Reductive Heck Reactions of N-Methyl-substituted Tricyclic Imides



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

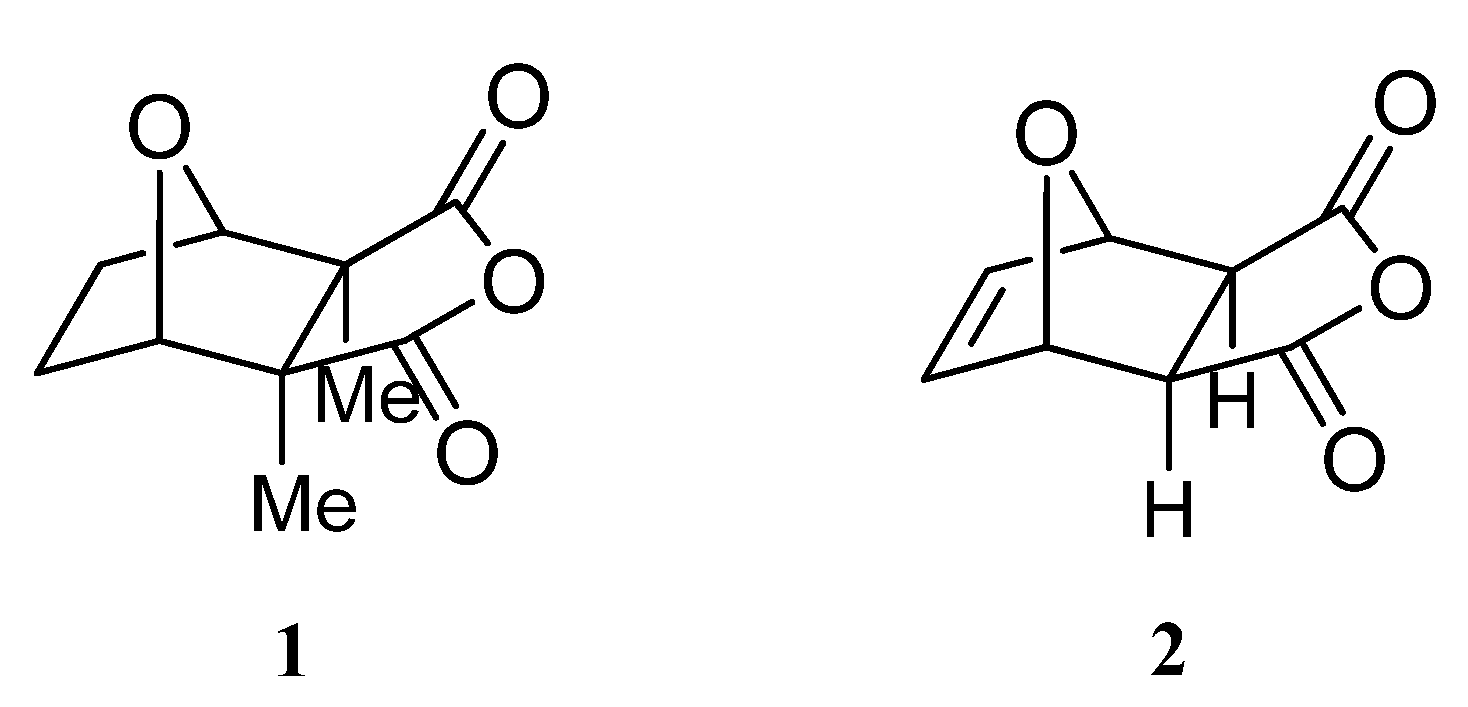
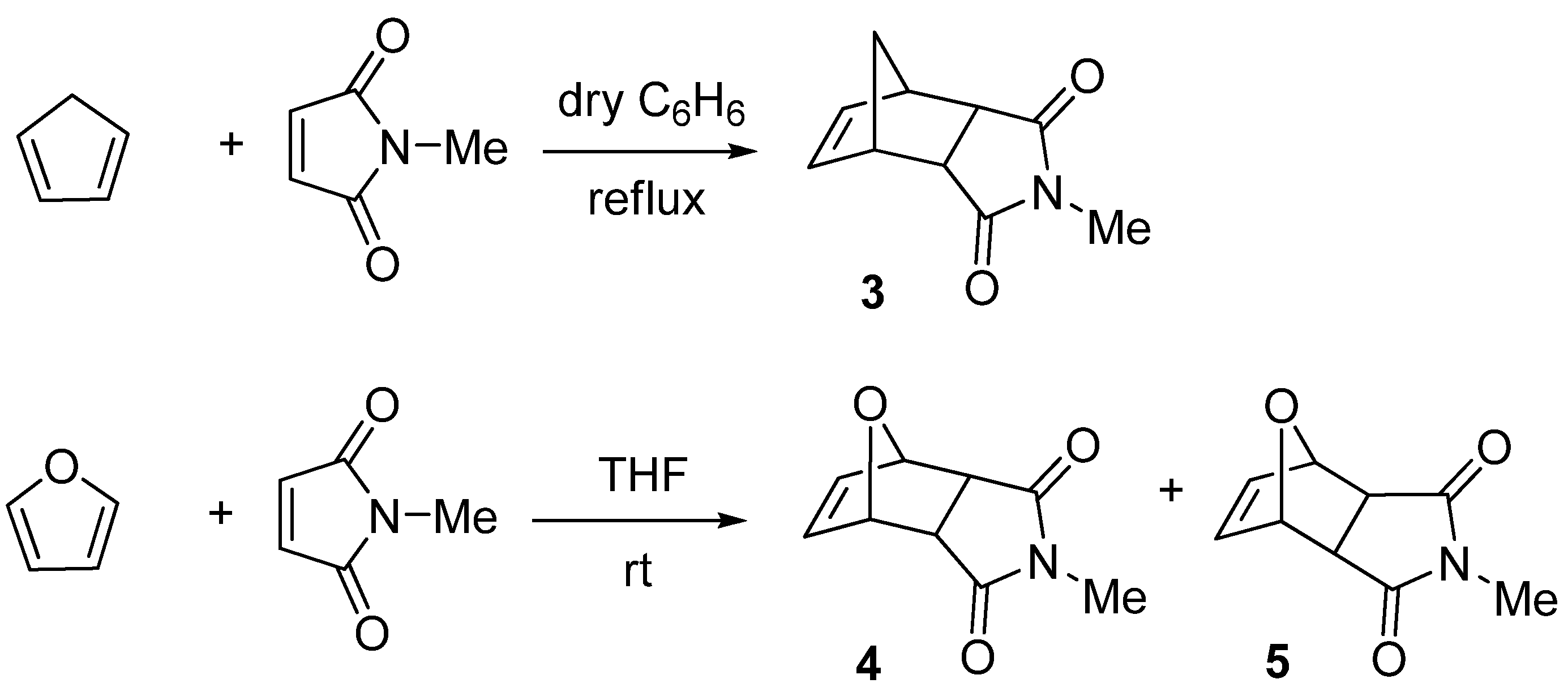
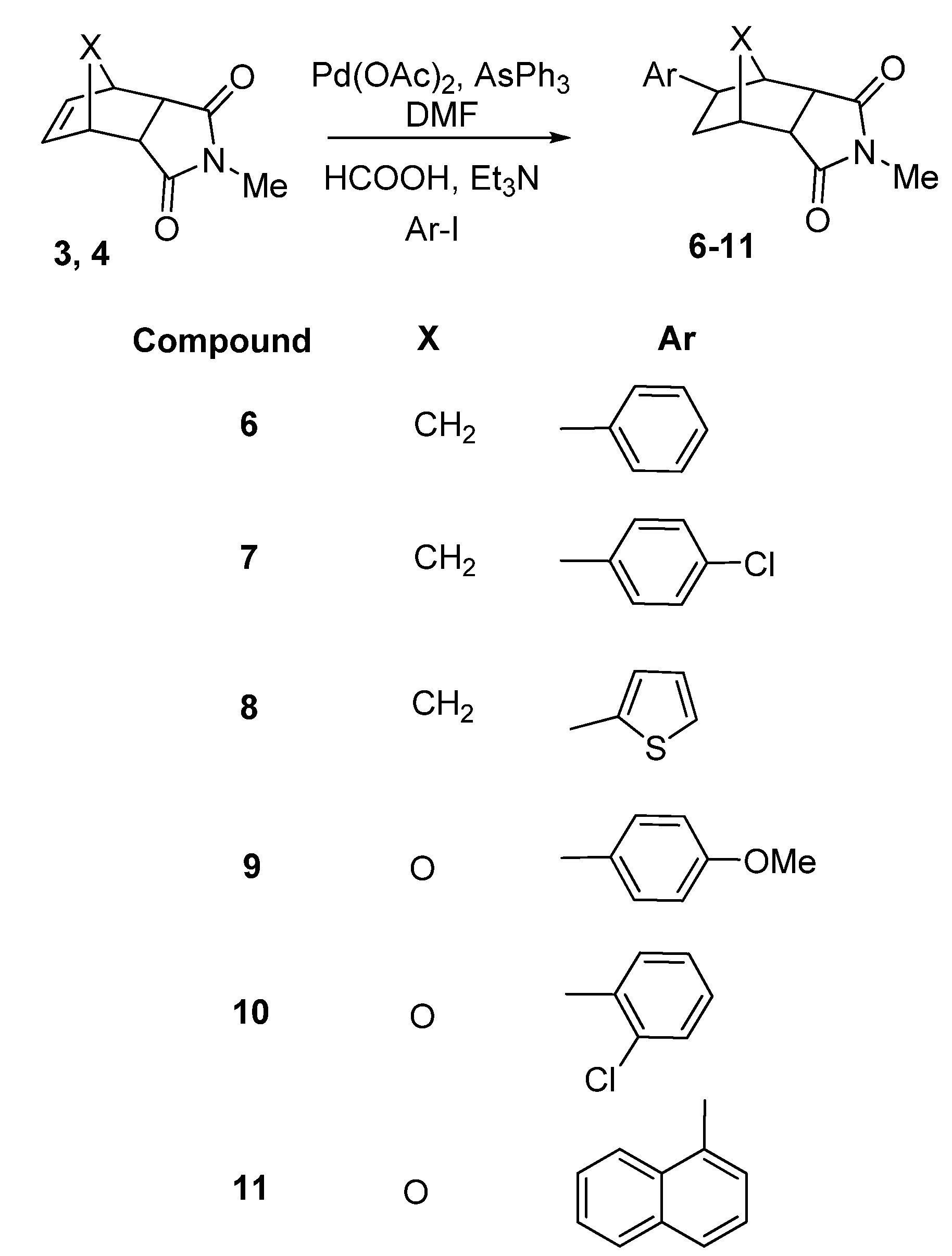
