Design and Use of Nanostructured Single-Site Heterogeneous Catalysts for the Selective Transformation of Fine Chemicals

Abstract
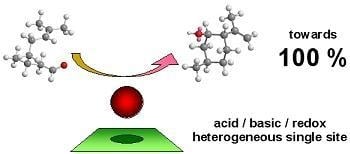
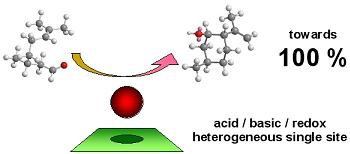
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Homogeneous Catalyst | Heterogeneous Catalyst |
|---|---|---|
| Form | metal complex | solid, often metal or metal oxide |
| Activity | high | variable |
| Selectivity | high | variable |
| Reaction Conditions | mild | drastic |
| Average Time of Life | variable | long |
| Sensitivity to Poisons | low | high |
| Problems of Diffusion | none | possible |
| Recycling | difficult (and expensive) | easy |
| Separation from Products | difficult | easy |
| Variations of steric and electronic features | possible | difficult |
| Intelligibility of the mechanism | possible | difficult |
2. Single-Site Heterogeneous Catalyst
- they consist of one or few atoms (as in the case of chemically defined metal clusters);
- are spatially isolated from one another;
- have all identical energy of interaction between the site itself and a reactant;
- are structurally well characterized.
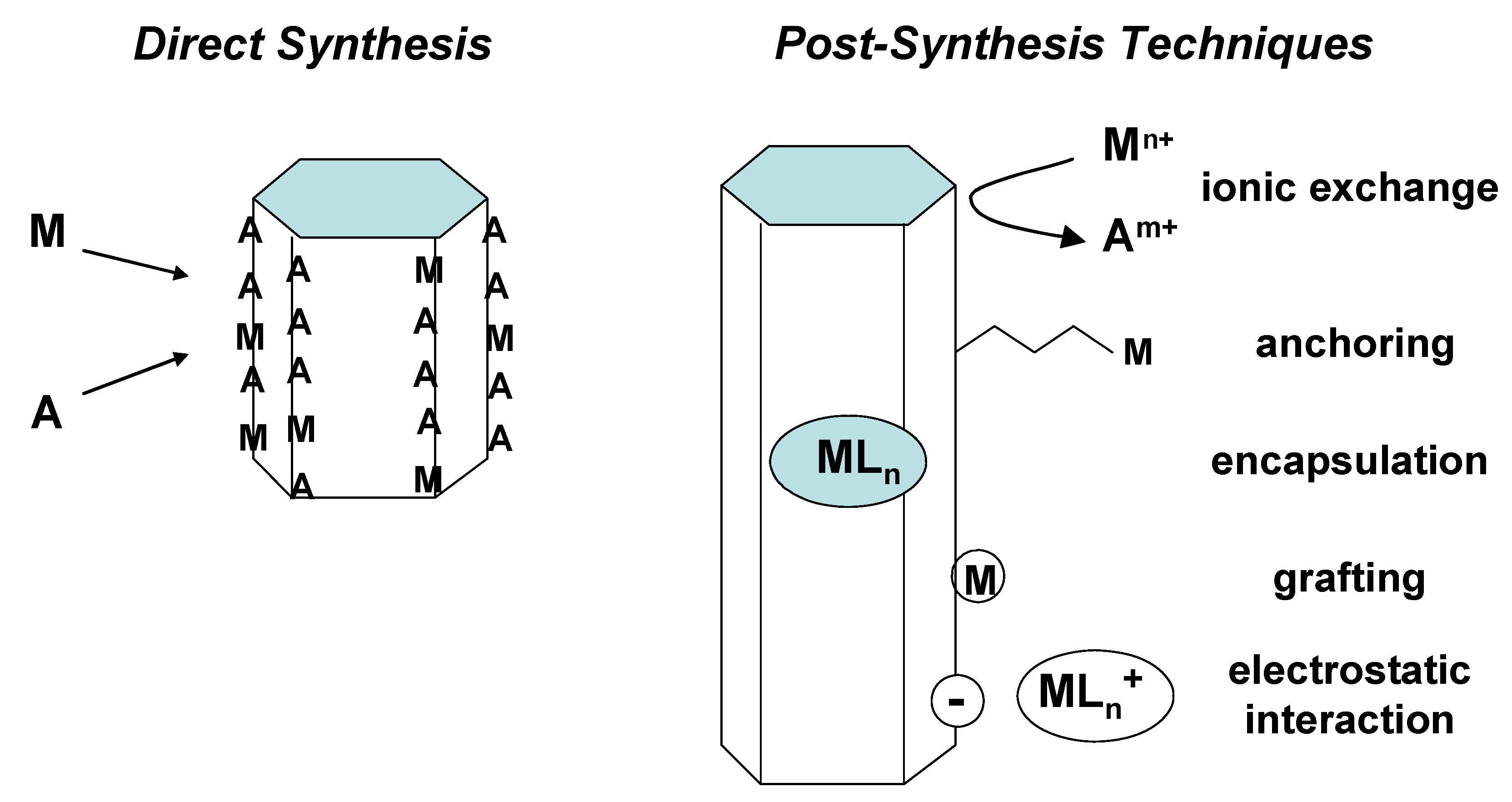
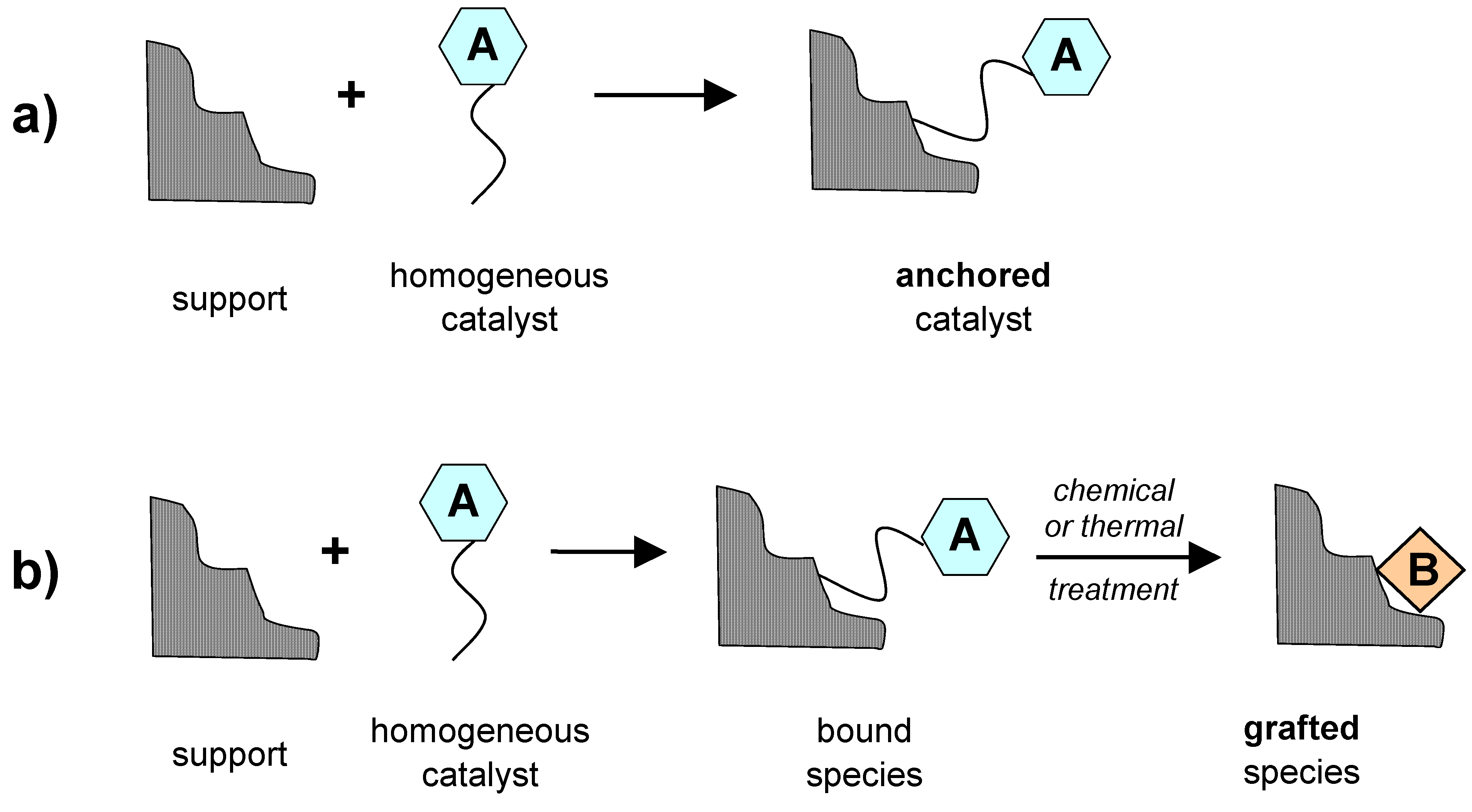
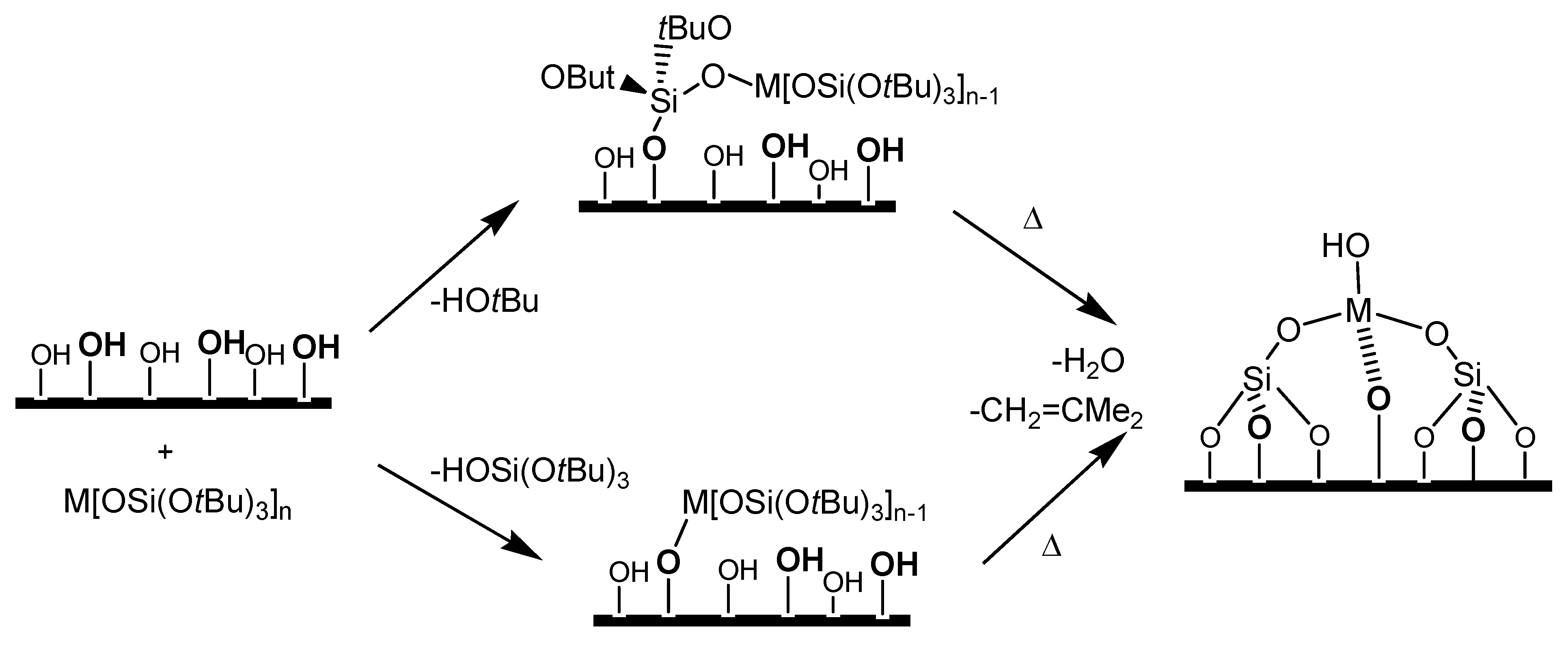
2.1. Preparation of single-site heterogeneous catalysts on inorganic supports

- 1)
- in-matrix synthesis,
- 2)
- post-synthetic covalent deposition,
- 3)
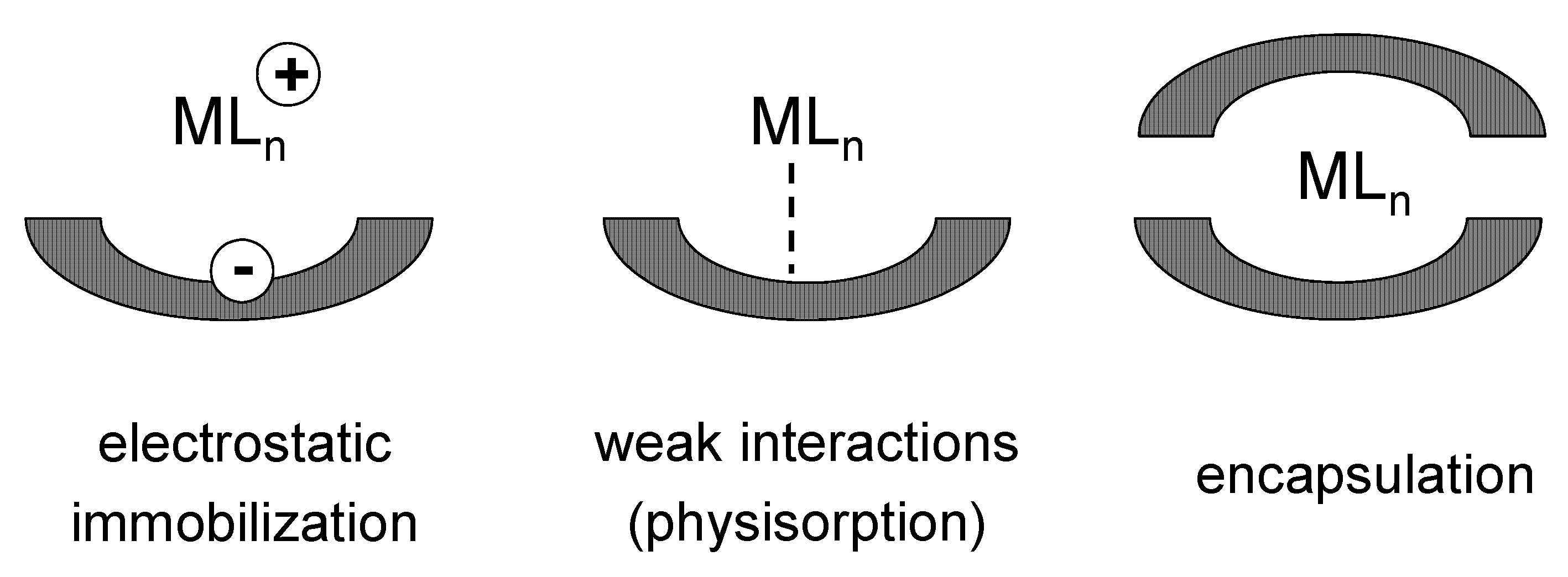
- post-synthetic non-covalent deposition.


