Synthesis of a New Chiral Pyrrolidine †
by

Mari Fe. Flores
,
Marta G. Núñez
,
Rosalina F. Moro
,
Narciso M. Garrido
,
Isidro S. Marcos
,
Enrique F. Iglesias
,
Pilar García
and
David Díez
*
Departamento de Química Orgánica, Universidad de Salamanca, Plaza de los Caídos 1-5, 37008, Salamanca, Spain
*
Author to whom correspondence should be addressed.
†
This paper is dedicated to Prof. Pelayo Camps on occasion of his 65th birthday.
Molecules 2010, 15(3), 1501-1512; https://doi.org/10.3390/molecules15031501
Submission received: 8 December 2009
/
Revised: 2 February 2010
/
Accepted: 5 March 2010
/
Published: 9 March 2010
Abstract
:The synthesis of a new chiral pyrrolidine has been performed using 2,3-O-iso-propylidene-D-erythronolactol as a suitable starting material.
1. Introduction
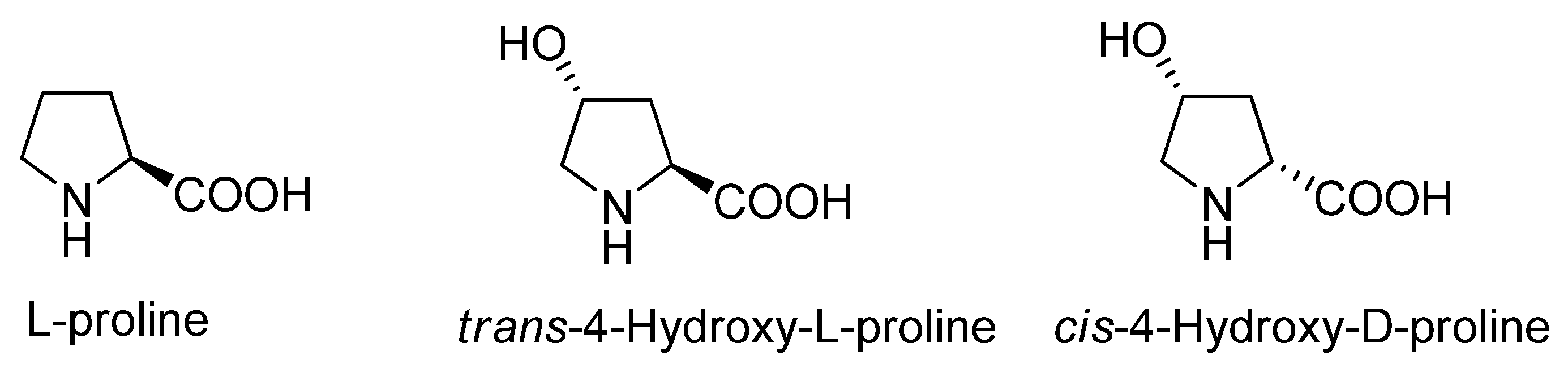
In the last years there has been a growing interest in organocatalysis [1,2,3,4,5,6], a new field which has quickly attracted researchers’ attention due to its potential for saving costs, time and energy compared to classic catalysis. Among the many known organocatalysts L-proline is perhaps the one which has been most studied. This fact has led to the appearance of many analogues [7,8,9,10,11,12,13,14]. In the seminal paper of List, Lerner and Barbas III [15], it is described how in the aldol reaction the catalytic activity of L-proline increases using trans-4-hydroxy-L-proline and also how the enantiomeric excess reverses using cis-4-hydroxy-D-proline, (Figure 1).
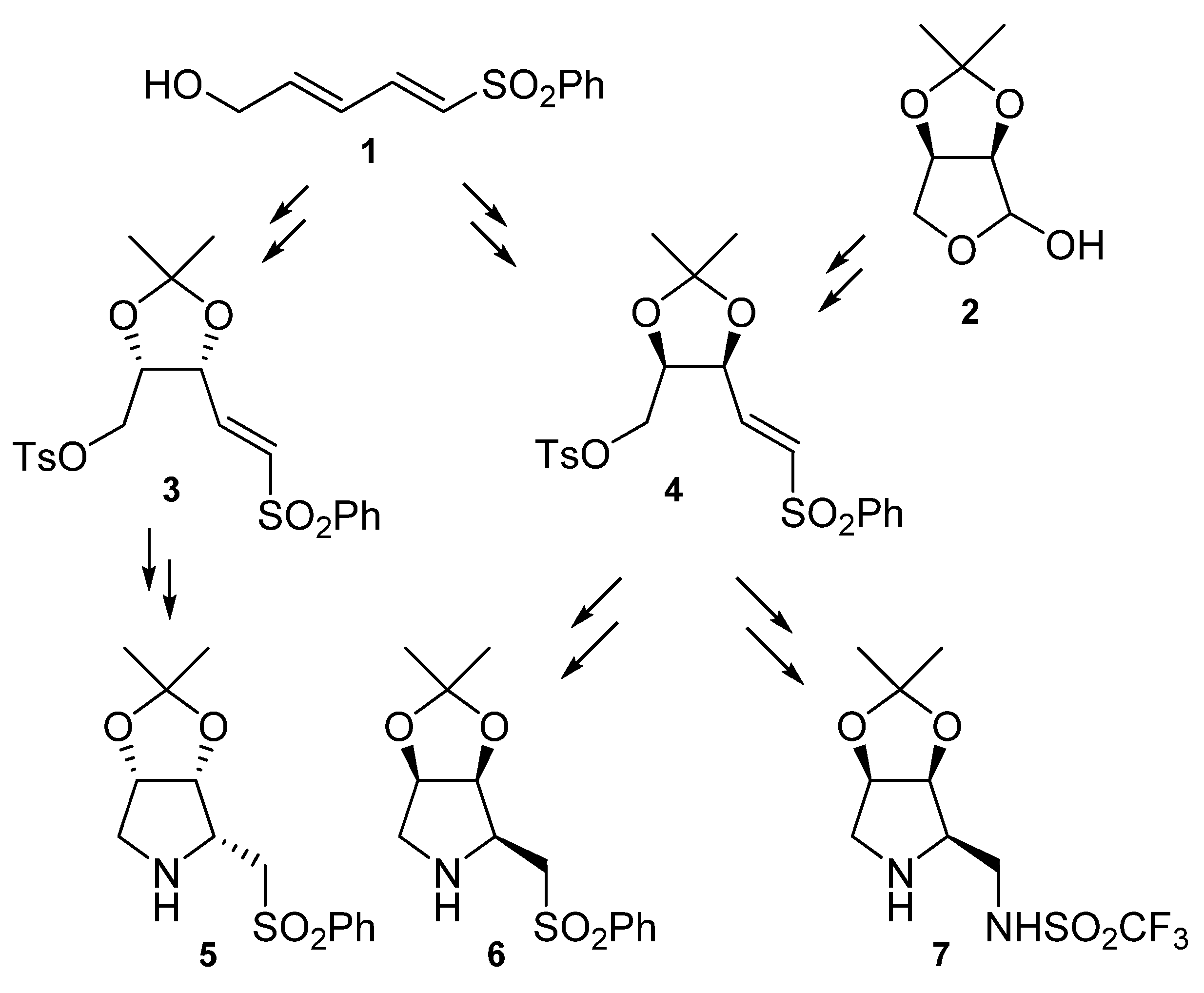
In our research group we have achieved the synthesis of several chiral pyrrolidines from sulfonylbutadiene 1 or 2,3-O-iso-propylidene-D-erythronolactol (2) (Scheme 1).
