholar] [CrossRef]
- Sabitha, G.; Sudhakar, K.; Reddy, N.M.; Rajkumar, M.; Yadav, J.S. Chelation-controlled reduction: An enantioselective synthesis of (-)-tarchonanthuslactone. Tetrahedron Lett. 2005, 46, 6567–6570. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Lei, H. Chelation-controlled reduction: Stereoselective formation of syn-1,3-diols and synthesis of compactin and mevinolin lactone. J. Org. Chem. 2002, 67, 8783–8788. [Google Scholar] [CrossRef]
- Ball, M.; Bradshaw, B.J.; Dumeunier, R.; Gregson, T.J.; MacCormick, S.; Omori, H.; Thomas, E.J. A preliminary evaluation of a metathesis approach to bryostatins. Tetrahedron Lett. 2006, 47, 2223–2227. [Google Scholar]
- Hiebel, M.; Pelotier, B.; Lhoste, P.; Piva, O. Synthesis of bistramide A and analogues, part 1: Stereoselective access to normethyl tetrahydropyran subunit. Synlett 2008, 1202–1204. [Google Scholar]
- Dupradeau, F.; Prandi, J.; Beau, J. Synthesis of 2,6-dideoxy-4-S-methyl-4-thio-D-ribo-hexopyranose, a component of the esperamycin oligosaccharide. Tetrahedron 1995, 51, 3205–3220. [Google Scholar]
- Gopalarathnam, A.; Nelson, S.G. Amphidinolide, B: Asymmetric synthesis of a C7-C20 synthon. Org. Lett. 2006, 8, 7–10. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Lei, H. An enantioselective synthesis of the core unit of the non-nucleoside reverse transcriptase inhibitor taurospongin A. Tetrahedron Asymmetry 2003, 14, 629–634. [Google Scholar] [CrossRef]
- Lebel, H.; Jacobsen, E.N. Enantioselective total synthesis of taurospongin A. J. Org. Chem. 1998, 63, 9624–9625. [Google Scholar]
- Roush, W.R.; Brown, R.J. Total synthesis of carbohydrates. 3. Efficient enantioselective syntheses of 2,6-dideoxyhexoses. J. Org. Chem. 1983, 48, 5093–5101. [Google Scholar] [CrossRef]
- Roush, W.R.; Adam, M.A. Directed openings of 2,3-epoxy alcohols via reactions with isocyanates: synthesis of (+)-erythro-dihydrosphingosine. J. Org. Chem. 1985, 50, 3752–3757. [Google Scholar]
- Martin, R.; Moyano, A.; Pericas, M.A.; Riera, A. A Concise enantioselective entry to the synthesis of deoxy-aza-sugars. Org. Lett. 2000, 2, 93–95. [Google Scholar] [CrossRef]
- Martin, R.; Murruzzu, C.; Pericas, M.A.; Riera, A. General approach to glycosidase inhibitors. Enantioselective synthesis of deoxymannojirimycin and swainsonine. J. Org. Chem. 2005, 70, 2325–2328. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Hermann, C.Y.W.; Dong, Q.; Shimizu, T.; Swaminathan, S.; Xi, N. Nitrogen heterocycles from furans. The aza-Achmatowicz reaction. Synlett 1998, 105–114. [Google Scholar]
- Shirai, M.; Okamoto, S.; Sato, F. Practical synthesis of optically active bicyclic oxazolidinylpiperidines, chiral building blocks for preparing 1-deoxyazasugars, from serine. Tetrahedron Lett. 1999, 40, 5331–5332. [Google Scholar] [CrossRef]
- Asano, K.; Hakogi, T.; Iwama, S.; Katsumura, S. New entry for asymmetric deoxyazasugar synthesis: Syntheses of deoxymannojirimycin, deoxyaltrojirimycin and deoxygalactostatin. Chem. Commun. 1999, 41–42. [Google Scholar]
- Al-Rawi, S.; Hinderlich, S.; Reutter, W.; Giannis, A. Sialic acids: Synthesis and biochemical properties of reversible inhibitors of UDP-N-acetylglucosamine 2-epimerase. Angew. Chem. Int. Ed. 2004, 43, 4366–4370. [Google Scholar] [CrossRef]