3. Selected Examples and Applications of SSHC
3.1. Titanosilicalite-1 (TS-1): the forerunner of in-matrix single-site catalysts


3.2. Grafted single-site transition metals supported onto high surface area silica


By varying the catalytically-active centre, it is possible to find applications for SSHC in a large choice of selective oxidations, following the guidelines of clean and sustainable chemistry. Figure 8 presents a range of single-site transition-metal ions grafted onto the surface of mesoporous silica [77,78,83,85,86].

3.3. Anchored organometallic complexes as SSHC



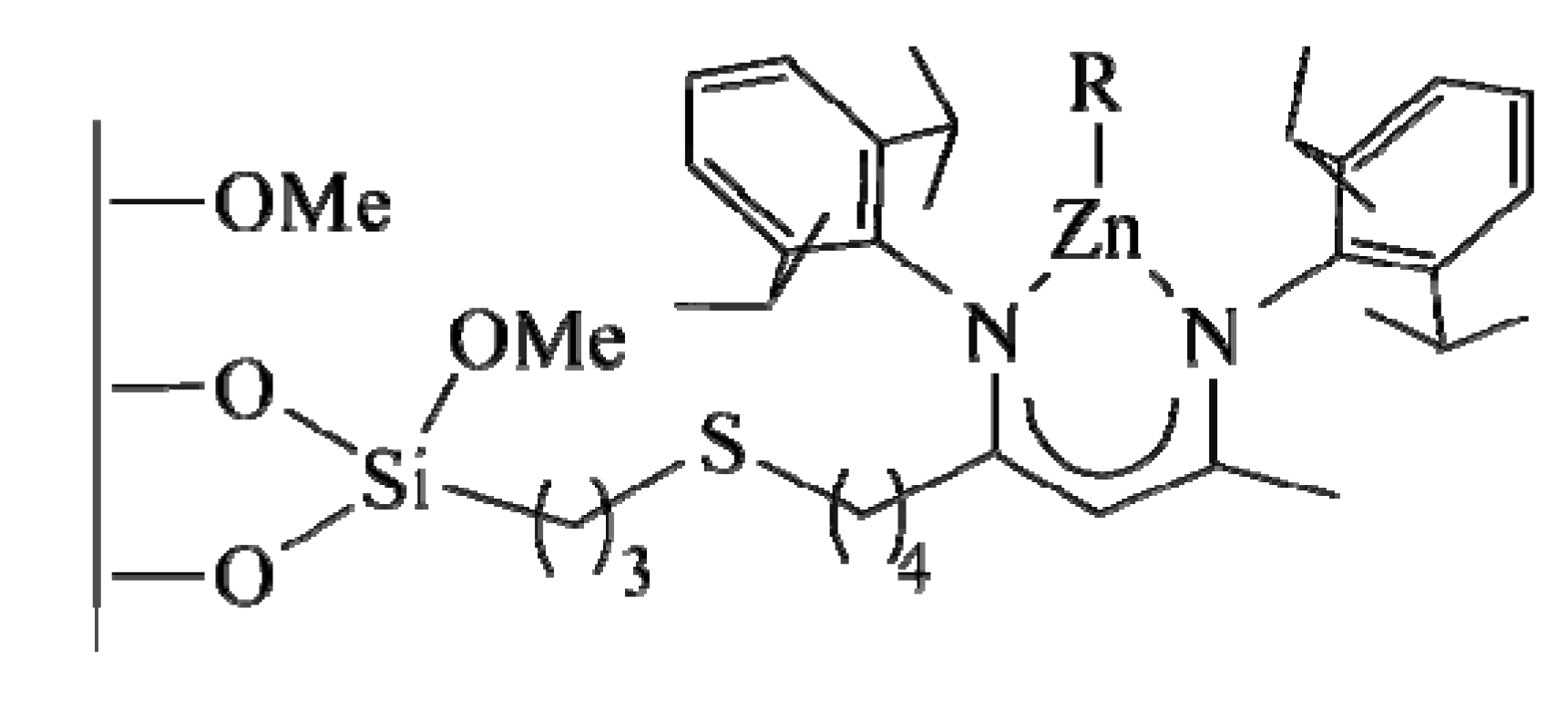
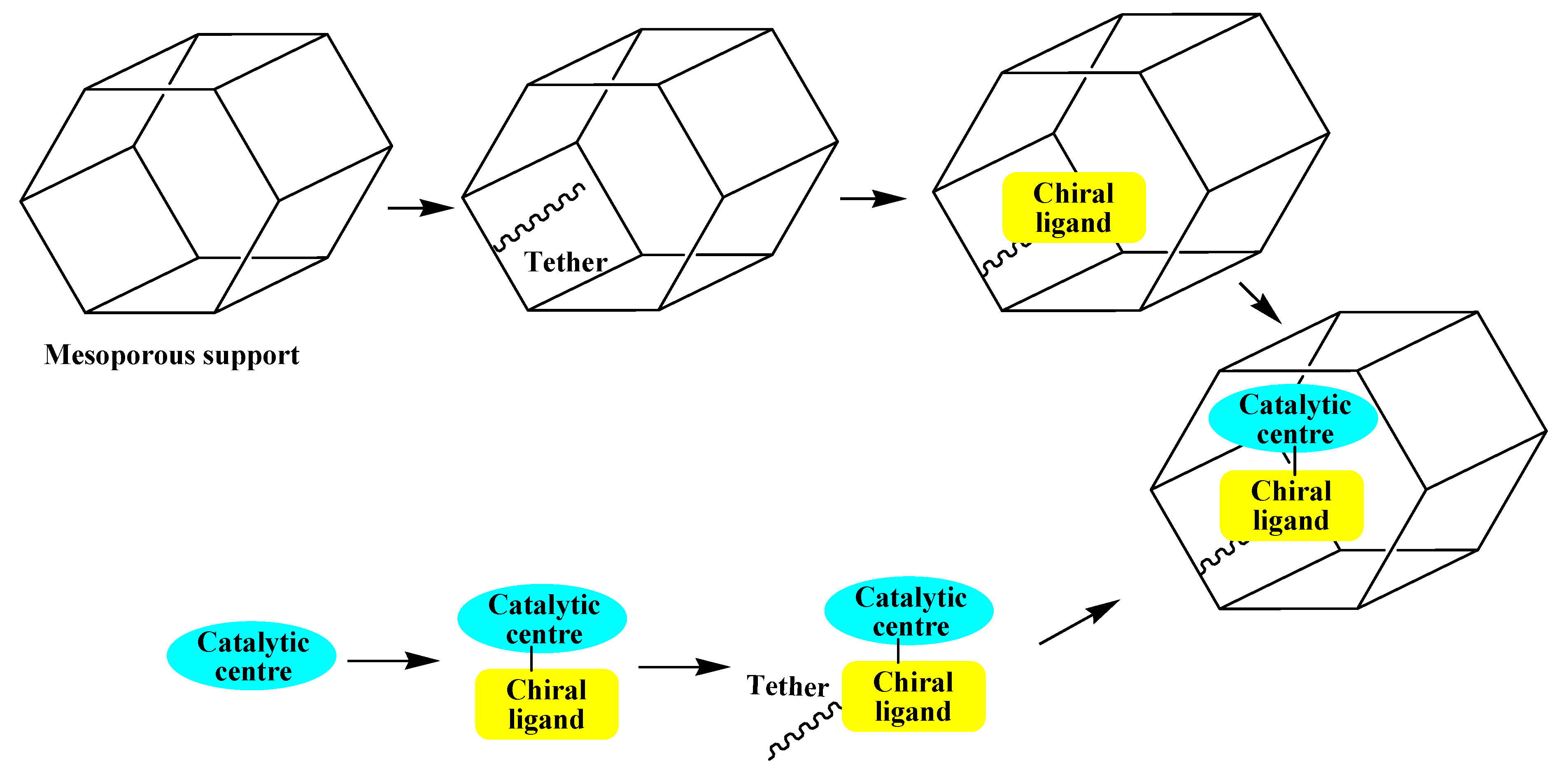
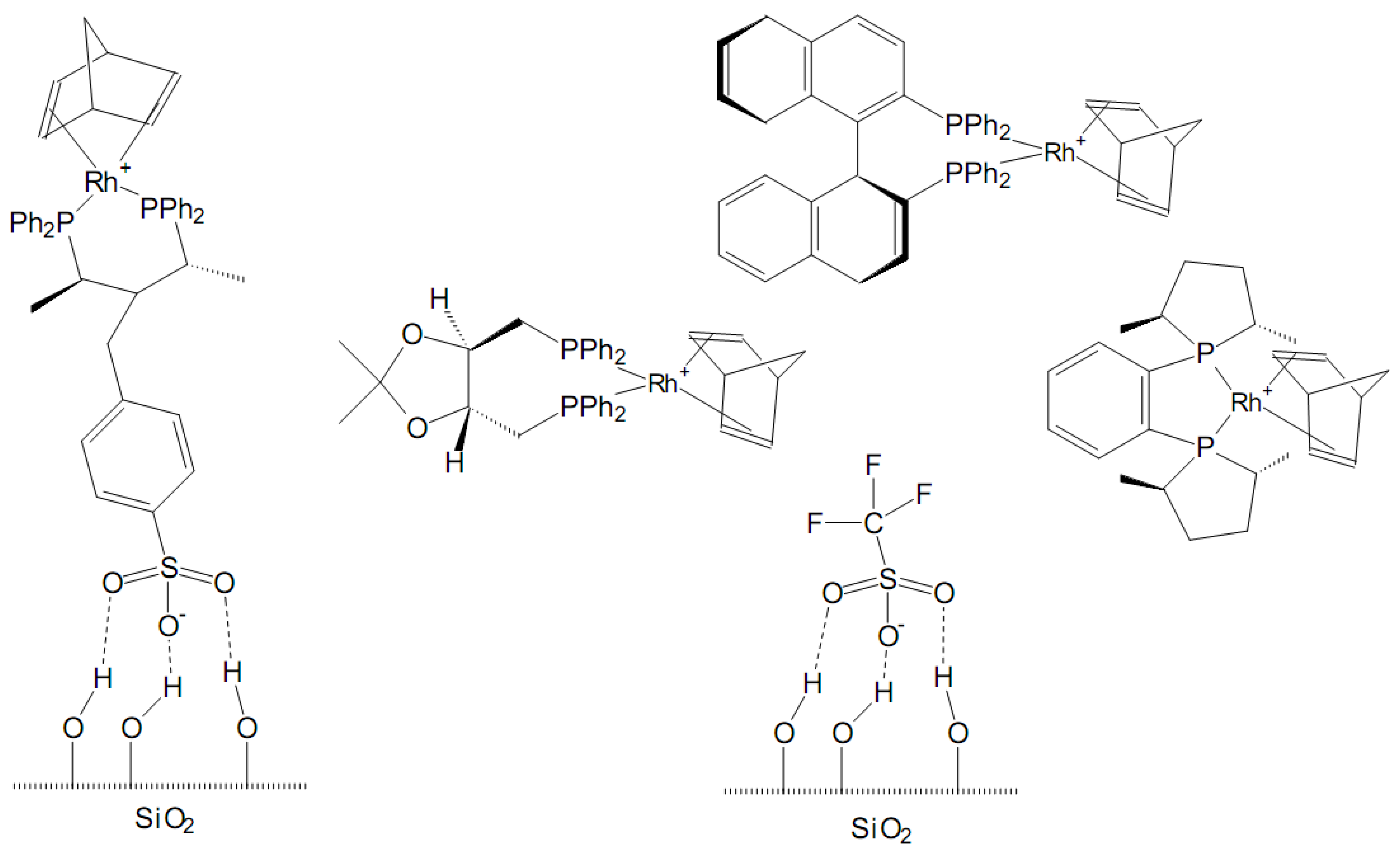
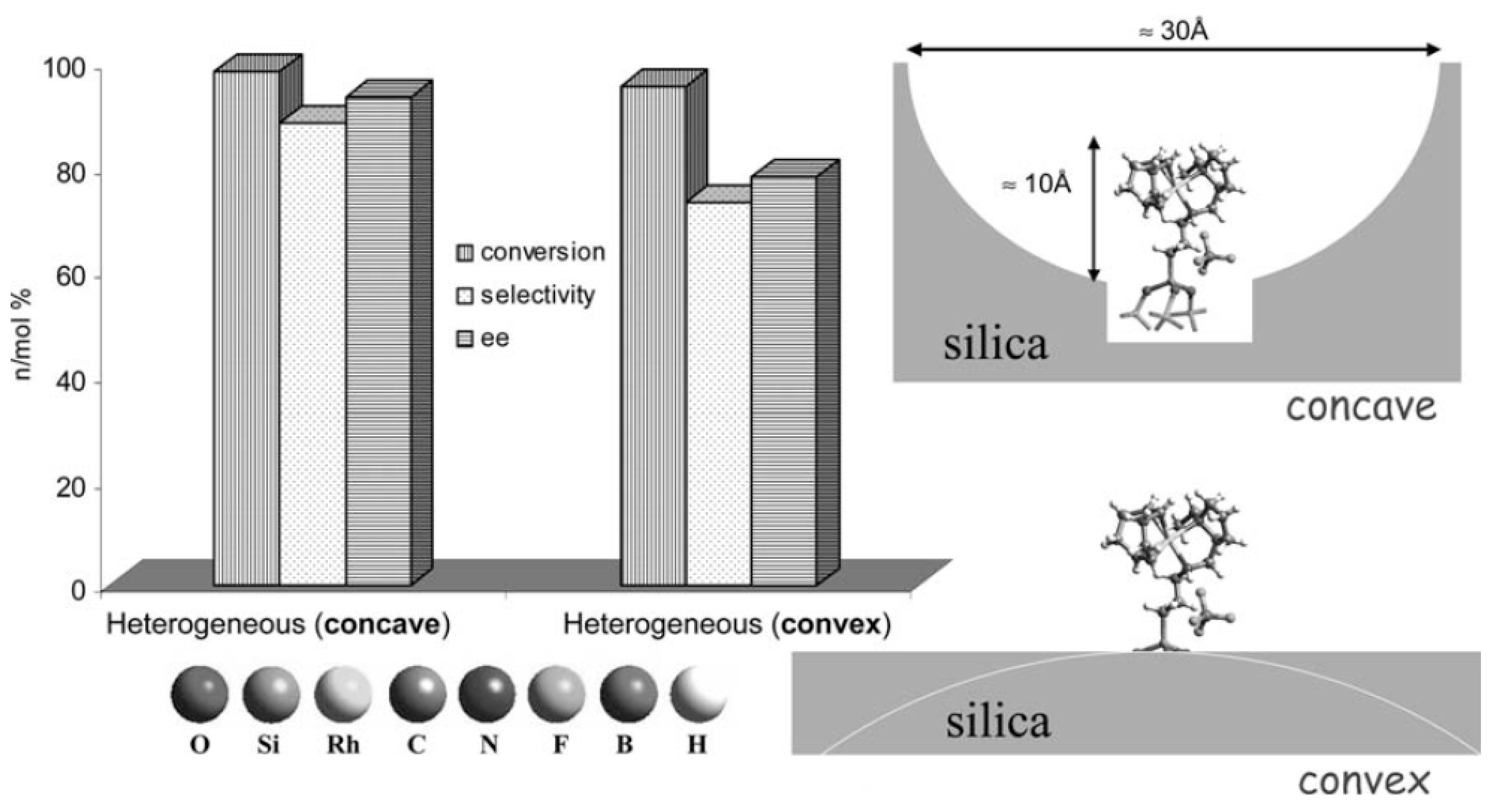
3.4. Single-site heterogeneous organometallic chiral catalysts



4. Hybrid Catalysts, a Synergistic Combination of Supported Metal Nanoparticles and SSHCs
5. Conclusions
Acknowledgements
References
- Centi, G.; Perathoner, S. Catalysis and sustainable (green) chemistry. Catal. Today 2003, 77, 287–297. [Google Scholar]
- Horvath, J.D.; Gellman, A.J. Naturally chiral surfaces. Top. Catal. 2003, 25, 9–15. [Google Scholar]
- Yeo, Y.Y.; Vattuone, L.; King, D.A. Calorimetric heats for CO and oxygen adsorption and for the catalytic CO oxidation reaction on Pt{111}. J. Chem. Phys. 1997, 106, 392–401. [Google Scholar]
- Copéret, C.; Chabanas, R.; Saint-Arroman, R.P.; Basset, J.M. Homogeneous and heterogeneous catalysis: Bridging the gap through surface organometallic chemistry. Angew. Chem. Int. Ed. 2003, 42, 156–181. [Google Scholar] [CrossRef]
- Thomas, J.M.; Raja, R.; Lewis, D.W. Single-Site Heterogeneous Catalysts. Angew. Chem. Int. Ed. 2005, 44, 6456–6482. [Google Scholar] [CrossRef]
- Thomas, J.M.; Raja, R. The advantages and future potential of single-site heterogeneous catalysts. Top. Catal. 2006, 40, 3–17. [Google Scholar] [CrossRef]
- Thomas, J.M.; Hernandez, J.C.; Raja, R.; Bell, R.G. Nanoporous oxidic solids: The confluence of heterogeneous and homogeneous catalysis. Phys. Chem. Chem. Phys. 2009, 11, 2799–2825. [Google Scholar]