Starting from sulfonylbutadiene 1, pyrrolidines 5 and 6 were obtained through vinyl sulfones 3 and 4, respectively [16,17]. In order to increase the yields, a new route for the synthesis of compound 6 was devised starting from 2,3-O-iso-propylidene-D-erythronolactol (2) through vinylsulfone 4 [18]. This pyrrolidine 6 has been proved to be an organocatalyst for the intramolecular oxa-Michael reaction [19,20]. Moreover, compound 7, which has been reported to catalyze Michael reactions [21], has been synthesized from intermediate vinylsulfone 4 (Scheme 1).
2. Results and Discussion
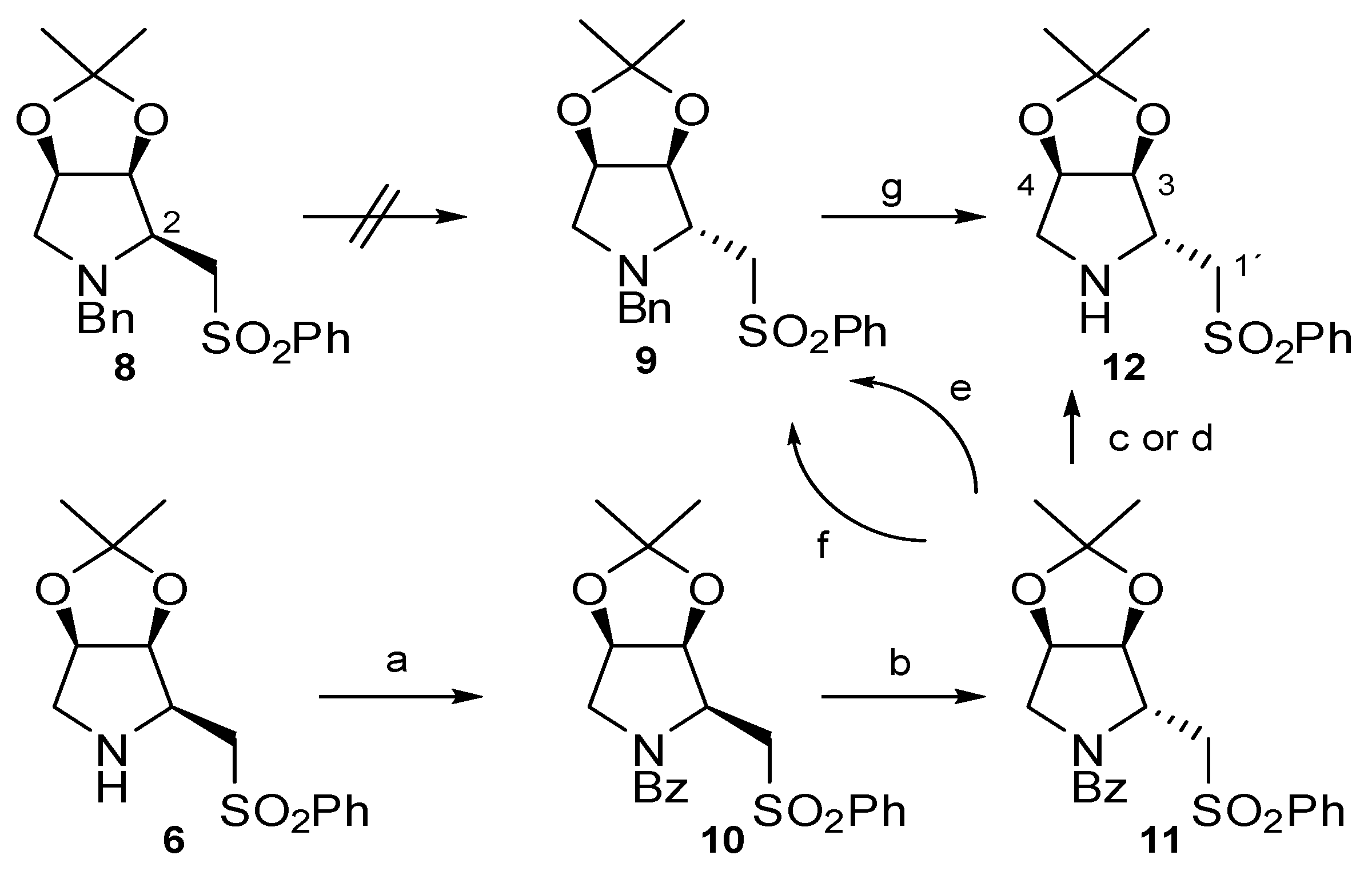
In this paper we describe our studies on the synthesis of the epimer at C-2 of pyrrolidine 6, as we are interested in comparing the properties of both diastereoisomers in organocatalytic reactions. In previous studies with PPY-derivatives, we had observed the epimerization of that stereogenic center when it was treated with bases [22]. Taking this into account, we first tried the epimerization at C-2 in compound 8, obtained directly from 4 by treatment with benzylamine. However, although several bases were used, none of them gave the epimerization. Therefore, we devised the following synthesis for the required compound 12 (Scheme 2).