- Murruzzu, C.; Alonso, M.; Canales, A.; Jimenez-Barbero, J.; Riera, A. Synthesis and NMR experiments of (4,5,6-13C)-deoxymannojirimycin. A new entry to 13C-labeled glycosidase inhibitors. Carbohydr. Res. 2007, 342, 1805–1812. [Google Scholar] [CrossRef]
- Alegret, C.; Ginesta, X.; Riera, A. Asymmetric synthesis of cis-4- and trans-3-hydroxypipecolic acids. Eur. J. Org. Chem. 2008, 1789–1796. [Google Scholar]
- Ginesta, X.; Pericas, M.A.; Riera, A. Enantioselective synthesis of erythro-β-hydroxyglutamic acid. Synth. Commun. 2005, 35, 289–297. [Google Scholar] [CrossRef]
- Suzuki, T.; Saimoto, H.; Tomioka, H.; Oshima, K.; Nozaki, H. Regio- and stereoselective ring opening of epoxy alcohols with organoaluminum compounds leading to 1,2-diols. Tetrahedron Lett. 1982, 23, 3597–3600. [Google Scholar] [CrossRef]
- Roush, W.R.; Adam, M.A.; Peseckis, S.M. Regioselectivity of the reactions of trialkylaluminum reagents with 2,3-epoxy alcohols: Application to the synthesis of alpha -chiral aldehydes. Tetrahedron Lett. 1983, 24, 1377–1380. [Google Scholar]
- Banwell, M.G.; McLeod, M.D.; Premraj, R.; Simpson, G.W. Improved synthetic route to enantiomerically pure samples of the tetrahydropyran-2-ylacetic acid core associated with the phytotoxic polyketide herboxidiene. Aust. J. Chem. 2000, 53, 659–664. [Google Scholar] [CrossRef]
- Balasubramaniam, R.P.; Moss, D.K.; Wyatt, J.K.; Spence, J.D.; Gee, A.; Nantz, M.H. Methylation-ring opening of 3,3-disubstituted 2,3-epoxy alcohols. Synthesis of chiral quaternary fragments for assembly of briaran diterpenes. Tetrahedron 1997, 53, 7429–7444. [Google Scholar]
- Caron, M.; Sharpless, K.B. Titanium isopropoxide-mediated nucleophilic openings of 2,3-epoxy alcohols. A mild procedure for regioselective ring-opening. J. Org. Chem. 1985, 50, 1557–1560. [Google Scholar] [CrossRef]
- Infante, I.; Bonini, C.; Lelj, F.; Righi, G. A first theoretical study on the origin of the metal-mediated regioselective opening of 2,3-epoxy alcohols. J. Org. Chem. 2003, 68, 3773–3780. [Google Scholar]
- Chini, M.; Crotti, P.; Macchia, F. Regioalternating selectivity in the metal salt catalyzed aminolysis of styrene oxide. J. Org. Chem. 1991, 56, 5939–5942. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Okazaki, M.; Miyamoto, K.; Nakata, T. Efficient phthalate-tethered ring-closing metathesis as a cross-coupling reaction. Tetrahedron Lett. 2001, 42, 7633–7636. [Google Scholar]
- McDonald, F.E.; Gleason, M.M. Asymmetric synthesis of nucleosides via molybdenum-catalyzed alkynol cycloisomerization coupled with stereoselective glycosylations of deoxyfuranose glycals and 3-amidofuranose glycals. J. Am. Chem. Soc. 1996, 118, 6648–6659. [Google Scholar] [CrossRef]
- McDonald, F.E.; Zhu, H.Y.H. Synthesis of pyranose glycals via tungsten and molybdenum pentacarbonyl-induced alkynol cyclizations. Tetrahedron 1997, 53, 11061–11068. [Google Scholar] [CrossRef]
- Poch, M.; Alcon, M.; Moyano, A.; Pericas, M.A.; Riera, A. A short enantioselective synthesis of N-Boc-α-amino acids from epoxy alcohols. Tetrahedron Lett. 1993, 34, 7781–7784. [Google Scholar] [CrossRef]