- Thomas, J.M.; Raja, R. Designed Open-structure Heterogeneous Catalysts for the Synthesis of Fine Chemicals and Pharmaceuticals. In From Zeolites to Porous MOF Materials; Xu, R., Gao, Z., Chen, J., Yan, W., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 19–40. [Google Scholar]
- Thomas, J.M. Uniform Heterogeneous Catalysts: The role of solid-state chemistry in their development and design. Angew. Chem. Int. Ed. 1988, 27, 1673–1691. [Google Scholar] [CrossRef]
- Ballard, D.G.H. Pi and Sigma Transition Metal Carbon Compounds as Catalysts for the Polymerization of Vinyl Monomers and Olefins. Adv. Catal. 1973, 23, 263–325. [Google Scholar] [CrossRef]
- Candlin, J.P.; Thomas, H. Supported Organometallic Catalysts. Adv. Chem. Ser. 1974, 132, 212–239. [Google Scholar] [CrossRef]
- Yermakov, Y.I.; Kuznetsov, B.N.; Zakharov, V.A. Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1981. [Google Scholar]
- Evans, J. Surface Organometallic Chemistry: Molecular Approaches to Surface Catalysis, NATO ASI series; Basset, J.M., Gates, B.C., Candy, J.P., Choplin, A., Leconte, M., Quignard, F., Santini, C., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1988. [Google Scholar]
- Wilson, S.T.; Lok, B.M.; Messina, C.A.; Cannan, T.R.; Flanigen, E.M. Aluminophosphate molecular sieves: A new class of microporous crystalline inorganic solids. J. Am. Chem. Soc. 1982, 104, 1146–1147. [Google Scholar] [CrossRef]
- Lok, B.M.; Messina, C.A.; Patton, R.L.; Gajek, R.T.; Cannan, T.R.; Flanigen, E.M. Silicoaluminophosphate molecular sieves: Another new class of microporous crystalline inorganic solids. J. Am. Chem. Soc. 1984, 106, 6092–6093. [Google Scholar] [CrossRef]
- Weisz, P.B.; Haag, W.O.; Lago, R.M. The active site of acidic aluminosilicate catalysts. Nature 1984, 309, 589–591. [Google Scholar] [CrossRef]
- Breck, D.W. Zeolite Molecular Sieves; John Wiley & Sons: New York, NY, USA, 1974; p. 320. [Google Scholar]
- Corma, A. From Microporous to Mesoporous Molecular Sieve Materials and Their Use in Catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef]
- van Bekkum, H.; Jacobs, P.A.; Flanigen, E.M.; Jansen, J.C. Introduction to Zeolite Science and Practice; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Xiao, F.S. Ordered mesoporous materials with improved stability and catalytic activity. Top. Catal. 2005, 35, 9–24. [Google Scholar]