Benzoylation of pyrrolidine 6 gave derivative 10 as outlined in Scheme 2. When this compound was submitted to treatment with n-BuLi, the C-2 epimer 11 was obtained in moderate yield. Once the required stereochemistry at C-2 in 11 was achieved, we proceeded to deprotect the nitrogen to obtain the desired pyrrolidine 12. This step was not as simple as it was thought initially, since the desired direct debenzoylation did not take place under several conditions. Finally, it was necessary to reduce the benzoyl to benzyl group and then deprotect under the usual conditions. Although, the final deprotection took place in good yield, the previous transformation from benzoyl to benzylderivative was only achieved in low yield, making useless this procedure to synthesise 12.
Therefore, we devised a new synthesis of compound 12 starting from 2,3-O-iso-propylidene-D-erythronolactol (2). Goti [23,24,25] and Wightman and Closa [26,27,28] have obtained nitrone 13 from compound 2. Besides, Merino and Goti have applied this versatile nitrone to the synthesis of iminocyclitols, pyrrolizidines and indolizidines [29,30], observing that the addition of organometallics to 13 took place to give the trans compounds. With this procedure in mind, we were able to achieve the desired compound 12 in a simple manner, as depicted in Scheme 3.
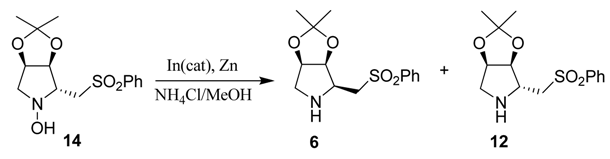
When compound 13 was treated with lithio(phenylsulfonyl)methane, only hydroxylamine 14 was obtained stereoselectively in moderate yield. The stereochemistry of 14 was established studying its NMR spectra (whose assignment is given in the Experimental section) and by the observation of the nOes that this molecule displays (Scheme 3). The nOes of the enantiomer of compound 6 has already been reported by our research group [16]. Once this compound 14 was synthesized, different reduction conditions to obtain the pyrrolidine ring were tested.
Hydrogenation under different conditions either Pd(OH)2 or Pd on carbon only leads to the pyrrolidine 6. When 14 was submitted to reduction with stoichiometric indium or with zinc, hydroxylamine 15 with inversion at C-2 was obtained. Thus, it was necessary to choose the adequate conditions to reduce the hydroxylamine to the required pyrrolidine 12 without epimerization at C-2. This was achieved using catalytic indium and stoichiometric Zn [29,30,31] (Table 1, entry 8).
Having obtained our desired compounds 6 and 12, they were tested as organocatalysts in Michael addition reactions of cyclohexanone to nitrostyrene, as shown in Table 2.
As can be observed, both compounds 6 and 12 are organocatalysts. However, their behaviour is rather different depending on the solvent chosen to carry out the reaction. It is worthy mentioning that compound 12 lead to the opposite enantiomer of 6 in the Michael addition, as a result of the stereochemistry change in C-2 position. The absolute configuration of the addition product was established by comparison of the HPLC data with the ones reported by us [21] and others [32,33].
3. Conclusions
The synthesis of a new chiral pyrrolidine 12, has been achieved from the same starting material, 2,3-O-iso-propylidene-D-erythronolactol through two different methodologies. In addition, the reduction of the chiral hydroxylamine into pyrrolidine has been studied under different conditions.
4. Experimental
4.1. General
1H-NMR and 13C-NMR spectra were recorded in CDCl3 at 200 and 400 MHz (1H) or 50 and 100 MHz (13C) on Varian 200 VX and BRUKER DRX 400 instruments, respectively. Multiplicities were determined by DEPT experiments. IR spectra were registered using a BOMEM 100 FTIR spectrophotometer. Optical rotations were determined using a Perkin-Elmer 241 polarimeter in a 1 dm cell and are given in units of 10-1 deg cm2 g−1. Concentrations are quoted in g per 100mL. The electron impact (EI) mass spectra were run on a VG-TS 250 spectrometer using a 70 eV ionizing voltage. HRMS were recorded using a VG Platform (Fisons) spectrometer using Chemical Ionization (ammonia as gas) or Fast Atom Bombardment (FAB) techniques. Thin layer chromatography (tlc) was performed on aluminum sheets coated with 60 F254 silica. Sheets were visualized using iodine, UV light or 1% aqueous KMnO4 solution. Column chromatography (CC) was performed with Merck silica gel 60 (70–230 mesh). Solvents and reagents were generally distilled prior to use: dichloromethane (DCM) from KOH.
4.2. Preparation of (2S,3S,4R)-N-Benzoyl-2-phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (10)
To a solution of pyrrolidine 6 (40 mg, 0.13 mmol) in pyridine (0.5 mL) at 0 °C was added PhCOCl (20 µL) and the mixture was stirred for 3 h. The reaction was quenched by the addition of water (0.2 mL) at 0 °C and then the mixture was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with aqueous solutions of CuSO4 (20%), NaHCO3 (5%) and brine. After drying (Na2SO4), filtering, and concentrating, the crude residue was purified by flash chromatography (silica gel, n-hexane/EtOAc 8:2) to give benzoyl derivative 10 (52 mg, 97%). IR (film) ν (cm−1) 3066, 2987, 2939, 1643, 1584, 1447, 1383, 1307, 1249, 1215, 1153, 1085; 1H-NMR (400 MHz) δ = 8.03–7.34 (m, 10H), 4.83 (t, J = 5.8 Hz, 1H), 4.66–4.61 (m, 2H), 3.98 (dd, J = 14.0 and 3.7 Hz, 1H), 3.84 (dd, J = 14.0 and 9.0 Hz, 1H), 3.59 (m, 2H), 1.54 (s, 3H), 1.32 (s, 3H); 13C-NMR (100 MHz) δ = 171.3, 139.7, 135.3, 133.6, 130.8, 129.1, 128.3, 128.1, 127.7, 113.1, 78.7, 77.5, 54.8, 54.5, 53.2, 27.1, 25.2; HRMS (ESI) C21H24NO5S requires (M+H+) 402.1369; found 402.1364. [α]D20 = −67.1 (c 1.4, CHCl3).