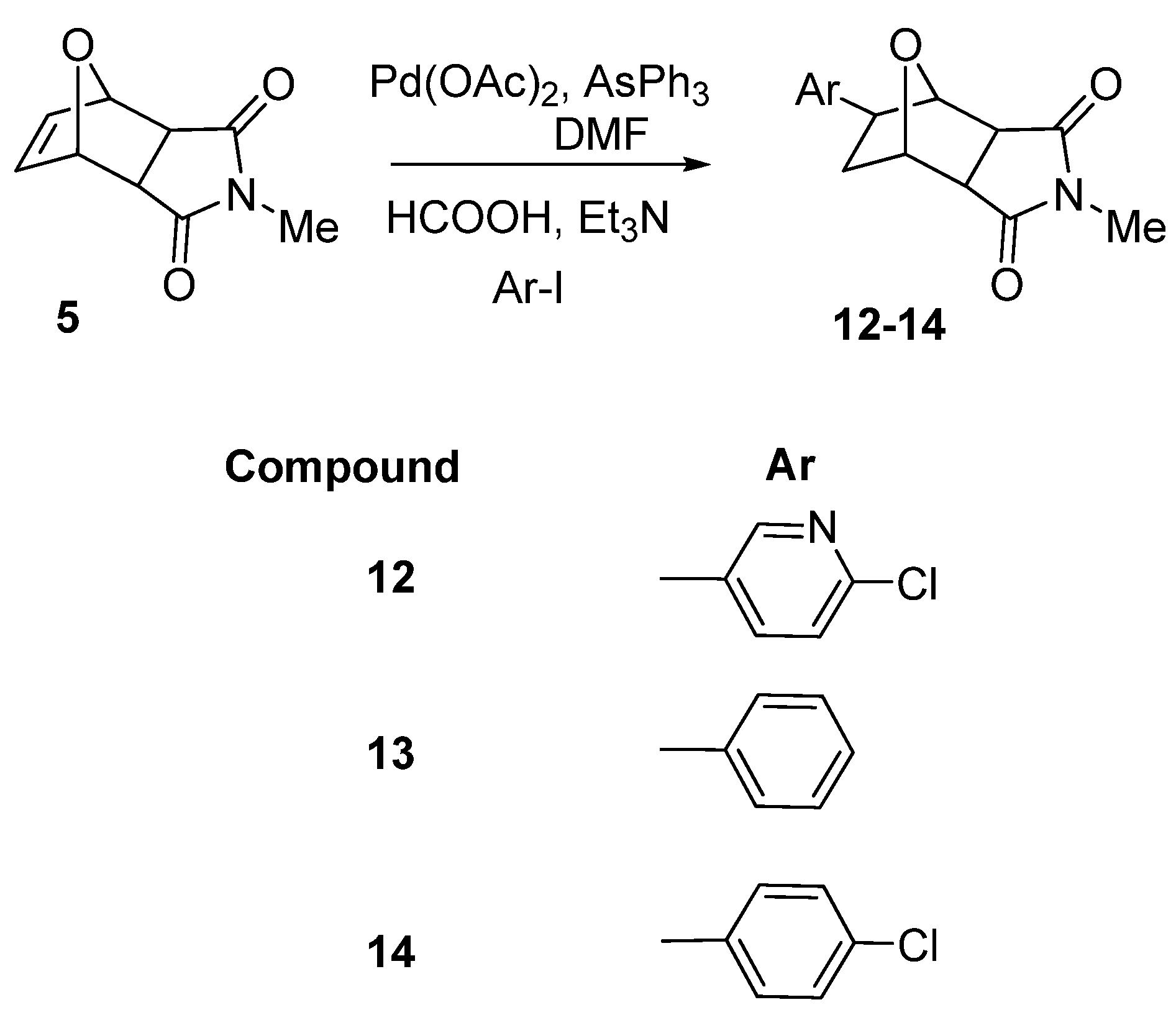
2. Results and Discussion



3. Experimental Section
3.1. General
3.2. General procedure for reductive Heck reactions

4. Conclusions
Acknowledgements
- Samples Availability: Samples of the compounds are available from the authors.