- Taguchi, A.; Schuth, F. Ordered mesoporous materials in catalysis. Microp. Mesop. Mater. 2005, 77, 1–45. [Google Scholar]
- Yang, Z.; Lu, Y.; Yang, Z. Mesoporous materials: Tunable structure, morphology and composition. Chem. Commun. 2009, 2270–2277. [Google Scholar]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710–712. [Google Scholar] [CrossRef]
- Voort, P.V.D.; Mathieu, M.; Mees, F.; Vansant, E.F. Synthesis of high-quality MCM-48 and MCM-41 by means of the GEMINI surfactant method. J. Phys. Chem. B 1998, 102, 8847–8851. [Google Scholar]
- Tanev, P.T.; Chibwe, M.; Pinnavaia, T.J. Titanium-containing mesoporous molecular sieves for catalytic oxidation of aromatic compounds. Nature 1994, 368, 321–323. [Google Scholar]
- Tanev, P.T.; Pinnavaia, T.J. A neutral templating route to mesoporous molecular sieves. Science 1995, 267, 865–867. [Google Scholar]
- Kleitz, F.; Choi, S.H.; Ryoo, R. Cubic Ia3d large mesoporous silica: Synthesis and replication to platinum nanowires, carbon nanorods and carbon nanotubes. Chem. Commun. 2003, 2136–2137. [Google Scholar]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Rigutto, M.S.; van Veen, R.; Huve, L. Application of mesoporous molecular sieves in catalysis and separation. Stud. Surf. Sci. Catal. 2007, 168, 837–854. [Google Scholar] [CrossRef]
- Wen, D.J.; Min, S.C.; Ling, Q.; Ming, S.J.; Zhong, X.X. Pilot synthesis and commercial application in FCC catalyst of MCM-41 zeolite. J. Porous Mater. 2008, 15, 189–197. [Google Scholar] [CrossRef]
- Ondrey, G. A commercial debut for a process to make mesoporous silica. Chem. Eng. 2008, 13. [Google Scholar]
- Guisnet, M.; Guidotti, M. One-pot Reactions on Bifunctional Catalysts. In Catalysts for Fine Chemical Synthesis; Derouane, E.G., Ed.; Wiley: Weinheim, Germany, 2006; pp. 157–169. [Google Scholar]
- Bisio, C.; Gatti, G.; Marchese, L.; Guidotti, M.; Psaro, R. Design and Applications of Multifunctional Catalysts Based on Inorganic Oxides. In Design and Applications of Multifunctional Catalysts Based on Inorganic Oxides; Pan Stanford Press: Singapore, in press.