4.3. Preparation of (2R,3S,4R)-N-Benzoyl-2-phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (11)
A solution of pyrrolidine 10 (40 mg, 0.10 mmol) in THF (1 mL) under argon at −78 °C, was added n-BuLi (75 µL, 1.6 M in hexanes). The resulting mixture was stirred for 3 h, allowing to warm to rt, whereupon the reaction was quenched with saturated aqueous solution of NH4Cl (0.2 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated. The resulting crude residue was purified by flash chromatography (silica gel, n-hexane/EtOAc 7:3) to obtain 11 (20 mg, 50%). IR (film) ν (cm−1) 3064, 2978, 2938, 1634, 1577, 1413, 1319, 1217, 1152, 1074, 1046, 858; 1H-NMR (400 MHz) δ = 7.98–7.38 (m, 10H), 5.25–5.23 (m, 1H), 4.85 (m, 1H), 4.79 (m, 1H), 3.89 (dd, J = 14.0 and 3.6 Hz, 1H), 3.78–3.71 (m, 2H), 3.44 (d, J = 14.0 Hz, 1H), 1.44 (s, 3H), 1.33 (s, 3H); 13C-NMR (100 MHz) δ = 170.0, 139.6, 135.5, 134.0, 130.2, 129.4, 128.3, 127.8, 127.2, 112.2, 82.5, 79.6, 60.2, 54.8, 54.7, 26.9, 24.9; HRMS (ESI) C21H23NO5S requires (M+Na) 424.1195; found 424.1189. [α]D20 = + 33.9 (c 1.1, CHCl3).
4.4. Preparation of (2R,3S,4R)-N-Benzyl-2-phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (9)
Scheme 2, step e: LiAlH4 (7 mg, 0.19 mmol) was added to a solution of 11 (38 mg, 0.1 mmol) in dry THF (1 mL) at 0 °C. The resulting mixture was stirred for 1h, allowing to warm to rt, whereupon the reaction was quenched with wet ether, dried (Na2SO4), filtered, and concentrated. The resulting crude residue was purified by flash chromatography (silica gel, n-hexane/EtOAc 8:2) to yield benzyl derivative 9 (10 mg, 30%). IR (film) ν (cm−1) 2983, 2942, 1454, 1377, 1307, 1209, 1148, 1086, 1054; 1H-NMR (400 MHz) δ = 7.90–7.15 (m, 10H), 4.70 (m, 1H), 4.64 (m, 1H), 3.81 (d, J = 13.3 Hz, 1H), 3.55 (d, J = 13.3 Hz, 1H), 3.32–3.27 (m, 2H), 3.13 (dd, J = 13.5 and 9.0 Hz, 1H), 2.87 (dd, J = 10.7 and 5.3 Hz, 1H), 2.68 (d, J = 10.7 Hz, 1H), 1.50 (s, 3H),1.28 (s, 3H); 13C-NMR (100 MHz) δ = 139.4, 137.0, 133.7, 129.2, 128.3, 128.1, 127.1, 112.4, 84.3, 78.7, 63.4, 57.3, 56.6, 55.1, 27.0, 25.0; HRMS (ESI) C21H25NO4S requires (M+Na) 410.1402; found 410.1397; [α]D20 = + 20.2 (c 1.0, CHCl3).
Scheme 2, step f: To a solution of 11 (38 mg, 0.1 mmol) in dry THF (1 mL) under argon at rt, was added BH3·THF (0.12 mL, 1M in THF). After stirring for 2 h at 40 °C, the reaction was quenched by the addition of water (0.3 mL) and extracted with Et2O (3 × 10 mL). The combined organic layers were washed with H2O and brine, dried (Na2SO4), filtered, and concentrated. The resulting crude residue was purified by flash chromatography (silica gel, n-hexane/EtOAc 8:2) to obtain benzyl derivative 9 (5 mg, 15%).
4.5. Preparation of (2R,3S,4R)-2-Phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (12)
Scheme 2, step g: To a solution of pyrrolidine 9 (10 mg, 0.03 mmol) in MeOH (0.5 mL) was hydrogenated in the presence of a catalytic amount of Pd/C with a H2 balloon at room temperature for 24 h. The catalyst was filtered through a pad of Celite® and washed with MeOH. After concentrating, compound 12 (6 mg, 80%) was obtained. IR (film) ν (cm−1) 3317, 2985, 2936, 1448, 1375, 1306, 1209, 1144, 1083, 1046, 865; 1H-NMR (400 MHz) δ = 7.93 (m, 2H), 7.65–7.54 (m, 3H), 4.67 (dd, J = 4.4 and 5.3 Hz, 1H), 4.57 (d, J = 5.3 Hz, 1H), 3.57 (t, J= 6.4 Hz, 1H), 3.13 (d, J = 13.2 Hz, 1H), 3.05 (d, J = 13.6 Hz, 1H), 2.69 (dd, J = 4.4 and 13.2 Hz, 1H), 1.44 (s, 3H), 1.27 (s, 3H); 13C-NMR (100 MHz) δ = 139.4, 133.8, 129.3, 128.1, 111.6, 85.0, 84.9, 60.3, 57.2, 51.8, 26.2, 24.1; HRMS (ESI) C14H20NO4S requires (M+H+) 298.1113; found 298.1115; [α]D20 = −13.1 (c 1.7, CHCl3).