- Pasto, M.; Moyano, A.; Pericas, M.A.; Riera, A. An enantioselective, stereodivergent approach to anti- and syn-α-hydroxy-β-amino acids from anti-3-amino-1,2-diols. Synthesis of the ready for coupling Taxotere side chain. Tetrahedron Asymmetry 1996, 7, 243–262. [Google Scholar] [CrossRef]
- Pasto, M.; Castejon, P.; Moyano, A.; Pericas, M.A.; Riera, A. A catalytic asymmetric synthesis of cyclohexylnorstatine. J. Org. Chem. 1996, 61, 6033–6037. [Google Scholar]
- Pasto, M.; Moyano, A.; Pericas, M.A.; Riera, A. A catalytic asymmetric synthesis of N-Boc-β-methylphenylalanines. J. Org. Chem. 1997, 62, 8425–8431. [Google Scholar]
- Medina, E.; Moyano, A.; Pericas, M.A.; Riera, A. Enantioselective syntheses of conformationally rigid, highly lipophilic mesityl-substituted amino acids. Helv. Chim. Acta 2000, 83, 972–988. [Google Scholar] [CrossRef]
- Martin, R.; Islas, G.; Moyano, A.; Pericas, M.A.; Riera, A. A new method for the enantioselective synthesis of N-Boc-α,α-disubstituted a-amino acids. Tetrahedron 2001, 57, 6367–6374. [Google Scholar] [CrossRef]
- Alcon, M.; Moyano, A.; Pericas, M.A.; Riera, A. Enantioselective synthesis of unsaturated amino acids using p-methoxybenzylamine as an ammonia equivalent. Tetrahedron Asymmetry 1999, 10, 4639–4651. [Google Scholar]
- Alcon, M.; Poch, M.; Moyano, A.; Pericas, M.A.; Riera, A. Enantioselective synthesis of (S)-vigabatrin. Tetrahedron Asymmetry 1997, 8, 2967–2974. [Google Scholar]
- Martin, R.; Alcon, M.; Pericas, M.A.; Riera, A. Ring-closing metathesis of chiral allylamines. Enantioselective synthesis of (2S,3R,4S)-3,4-dihydroxyproline. J. Org. Chem. 2002, 67, 6896–6901. [Google Scholar] [CrossRef]
- Murruzzu, C.; Riera, A. Enantioselective synthesis of hydroxylated pyrrolidines via sharpless epoxidation and olefin metathesis. Tetrahedron Asymmetry 2007, 18, 149–154. [Google Scholar]
- Ginesta, X.; Pericas, M.A.; Riera, A. Straightforward entry to the pipecolic acid nucleus. Enantioselective synthesis of baikiain. Tetrahedron Lett. 2002, 43, 779–782. [Google Scholar] [CrossRef]
- Alegret, C.; Riera, A. Enantioselective synthesis of indolizidine alkaloid trans-209D. J. Org. Chem. 2008, 73, 8661–8664. [Google Scholar]
- Alegret, C.; Santacana, F.; Riera, A. Enantioselective synthesis of trans-4-methylpipecolic acid. J. Org. Chem. 2007, 72, 7688–7692. [Google Scholar] [CrossRef]
- Ginesta, X.; Pasto, M.; Pericas, M.A.; Riera, A. New stereodivergent approach to 3-amino-2,3,6-trideoxysugars. Enantioselective synthesis of daunosamine, ristosamine, acosamine, and epi-daunosamine. Org. Lett. 2003, 5, 3001–3004. [Google Scholar] [CrossRef]
- Alegret, C.; Benet-Buchholz, J.; Riera, A. Stereodivergent syntheses of conduramines and aminocyclitols. Org. Lett. 2006, 8, 3069–3072. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2010 by the authors;
Share and Cite
MDPI and ACS Style
Riera, A.; Moreno, M. Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation. Molecules 2010, 15, 1041-1073. https://doi.org/10.3390/molecules15021041
AMA Style
Riera A, Moreno M. Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation. Molecules. 2010; 15(2):1041-1073. https://doi.org/10.3390/molecules15021041
Chicago/Turabian StyleRiera, Antoni, and María Moreno. 2010. "Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation" Molecules 15, no. 2: 1041-1073. https://doi.org/10.3390/molecules15021041