- Maishal, T.K.; Alauzun, J.; Basset, J.M.; Copéret, C.; Corriu, R.J.P.; Jeanneau, E.; Mehdi, A.; Reyé, C.; Veyre, L.; Thieuleux, C. A tailored organometallic–inorganic hybrid mesostructured material: A route to a well-defined, active, and reusable heterogeneous iridium-NHC catalyst for H/D exchange. Angew. Chem. Int. Ed. 2008, 47, 8654–8656. [Google Scholar]
- Campbell, I.M. The Constitution of the Catalytic Surfaces. In Catalysis at Surfaces; Chapman & Hall: London, UK, 1988; p. 64. [Google Scholar]
- Louis, C.; Che, M. Anchoring and Grafting of Coordination Metal Complexes onto Oxide Supports. In Preparation of Solid Catalysts; Ertl, G., Knözinger, H., Weitkamp, J., Eds.; Wiley-VCH: Weinheim, Germany, 1999; p. 341. [Google Scholar]
- Heinrichs, C.; Hölderich, W.F. Novel zeolitic hosts for “ship-in-a-bottle” catalysts. Catal. Lett. 1999, 58, 75–80. [Google Scholar] [CrossRef]
- Fukoka, A.; Higashimoto, N.; Sakamoto, Y.; Sasaki, M.; Sugimoto, N.; Inagaki, S.; Fukoshima, Y.; Ichikawa, M. Ship-in-bottle synthesis and catalytic performances of platinum carbonyl clusters, nanowires, and nanoparticles in micro- and mesoporous materials. Catal. Today 2001, 66, 23–31. [Google Scholar]
- De Vos, D.; Hermans, I.; Sels, B.; Jacobs, P. Hybrid Oxidation Catalysts from Immobilized Complexes on Inorganic Microporous Supports. In Catalysts for Fine Chemical Synthesis; Derouane, E.G., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 207–240. [Google Scholar]
- Taramasso, M.; Perego, G.; Notari, B. Preparation of porous crystalline synthetic material comprised of silicon and titanium oxides. U.S. Patent 4,410,501, 18 October 1983. [Google Scholar]
- Neri, C.; Anfossi, B.; Esposito, A.; Buonomo, F. Process for the epoxidation of olefinic compounds. Patent IT 22608, 31 December 1982. EP 100119, 13 July 1986. [Google Scholar]
- Cleric, M.G. Titanium Silicalite-1. In Metal Oxide Catalysis; Jackson, S.D., Hargreaves, J.S.J., Eds.; Wiley-VCH: Weinheim, Germany, 2009; p. 705. [Google Scholar]
- Guisnet, M.; Guidotti, M. Applications in Synthesis of Commodities and Fine Chemicals. In Zeolite Chemistry and Catalysis. An Integrated Approach and Tutorial; Chester, A.W., Derouane, E.G., Eds.; Springer: Dordrecht, The Nederlands, 2009; p. 275. [Google Scholar]
- Cavani, F.; Teles, J.H. Sustainability in catalytic oxidation: An alternative approach or a structural evolution. Chem. Sus. Chem. 2009, 2, 508–534. [Google Scholar]
- Perego, G.; Bellussi, G.; Corno, C.; Taramasso, M.; Buonomo, F.; Esposito, A. Titanium-Silicalite: a Novel Derivative in the Pentasil Family. Stud. Surf. Sci. Catal. 1986, 28, 129–136. [Google Scholar]
- Petrini, G.; Leofanti, G.; Mantegazza, M.A.; Pignataro, F. Caprolactam via ammoximation. ACS Symp. Ser. 1996, 626, 33–48. [Google Scholar]
- Ratton, S. Heterogeneous catalysis in the fine chemicals industry. From dream to reality. Chem. Today 1998, 16, 33–37. [Google Scholar]
- Sheldon, R.A.; Wallau, M.; Arends, I.W.C.E.; Schuchardt, V. Heterogeneous catalysts for liquid-phase oxidations: Philosophers' Stones or Trojan Horses. Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar] [CrossRef]
- Boronat, M.; Concepciòn, P.; Corma, A.; Renz, M. Peculiarities of Sn-Beta and potential industrial applications. Catal. Today 2007, 121, 39–44. [Google Scholar]
- Zhu, Y.; Liu, S.; Jaenicke, S.; Chuah, G. Zirconia catalysts in Meerwein-Ponndorf-Verley reduction of citral. Catal. Today 2004, 97, 249–255. [Google Scholar]
- Pastore, H.O.; Coluccia, S.; Marchese, L. Porous Aluminophosphates: From molecular sieves to designed acid catalysts. Ann. Rev. Mater. Res. 2005, 35, 351–395. [Google Scholar] [CrossRef]
- Corma, A.; Nemeth, L.T.; Renz, M.; Valencia, S. Sn-zeolite beta as a heterogeneous chemoselective catalyst for Baeyer–Villiger oxidations. Nature 2001, 412, 423–425. [Google Scholar] [CrossRef]
- Yongzhong, Z.; Yuntong, N.; Jaenicke, S.; Chuah, G.K. Cyclisation of citronellal over zirconium zeolite beta; a highly diastereoselective catalyst to (±)-isopulegol. J. Catal. 2005, 229, 404–413. [Google Scholar] [CrossRef]
- Zhu, Y.; Chuah, G.K.; Jaenicke, S. Selective Meerwein-Ponndorf-Verley reduction of α,β-unsaturated aldehydes over Zr-zeolite beta. J. Catal. 2006, 241, 25–33. [Google Scholar]
- Raja, R.; Sankar, G.; Thomas, J.M. Bifunctional molecular sieve catalysts for the benign ammoximation of cyclohexanone: One-step, solvent-free production of oxime and ε-caprolactam with a mixture of air and ammonia. J. Am. Chem. Soc. 2001, 123, 8153–8154. [Google Scholar]
- Thomas, J.M.; Raja, R. Design of a “green” one-step catalytic production of ε-caprolactam (precursor of nylon-6). Proc. Nat. Acad. Sci. USA 2005, 102, 13732–13736. [Google Scholar]
- Raja, R.; Thomas, J.M.; Greenhill-Hooper, M.; Ley, S.V.; Almeida-Paz, F.A. Facile, one-step production of niacin (Vitamin B3) and other nitrogen-containing pharmaceutical chemicals with a single-site heterogeneous catalyst. Chem. Eur. J. 2008, 14, 2340–2348. [Google Scholar] [CrossRef]
- Basset, J.M.; Choplin, A. Surface organometallic chemistry: A new approach to heterogeneous catalysis. J. Mol. Catal. 1983, 21, 95–108. [Google Scholar]
- Basset, J.M.; Ugo, R. Modern Surface Organometallic Chemistry; Basset, J.M., Psaro, R., Roberto, D., Ugo, R., Eds.; Wiley-VCH: Weinheim, Germany, 2009; p. 1. [Google Scholar]