4.6. Preparation of (3S,4R)-3,4-Isopropylidenedioxypyrroline-1-oxide (13)
To a solution of lactol 2 (1.34 g, 8.54 mmol) in dry pyridine (8.6 mL) containing 3 Å activated molecular sieves (pellets, 10 g) was added a solution of NH2OSiMe2t-Bu (1.51 g, 10.25 mmol) in pyridine (8.6 mL) and the mixture was stirred at rt for 16 h. The reaction mixture was cooled to 0 °C, and methanesulfonyl chloride (0.8 mL, 10.25 mmol) was added slowly during 40 min. The reaction was stirred for 2 h at 0 °C, warmed to rt and stirred for 4 h. The mixture was then dilueted with CH2Cl2 (9 mL), filtered through Celite®, and concentrated. The resulting crude residue was purified by flash chromatography (silica gel, CH2Cl2/EtOAc/MeOH 15/7/1) to give pure nitrone 13 (635 mg, 50%). IR (film) ν (cm−1) 3084, 2993, 2980, 1579; 1H-NMR (400 MHz) δ = 6.82 (q, J= 1.5 Hz, 1H), 5.24 (d, J = 6.2, 1H), 4.86 (ddd, J= 6.2, 5.1 and 1.5 Hz, 1H), 4.07–3.99 (m, 2H), 1.40 (s, 3H), 1.31 (s, 3H); HRMS (ESI) C7H12NO3 requires (M+H+) 158.1785; found 158.0822; [α]D20 = −26.9 (c 1.1, CH2Cl2).
4.7. Preparation of (2R,3S,4R)-2-Phenylsulfonylmethyl-1-hydroxy-3,4-isopropylidenedioxypyrrolidine (14)
To a stirred solution of MeSO2Ph (520 mg, 2.58 mmol) in THF (4.7 mL) was added slowly n-BuLi (1.9 mL, 2.58 mmol) and the mixture was reacted at 0 °C for 10 min. Afterwards, the reaction mixture was cooled to −78 °C and stirred for 10 min at this temperature. Then, it was added into a solution of nitrone 13 (320 mg, 2.03 mmol) in THF (6.7 mL) and the mixture was stirred at −78 °C for 30 min and then for 2 h allowing to warm to rt. The reaction was quenched with saturated aqueous solution of NH4Cl and the product was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated. The resulting crude residue was purified by flash chromatography (silica gel, n-hexane/EtOAc 1:1) to obtain hydroxylamine 14 (300 mg, 48%). IR (film) ν (cm−1) 3428, 3207, 2987, 2925, 2856, 1581, 1442, 1385, 1295, 1131, 1074, 845; 1H-NMR (400 MHz) δ = 7.96 (m, 2H), 7.67–7.53 (m, 3H), 6.25 (s, 1H), 4.75 (m, 1H), 4.62 (dd, J = 4.2 and 6.0 Hz, 1H), 3.69 (dd, J = 6.4 and 14.0 Hz, 1H), 3.44 (m, 1H), 3.37 (m, 1H), 3.25 (dd, J = 6.8 and 14.0 Hz, 1H), 3.12 (dd, J = 4.4 and 12.4 Hz, 1H), 1.43 (s, 3H), 1.27 (s, 3H); 13C-NMR (100 MHz) δ = 139.4, 133.9, 129.3, 128.1, 113.7, 82.2, 77.2, 67.3, 62.3, 54.1, 26.7, 24.7; HRMS (ESI) C14H19NO5S requires (M + Na) 336.0882; found 336.0877; [α]D20 = −20.8 (c 2.2, CHCl3).
4.8. Preparation of (2S,3S,4R)-2-Phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (6)
Scheme 3, step c: A stirred solution of hydroxylamine 14 (50 mg, 0.16 mmol) in a 12:1 solution of HCl/MeOH (1 mL) was hydrogenated in the presence of a catalytic amount of Pd (OH)2/C, at a hydrogen pressure of 4 atm for 48 h. The catalyst was filtered through a pad of Celite,® washed with MeOH and concentrated. The product was purified by flash chromatography (silica gel, n-hexane/EtOAc 1:1) to provide pyrrolidine 6 (13 mg, 27%). IR (film) ν (cm−1) 3000, 2936, 1447, 1381, 1308, 1148, 1086, 650 cm−1; 1H-NMR (400 MHz) δ = 7.93 (m, 2H), 7.69–7.50 (m, 3H), 4.67 (dd, J = 4.0 and 5.3 Hz, 1H), 4.54 (dd, J = 4.0 and 5.3 Hz, 1H), 3.57 (dd, J = 5.0 and 14.0 Hz, 1H), 3.36 (dd, J = 7.0 and 14.0 Hz, 1H), 3.22 (m, 1H), 3.13 (d, J = 12.7 Hz, 1H), 2.69 (dd, J = 4.0 and 12.7 Hz, 1H), 2.20 (s, 1H), 1.41 (s, 3H) and 1.25 (s, 3H); 13C-NMR (100 MHz) δ = 139.7, 133.7, 129.2, 127.2, 111.1, 81.0, 80.8, 57.1, 55.9, 52.6, 25.7, 24.0; HRMS (ESI) C14H20NO4S requires (M+H+) 298.1113; found 298.1128; [α]D20 = −37.3 (c 0.5, CHCl3).
(Scheme 3, step d): A stirred solution of hydroxylamine 14 (66 mg, 0.21 mmol) in 1 mL of MeOH was hydrogenated in the presence of a catalytic amount of Pd/C with a H2 balloon at room temperature for 24 h. The catalyst was filtered through a pad of Celite,® washed with MeOH and concentrated. The product was purified by flash chromatography (silica gel, n-hexane/EtOAc 1:1) to provide pyrrolidine 6 (24 mg, 39%).
4.9. Preparation of (2R,3S,4R)-2-Phenylsulfonylmethyl-1-hydroxy-3,4-isopropylidenedioxypyrrolidine (15)
(Scheme 3, step e): To a stirred solution of hydroxylamine 14 (32.1 mg, 0.10 mmol) in a 2:1 solution of EtOH /saturated aqueous NH4Cl (18.5 mL), powdered indium (14 g, 0.12 mmol) was added and the mixture was heated under reflux. After 24 h the reaction mixture was cooled, filtered through Celite,® and concentrated under reduced pressure. A saturated aqueous Na2CO3 solution (5 mL) was then added, and the product was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated. The resulting crude residue was purified by flash chromatography (silica gel, hexane/EtOAc 1:1) to obtain hydroxylamine 15 (28.8 mg, 92%). IR (film) ν (cm−1) 3448, 2985, 2933, 2854, 1448, 1383, 1306, 1085, 856; 1H-NMR (400 MHz) δ = 7.97 (m, 2H), 7.67–7.52 (m, 3H), 6.27 (s, 1H), 4.60 (m, 2H), 3.70 (dd, J = 8.4 and 14.o Hz, 1H), 3.50–3.38 (m, 2H), 3.05 (m, 1H), 2.70 (dd, J = 4.4 and 11.0 Hz, 1H), 1.37 (s, 3H), 1.22 (s, 3H); 13C-NMR (100 MHz) δ = 139.9, 134.1, 129.5, 128.3, 111.1, 77.2, 76.4, 65.9, 62.2, 54.5, 25.9, 24.4; HRMS (ESI) C14H19NO5S requires (M + Na) 336.0882; found 336.0895; [α]D20 = −10.4 (c 1.3, CHCl3).