References and Notes
- Brana, M.F.; Gradillas, A.; Gomez, A.; Acero, N.; Llinares, F.; Munoz-Mingarrro, D.; Abradelo, C.; Rey-Stolle, F.; Yuste, M.; Campos, J.; Gallo, M.A.; Espinosa, A. Synthesis, biological activity, and quantitative structure−activity relationship study of azanaphthalimide and arylnaphthalimide derivatives. J. Med. Chem. 2004, 47, 2236–2242. [Google Scholar]
- Sondhi, S.M.; Rani, R.; Roy, P.; Agrawal, S.K.; Saxena, A.K. Microwave-assisted synthesis of N-substituted cyclic imides and their evaluation for anticancer and anti-inflammatory activities. Bioorg. Med. Chem. Lett. 2009, 19, 1534–1538. [Google Scholar]
- Kossakowski, J.; Jarocka, M. Synthesis of new N-substituted cyclic imides with an expected anxiolytic activity. XVII. Derivatives of 1-ethoxybicyclo[2.2.2]oct-5-one-2,3-dicarboximide. II Farmaco 2001, 56, 785–789. [Google Scholar]
- Hart, M.E.; Chamberlin, A.R.; Walkom, C.; Sakoff, J.A.; McCluskey, A. Modified norcantharidins: synthesis, protein phosphatases 1 and 2A inhibition, and anticancer activity. Bioorg. Med. Chem. Lett. 2004, 14, 1969–1973. [Google Scholar] [CrossRef]
- Deng, L.; Yongzhou, H. 1,3-Dipolar cycloaddition reaction: synthesis and configuration of norcantharidin derivatives of substituted aromatic amines. J. Heterocycl. Chem. 2007, 44, 597–601. [Google Scholar] [CrossRef]
- Deng, L.-P.; Liu, F.-M.; Wang, H.-Y. 1,3-Dipolar cycloaddition reaction: synthesis of novel 5,6-dehydronorcantharidin derivatives of substituted aromatic amines with potential antitumor activities. J. Heterocycl. Chem. 2005, 42, 13–18. [Google Scholar] [CrossRef]
- Namyslo, J.C.; Kaufmann, D.E. Triphenylarsine as an efficient ligand in the Pd-catalyzed synthesis of Epibatidine and analogs. Synlett 1999, 114–116. [Google Scholar]
- Goksu, G.; Gul, M.; Ocal, N.; Kaufmann, D.E. Hydroarylation of bicyclic, unsaturated dicarboximides: access to aryl-substituted, bridged perhydroisoindoles. Tetrahedron Lett. 2008, 49, 2685–2688. [Google Scholar] [CrossRef]
- Celik, C.; Kulu, I.; Ocal, N.; Kaufmann, D.E. Domino-Heck reactions of carba- and oxabicyclic, unsaturated dicarboximides: Synthesis of aryl-substituted, bridged perhydroisoindole derivatives. Helv. Chim. Acta 2009, 92, 1092–1101. [Google Scholar] [CrossRef]
- Birney, D.; Lim, T.-K.; Koh, Joanne H.P.; Pool, B.R.; White, J.M. Structural investigations into the retro-Diels-Alder reaction. Experimental and theoretical studies. J. Am. Chem. Soc. 2002, 124, 5091–5099. [Google Scholar]
- By, G.; Yit, W.; Pool, B.R.; White, J.M. Structural studies on cycloadducts of furan, 2-methoxyfuran, and 5-trimethylsilylcyclopentadiene with maleic anhydride and N-methylmaleimide. J. Org. Chem. 2008, 73, 151–156. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goksu, G.; Ocal, N.; Kaufmann, D.E. Reductive Heck Reactions of N-Methyl-substituted Tricyclic Imides. Molecules 2010, 15, 1302-1308. https://doi.org/10.3390/molecules15031303
Goksu G, Ocal N, Kaufmann DE. Reductive Heck Reactions of N-Methyl-substituted Tricyclic Imides. Molecules. 2010; 15(3):1302-1308. https://doi.org/10.3390/molecules15031303
Chicago/Turabian StyleGoksu, Gokce, Nuket Ocal, and Dieter E. Kaufmann. 2010. "Reductive Heck Reactions of N-Methyl-substituted Tricyclic Imides" Molecules 15, no. 3: 1302-1308. https://doi.org/10.3390/molecules15031303