- Ugo, R.; Psaro, R.; Zanderighi, G.M.; Basset, J.M.; Theolier, A.; Smith, A.K. Fundamental Research in Homogeneous Catalysis; Tsutsui, M., Ed.; Plenum Press: New York, NY, USA, 1979; Volume 3, pp. 579–601. [Google Scholar]
- Besson, B.; Moraweck, B.; Smith, A.K.; Basset, J.M.; Psaro, R.; Fusi, A.; Ugo, R. IR and EXAFS characterisation of a supported osmium cluster carbonyl. J. Chem. Soc. Chem. Commun. 1980, 569–571. [Google Scholar]
- Lecuyer, C.; Quignard, F.; Choplin, A.; Olivier, D.; Basset, J.M. Surface organometallic chemistry on oxides: Selective catatalytic low-temperature hydrogenolysis of alkenes by a highly electrophilic zirconium hydride complex supported on silica. Angew. Chem. Int. Ed. 1991, 30, 1660–1661. [Google Scholar] [CrossRef]
- Chabanas, M.; Vidal, V.; Copéret, C.; Thivolle-Cazat, J.; Basset, J.M. Low-temperature hydrogenolysis of alkanes catalyzed by a silica-supported tantalum hydride complex, and evidence for a mechanistic switch from Group IV to Group V Metal Surface Hydride Complexes. Angew. Chem. Int. Ed. 2000, 39, 1962–1965. [Google Scholar] [CrossRef]
- Corma, A.; Navarro, M.T.; Parente, J.P. Synthesis of an ultralarge pore titanium silicate isomorphous to MCM-41 and its application as a catalyst for selective oxidation of hydrocarbons. J. Chem. Soc. Chem. Commun. 1994, 147–148. [Google Scholar]
- Sankar, G.; Rey, F.; Thomas, J.M.; Greaves, G.N.; Corma, A.; Dobson, B.R.; Dent, A.J. Probing active sites in solid catalysts for the liquid-phase epoxidation of alkenes. J. Chem. Soc. Chem. Commun. 1994, 2279–2280. [Google Scholar]
- Thomas, J.M. The chemistry of crystalline sponges. Nature 1994, 368, 289–290. [Google Scholar] [CrossRef]
- Maschmeyer, T.; Rey, F.; Sankar, G.; Thomas, J.M. Heterogeneous catalysts obtained by grafting metallocene complexes onto mesoporous silica. Nature 1995, 378, 159–162. [Google Scholar]
- Guidotti, M.; Ravasio, N.; Psaro, R.; Ferraris, G.; Moretti, G. Epoxidation on titanium-containing silicates: Do structural features really affect the catalytic performance. J. Catal. 2003, 214, 242–250. [Google Scholar] [CrossRef]
- Ravasio, N.; Zaccheria, F.; Guidotti, M.; Psaro, R. Mono- and bi-functional heterogeneous catalytic transformation of terpenes and terpenoids. Top. Catal. 2004, 27, 157–168. [Google Scholar]
- Guidotti, M.; Ravasio, N.; Psaro, R.; Gianotti, E.; Marchese, L.; Coluccia, S. Epoxidation of unsaturated FAMEs obtained from vegetable source over Ti(IV)-grafted silica catalysts: A comparison between ordered and non-ordered mesoporous materials. J. Mol. Catal. A Chem. 2006, 250, 218–225. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Guidotti, M.; Pirovano, C.; Ravasio, N.; Barmatova, M.V.; Chesalov, Y.A. Highly selective oxidation of alkylphenols to benzoquinones with Hydrogen peroxide over silica-supported titanium catalysts: Titanium cluster site versus titanium single site. Adv. Synth. Catal. 2009, 351, 1877–1889. [Google Scholar]
- Guidotti, M.; Pirovano, C.; Ravasio, N.; Lázaro, B.; Fraile, J.M.; Mayoral, J.A.; Coq, B.; Galarneau, A. The use of H2O2 over titanium-grafted mesoporous silica catalysts: A step further towards sustainable epoxidation. Green Chem. 2009, 11, 1421. [Google Scholar]
- Cativiela, C.; Fraile, J.M.; Garcia, J.I.; Mayoral, J.A. A new titanium-silica catalyst for the epoxidation of alkenes. J. Mol. Catal. A 1996, 112, 259–267. [Google Scholar] [CrossRef]
- Fraile, J.M.; Garcia, J.I.; Mayoral, J.A.; Vispe, E. Silica-supported titanium derivatives as catalysts for the epoxidation of alkenes with hydrogen peroxide: A new way to tunable catalytic activity through ligand exchange. J. Catal. 2000, 189, 40–51. [Google Scholar]
- Fraile, J.M.; Garcia, J.I.; Mayoral, J.A.; Vispe, E. Effect of the reaction conditions on the epoxidation of alkenes with hydrogen peroxide catalyzed by silica-supported titanium derivatives. J. Catal. 2001, 204, 146–156. [Google Scholar] [CrossRef]
- Fraile, J.M.; Garcia, J.I.; Mayoral, J.A.; Vispe, E. Optimization of cyclohexene epoxidation with dilute hydrogen peroxide and silica-supported titanium catalysts. Appl. Catal. A 2003, 245, 363–376. [Google Scholar] [CrossRef]
- Thomas, J.M.; Raja, R. Significance of mesoporous catalysts for catalytic application. Stud. Surf. Sci. Catal. 2004, 148, 163–211. [Google Scholar] [CrossRef]
- Sakthivel, A.; Zhao, J.; Kühn, F.E. Cyclopentadienyl molybdenum complexes grafted on zeolites – synthesis and catalytic application. Catal. Lett. 2005, 102, 115–119. [Google Scholar] [CrossRef]
- Cativiela, C.; Fraile, J.M.; Garcia, J.I.; Mayoral, J.A.; Pires, E.; Royo, A.J. Silica and alumina modified by Lewis acids as catalysts in Diels-Alder reaction of carbonyl-containing dienophiles. Tetrahedron 1993, 49, 4073–4084. [Google Scholar] [CrossRef]
- Fraile, J.M.; Garcia, J.I.; Mayoral, J.A.; Pires, E.; Salvatella, L.; Ten, M. On the nature of Lewis acid sites of aluminium-modified silica. A theoretical and experimental study. J. Phys. Chem. B 1999, 103, 1664–1670. [Google Scholar]
- Boronat, M.; Concepcion, P.; Corma, A.; Navarro, M.T.; Renz, M.; Valencia, S. Reactivity in the confined spaces of zeolites: The interplay between spectroscopy and theory to develop structure-activity relationships for catalysis. Phys. Chem. Chem. Phys. 2009, 11, 2876–2884. [Google Scholar]
- Jarupatrakorn, J.; Tilley, J.D. Silica-supported, single-site titanium catalysts for olefin epoxidation. A molecular precursor strategy for control of catalyst structure. J. Am. Chem. Soc. 2002, 124, 8380–8388. [Google Scholar] [CrossRef]