Scheme 3, step f: To a stirred solution of hydroxylamine 14 (69 mg, 0.22 mmol) in a solution of MeOH/NH4Clsat (3.4 /5 mL), powdered Zn (29 mg, 0.44 mmol) was added at 20 °C and the mixture was stirred for 6h. The solvent was evaporated under vacuum and a saturated aqueous solution of Na2CO3 (1.5 mL) was added. The mixture was extracted with EtOAc (3 × 15 mL) and the combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated. The resulting crude residue was purified by flash chromatography (silica gel, n-hexane/EtOAc 1:1) to obtain hydroxylamine 15 (34 mg, 50%).
4.10. Preparation of (2R,3S,4R)-2-Phenylsulfonylmethyl-3,4-isopropylidenedioxypyrrolidine (12)
Scheme 3, step g, Table 1, entry 8, as example: To a stirred solution of hydroxylamine 14 (852 mg, 2.58 mmol), in methanol (37 mL), a saturated solution of NH4Cl (56 mL), powdered Zn (712 mg, 10.9 mmol) and a catalytic amount of indium dust (20 mg) were added at 20 °C. The mixture was stirred for 6h. The solvent was evaporated under vacuum and a saturated aqueous solution of Na2CO3 (5 mL) was added. The mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated. The resulting crude residue was purified by flash chromatography (silica gel, n-hexane/EtOAc 1:1) to obtain pyrrolidines 12 (248 mg, 35%) and 6 (13 mg, 2%).
4.11. (2S,1´R)-2-[1´-Phenyl-2´-nitroethyl]-cyclohexanone
To a suspension of catalyst 6 (10 mg, 15%) and 0.438 mL (4.40 mmol) of cyclohexanone in 3.5 mL of DMSO was added 33 mg (0.22 mmol) of trans-β nitrostyrene. The resulting mixture was allowed to stir at room temperature for 16 h, whereupon the reaction was quenched with saturated aqueous ammonium chloride (2 mL) and the aqueous layers were extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo and the resulting residue was purified by flash column chromatography using (hexane/EtOAc, 9/1) to give the title compound, as a white solid (11mg, 20%). 1H-NMR (400 MHz) δ 7.34–7.16 (m, 5H, Ph), 4.93 (dd, J = 12.5 and 4.5 Hz, 1H, CH2), 4.60 (dd, J = 12.5 and 9.9 Hz, 1H, CH2), 3.76 (m, 1H, CH), 2.69 (m, 1H, CH), 2.50–1.52 (m, 8H, 4CH2). The absolute configuration was established by comparison with reported HPLC data [21]. HPLC: Daicel Chiralpak AD; n-hexane/iPrOH: 0.45 mL min−1; λmax230 nm: tR (minor)-10.8 min; tR (major)-13.1 min.
4.12. (2R,1´S)-2-[1´-Phenyl-2´-nitroethyl]-cyclohexanone
To a suspension of catalyst 12 (10 mg, 15%) and 0.438 mL (4.40 mmol) of cyclohexanone in 3.5 mL of CHCl3 was added 33 mg (0.22 mmol) of trans-β nitrostyrene. The resulting mixture was allowed to stir at room temperature for 16 h, whereupon the reaction was quenched with saturated aqueous ammonium chloride (2 mL) and the aqueous layers were extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered and evaporated in vacuo and the resulting residue was purified by flash column chromatography using (n-hexane/EtOAc, 9/1) to give the title compound, as a white solid (22mg, 35%). The absolute configuration was established by comparison with reported HPLC data [21]. HPLC: Daicel Chiralpak AD; n-hexane/iPrOH: 0.45 mL min−1; λmax230 nm: tR (major)-10.5 min; tR (minor)-12.9 min.
Acknowledgments
Financial support for this work came from FSE, Spanish MEC (CTQ2006-08296/BQU, CTQ2009-11172/BQU) and Junta de Castilla y León (Spain) GR-178, (SA001A09). The authors thank also A. M. Lithgow for the NMR spectra and César Raposo for the mass spectra. M.G.N. is grateful for a FPU doctoral fellowship of the Spanish MEC and M.F.F. to Junta de Castilla y León.
References and Notes
- Dalko, P.I.; Moisan, L. Enantioselective Organocatalysis. Angew. Chem. Int. Ed. Engl. 2001, 113, 3726–3748. [Google Scholar] [CrossRef]
- List, B. Asymmetric Aminocatalysis. Synlett 2001, 1675–1686. [Google Scholar] [CrossRef]
- Brown, S.P.; Brochu, M.P.; Sinz, C.J.; MacMillan, D.W.C. The Direct and Enantioselective Organocatalytic -Oxidation of Aldehydes. J. Am. Chem. Soc. 2003, 125, 10808–10809. [Google Scholar] [CrossRef] [PubMed]
- Pidathala, C.; Hoang, L.; Vignola, N.; List, B. Direct Catalytic Asymmetric Enolexo Aldolizations. Angew. Chem. Int. Ed. Engl. 2003, 42, 2785–2788. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis. From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Mase, N.; Barbas, C.F. Design and Use of Fluorogenic Aldehydes for Monitoring the Progress of Aldehyde Transformations. J. Am. Chem. Soc. 2004, 126, 3692–3693. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, Y.; Sasaoka, A.; Shimomoto, A.; Fujioka, S.; Kotsuki, H. High-Pressure-Promoted Asymmetric Aldol Reactions of Ketones with Aldehydes Catalyzed by L-Proline. Synlett 2003, 1655–1658. [Google Scholar]
- Hayashi, Y.; Yamaguchi, J.; Sumiya, T.; Shoji, M. Direct Proline-Catalyzed Asymmetric -Aminoxylation of Ketones. Angew. Chem. Int. Ed. Engl. 2004, 43, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
- Mangion, I.K.; Northrup, A.B.; MacMillan, D.W.C. The Importance of Iminium Geometry Control in Enamine Catalysis: Identification of a New Catalyst Architecture for Aldehyde-Aldehyde Couplings. Angew. Chem. Int. Ed. Engl. 2004, 43, 6722–6724. [Google Scholar] [CrossRef] [PubMed]
- Cobb, A.J.A.; Longbottom, D.A.; Shaw, D.M.; Ley, S.V. 5-Pyrrolidin-2-yltetrazole as an asymmetric organocatalyst for the addition of ketones to nitro-olefins. Chem. Commun. 2004, 1808–1809. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, J.; Li, H. Direct, Highly Enantioselective Pyrrolidine Sulfonamide Catalyzed Michael Addition of Aldehydes to Nitrostyrenes. Angew. Chem. Int. Ed. Engl. 2005, 44, 1369–1371. [Google Scholar] [CrossRef] [PubMed]
- Andrey, O.; Alexakis, A.; Tomassini, A.; Bernardinelli, G. The Use of N-Alkyl-2,2´-bipyrrolidine Derivatives as Organocatalysts for the Asymmetric Michael Addition of Ketones and Aldehydes to Nitroolefins. Adv. Synth. Catal. 2004, 346, 1147–1168. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Diez, D.; Beneitez, M.T.; Marcos, I.S.; Garrido, N.M.; Basabe, P.; Urones, J.G. Enantioselective Synthesis of a 2,3,4-Trisubstituted Pyrrolidine from 1-Hydroxymethyl-4-phenylsulfonylbutadiene. Synlett 2001, 655–657. [Google Scholar] [CrossRef]
- Diez, D.; Beneitez, M.T.; Moro, R.F.; Marcos, I.S.; Basabe, P.; Garrido, N.M.; Urones, J.G. Regio- and stereoselective ring opening of epoxides Enantioselective synthesis of 2,3,4-trisubstituted five-membered heterocycles. Tetrahedron Asymmetry 2002, 13, 639–646. [Google Scholar] [CrossRef]
- Diez, D.; Beneitez, M.T.; Moro, R.F.; Marcos, I.S.; Basabe, P.; Garrido, N.M.; Urones, J.G. Synthesis of Vinylsulfone Derivatives of Sugars: An Easy Preparation of (2R,3S,4E)-5-Benzenesulfonyl-2,3-iso-propylidene-dioxy-pent-4-en-1-yl-tosylate. Synlett 2003, 729–731. [Google Scholar] [CrossRef]
- Diez, D.; Núñez, M.G.; Moro, R.F.; Marcos, I.S.; Basabe, P.; Broughton, H.B.; Urones, J.G. Organocatalytic Synthesis of an Alkyltetrahydropyran. Synlett 2009, 390–394. [Google Scholar] [CrossRef]
- Nising, C.F.; Bräse, S. The oxa-Michael reaction: From recent developments to applications in natural product synthesis. Chem. Soc. Rev. 2008, 37, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Diez, D.; Gil, M.J.; Moro, R.F.; Marcos, I.S.; García, P.; Basabe, P.; Garrido, N.M.; Broughton, H.B.; Urones, J.G. A new class of chiral pyrrolidine for asymmetric Michael addition reactions. New mechanism via simple 4+2 type attack of the enamine on the trans-nitrostyrene. Tetrahedron 2007, 63, 740–747. [Google Scholar] [CrossRef]
- Diez, D.; Gil, M.J.; Moro, R.F.; Garrido, N.M.; Marcos, I.S.; Basabe, P.; Sanz, F.; Broughton, H.B.; Urones, J.G. Chemistry of sulfones: Synthesis of a new chiral nucleophilic catalyst. Tetrahedron Asymmetry 2005, 16, 2980–2985. [Google Scholar] [CrossRef]
- Cicchi, S.; Marradi, M.; Vogel, P.; Goti, A. One-Pot Synthesis of cyclic Nitrones and Their Conversion to Pyrrolizidines: 7a-epi-Crotanecine Inhibits -Mannosidases. J. Org. Chem. 2006, 71, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Cicchi, S.; Corsi, M.; Brandi, A.; Goti, A. Straigtforward Synthesis of Enantiomerically Pure (3S,4R)-and (3R,4S)-3,4-Isopropylidendioxypyrroline 1-Oxide, Precursors of Fucntionalized cis-Dihydroxy Azaheterocycles, by a Novel “One-Pot” Procedure. J. Org. Chem. 2002, 67, 1678–1681. [Google Scholar] [CrossRef]
- Revuelta, J.; Cicchi, S.; Goti, A.; Brandi, A. Enantiopure Cyclic Nitrones: A Useful Class of Building Blocks for Asymmetric Syntheses. Synthesis 2007, 485–504. [Google Scholar] [CrossRef]
- McCaig, A.E.; Meldrum, K.P.; Wightman, R.H. Synthesis of Trihydroxylated Pyrrolizidines using Cycloaddition Reactions of Functionalized Cyclic Nitrones, and the Synthesis of (+)- and (-)-Lentiginosine. Tetrahedron 1998, 54, 9429–9446. [Google Scholar] [CrossRef]
- Hall, A.; Meldrum, K.P.; Therond, P.R.; Wightman, R.H. Synthesis of Hydroxylated Pyrrolizidines Related to Alexine using Cycloaddition Reactions of Functionalized Cyclic Nitrones. Synlett 1997, 123–125. [Google Scholar]
- Closa, M.; Wightman, R.H. Synthesis of (3S,4R)-3,4-Isopropylidenedioxy-1-pyrroline-N-oxide, an Enantiopure Functionalized cyclic nitrone; Cycloaddition reactions with Dimethyl maleate and Dimethyl fumarate. Synth. Commun. 1998, 28, 3443–3450. [Google Scholar] [CrossRef]
- Cardona, F.; Moreno, G.; Guarna, F.; Vogel, P.; Schuetz, C.; Merino, P.; Goti, A. New Concise Total Synthesis of (+)-Lentiginosine and Some Structural Analogues. J. Org. Chem. 2005, 70, 6552–6555. [Google Scholar] [CrossRef] [PubMed]
- Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F.M.; Goti, A. Stereocontrolled Cyclic Nitrone Cycloaddition Strategy for the Synthesis of Pyrrolizidine and Indolizine Alkaloids. Chem. Eur. J. 2009, 15, 7808–7821. [Google Scholar] [CrossRef] [PubMed]
- Cicchi, S.; Bonami, M.; Cardona, F.; Revuelta, J.; Goti, A. Indium-Mediated Reduction of Hydroxylamines to Amines. Org. Lett. 2003, 5, 1773–1776. [Google Scholar] [CrossRef] [PubMed]
- Cobb, A.J.A.; Shaw, D.M.; Longbottom, D.A.; Gold, J.B.; Ley, S.V. Organocatalysis with proline derivatives: Improved catalysts for the asymmetric Mannich, nitro-Michael and aldol reactions. Org. Biomol. Chem. 2005, 3, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Alza, E.; Cambeiro, X.C.; Jimeno, C.; Pericàs, M.A. Highly Enantioselective Michael Additions in Water Catalyzed by a PS-Supported Pyrrolidine. Org. Lett. 2007, 9, 3717–3720. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 6 and 12 are available from the authors. |
Figure 1.
L-proline and two analogues.

Scheme 1.
Synthesis of several pyrrolidines from a sulfonylbutadiene or 2,3-O-iso-propylidene-D-erythronolactol.
Scheme 1.
Synthesis of several pyrrolidines from a sulfonylbutadiene or 2,3-O-iso-propylidene-D-erythronolactol.

Scheme 2.
Epimerization of C-2 in compound 8.

a. PhCOCl, Py., 0 °C-r.t., 97%; b. n-BuLi, THF, −78 °C-r.t., 50%; c NH2-NH2, EtOH, reflux, 0%; d. HCl 6M, reflux, 0%; e. LiAlH4, THF, 0 °C-r.t., 30%; f. BH3·THF, THF, 0 °C-40 °C, 15%; g. H2, Pd/C, MeOH, r.t., 80%.
Scheme 3.
Synthesis of 12 from 2,3-O-iso-propylidene-D-erythronolactol.

a. 1. NH2OSiMe2t-Bu, Py., r.t., 15h.; 2. MsCl, 0 °C, 2h, 50%; b. MeSO2Ph, n-BuLi, THF, −78 °C, 48%; c. H2, Pd(OH)2/C, HCl / MeOH, 4atm, 27%; d. H2, Pd/C, MeOH, 39%; e. In, NH4Cl/EtOH, reflux, 24h, 92%; f. Zn, NH4Cl/MeOH, reflux, 2h, 50%; g. In(cat), Zn, NH4Cl/MeOH reflux or r.t., see Table 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Reduction conditions of hydroxylamine 14 to pyrrolidines 6 and 12.
| Entry | Zn Equiv. | Ta | t(h) | η (%) | Ratio 6/12 |
|---|---|---|---|---|---|
| 1 | 6 | Reflux | 1 | 15 | 40/60 |
| 2 | 4 | Reflux | 2 | 10 | 36/64 |
| 3 | 4 | Reflux | 1 | 15 | 30/70 |
| 4 | 4 | Reflux | 0.5 | NDa | NDa |
| 5 | 2 | Reflux | 1 | NDa | NDa |
| 6 | 2 | Reflux | 1.5 | 10 | 40/60 |
| 7 | 2 | Reflux | 2 | 20 | 15/85 |
| 8 | 4 | r.t. | 5.5 | 40 | 5/95 |
| 9 | 3 | r.t. | 4 | 8 | 45/55 |
| 10 | 2 | r.t. | 4 | NDa | NDa |
a Not determined.
Table 2.
Solvent effects on the asymmetric Michael addition of cyclohexanone to trans-β-nitrostyrene with catalysts 6 and 12.
Table 2.
Solvent effects on the asymmetric Michael addition of cyclohexanone to trans-β-nitrostyrene with catalysts 6 and 12.
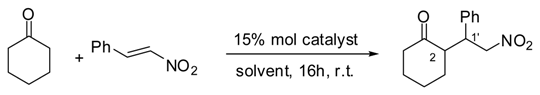
| Entry[a] | Solv. | Catalyst | Yield [%][b] | d.r.[%] [c] | ee[%][d] | Conf. |
|---|---|---|---|---|---|---|
| 1 | CHCl3 | 6 | - | - | - | - |
| 2 | CHCl3 | 12 | 35 | >95 | 38 | 2R, 1’S |
| 3 | DMSO | 6 | 20 | >95 | 69 | 2S, 1’R |
| 4 | DMSO | 12 | - | - | - | - |
[a] For experimental conditions see the Experimental section. [b] Yield of the isolated product. [c] Determined by 1H-NMR spectroscopic analysis. [d] Determined by chiral high-performance liquid chromatography (HPLC) analysis (Daicel Chiralpak AD, 25 cm/4.6 mm/5 μ).
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Flores, M.F.; Núñez, M.G.; Moro, R.F.; Garrido, N.M.; Marcos, I.S.; Iglesias, E.F.; García, P.; Díez, D. Synthesis of a New Chiral Pyrrolidine. Molecules 2010, 15, 1501-1512. https://doi.org/10.3390/molecules15031501
AMA Style
Flores MF, Núñez MG, Moro RF, Garrido NM, Marcos IS, Iglesias EF, García P, Díez D. Synthesis of a New Chiral Pyrrolidine. Molecules. 2010; 15(3):1501-1512. https://doi.org/10.3390/molecules15031501
Chicago/Turabian StyleFlores, Mari Fe., Marta G. Núñez, Rosalina F. Moro, Narciso M. Garrido, Isidro S. Marcos, Enrique F. Iglesias, Pilar García, and David Díez. 2010. "Synthesis of a New Chiral Pyrrolidine" Molecules 15, no. 3: 1501-1512. https://doi.org/10.3390/molecules15031501