- Fujdala, K.L.; Tilley, T.D. Design and synthesis of heterogeneous catalysts: The thermolytic molecular precursor approach. J. Catal. 2003, 216, 265–275. [Google Scholar] [CrossRef]
- Drake, I.J.; Fujdala, K.L.; Bell, A.T.; Tilley, T.D. Dimethyl carbonate production via the oxidative carbonylation of methanol over Cu/SiO2 catalysts prepared via molecular precursor grafting and chemical vapor deposition approaches. J. Catal. 2005, 230, 14–27. [Google Scholar]
- Bell, A.T. The impact of nanoscience on heterogeneous catalysis. Science 2003, 299, 1688–1691. [Google Scholar]
- Burcham, L.J.; Briand, L.E.; Wachs, I.E. Quantification of active sites for the determination of methanol oxidation turn-over frequencies using methanol chemisorption and in situ infrared techniques. 1. Supported metal oxide catalysts. Langmuir 2001, 17, 6164–6174. [Google Scholar] [CrossRef]
- Thomas, J.M.; Johnson, B.F.G.; Raja, R.; Sankar, G.; Midgley, P.A. High-performance nanocatalysts for single-step hydrogenation. Acc. Chem. Res. 2003, 36, 20–30. [Google Scholar]
- Hermans, S.; Raja, R.; Thomas, J.M.; Johnson, B.F.G.; Sankar, G.; Gleeson, D. Solvent-free, low-temperature, selective hydrogenation of polyenes using a bimetallic nanoparticle Ru-Sn catalyst. Angew. Chem. Int. Ed. 2001, 40, 1211–1215. [Google Scholar] [CrossRef]
- Raja, R.; Khimyak, T.; Thomas, J.M.; Hermans, S.; Johnson, B.F.G. Single-step, highly active and highly selective nanoparticle catalysts for the hydrogenation of key organic compounds. Angew. Chem. Int. Ed. 2001, 40, 4638–4642. [Google Scholar] [CrossRef]
- Chabanas, M.; Baudouin, A.; Coperet, C.; Basset, J.M. A highly active well-defined Rhenium heterogeneous catalyst for olefin metathesis prepared via surface organometallic chemistry. J. Am. Chem. Soc. 2001, 123, 2062–2063. [Google Scholar] [CrossRef]
- Chabanas, M.; Coperet, C.; Basset, J.M. Re-Based Heterogeneous Catalysts for Olefin Metathesis Prepared by Surface Organometallic Chemistry: Reactivity and Selectivity. Eur. J. Chem. 2003, 9, 971–975. [Google Scholar] [CrossRef]
- Blanc, F.; Chabanas, M.; Coperet, C.; Fenet, B.; Herdweck, E. Reactivity differences between molecular and surface silanols in the preparation of homogeneous and heterogeneous olefin metathesis catalysts. J. Organom. Chem. 2005, 690, 5014–5026. [Google Scholar]
- Le Roux, E.; Taoufik, M.; Chabanas, M.; Alcor, D.; Baudouin, A.; Coperet, C.; Thivolle-Cazat, J.; Basset, J.M.; Lesage, A.; Hediger, S.; Emsley, L. Well-defined surface tungstenocarbyne complexes through the reaction of [W(≡CtBu)(CH2tBu)3] with silica. Organometallics 2005, 24, 4274–4279. [Google Scholar]
- Le Roux, E.; Taoufik, M.; Coperet, C.; de Mallmann, A.; Thivolle-Cazat, J.; Basset, J.M.; Maunders, B.M.; Sunley, G.J. Development of Tungsten-based heterogeneous alkane metathesis catalysts through a structure–activity relationship. Angew. Chem. Int. Ed. 2005, 44, 6755–6758. [Google Scholar]
- Copéret, C.; Basset, J.M. Strategies to immobilize well-defined olefin metathesis catalysts: Supported Homogeneous Catalysis vs. Surface Organometallic Chemistry. Adv. Synth. Catal. 2007, 349, 78–92. [Google Scholar] [CrossRef]
- Jones, C.W.; McKittrick, M.W.; Nguyen, J.V.; Yu, K. Design of silica-tethered metal complexes for polymerization catalysis. Top. Catal. 2005, 34, 67–76. [Google Scholar]
- Yu, K.; Jones, C.W. Silica immobilized Zinc β-diiminate catalysts for the copolimerization of epoxides and carbon dioxide. Organometallics 2003, 22, 2571–2580. [Google Scholar] [CrossRef]
- McKittrick, M.W.; Jones, C.W. Toward single-site, immobilized molecular catalysts: Site-isolated Ti ethylene polymerization catalysts supported on porous silica. J. Am. Chem. Soc. 2004, 126, 3052–3053. [Google Scholar] [CrossRef]
- McKittrick, M.W.; Jones, C.W. Effect of site isolation on the preparation and performance of silica-immobilized Ti CGC-inspired ethylene polymerization catalysts. J. Catal. 2004, 227, 186–201. [Google Scholar]
- Yu, K.; McKittrick, M.W.; Jones, C.W. Role of amine structure and site isolation on the performance of amminosilica-immobilized zirconium CGC-inspired ethylene polymerization catalysts. Organometallics 2004, 23, 4089–4096. [Google Scholar] [CrossRef]
- Carniato, F.; Bisio, C.; Boccaleri, E.; Guidotti, M.; Gavrilova, E.; Marchese, L. Titanosilsesquioxane anchored on mesoporous silica. A novel approach for the preparation of heterogeneous catalysts for selective oxidations. Chem. Eur. J. 2008, 14, 8098–8101. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Thomas, J.M.; Willock, D.J. Preface. Top. Catal. 2003, 25, 1. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Bekkum, H.V. Fine Chemicals Through Heterogeneous Catalysis; Wiley-VCH: Weinheim, Germany, 2001; p. 1. [Google Scholar]
- De Vos, D.E.; Vankelecom, I.F.J.; Jacobs, P.A. Chiral Catalyst Immobilization and Recycling; Wiley-VCH: Weinheim, Germany, 2000; p. 1. [Google Scholar]
- McMorn, P.; Hutchings, G.J. Heterogeneous enantioselective catalysts: Strategies for the immobilisation of homogeneous catalysts. Chem. Soc. Rev. 2004, 33, 108–122. [Google Scholar] [CrossRef]
- Li, C. Chiral synthesis on catalysts immobilized in microporous and mesoporous materials. Catal. Rev. 2004, 46, 419–492. [Google Scholar] [CrossRef]
- Li, C.; Zhang, H.; Jiang, D.; Yang, Q. Chiral catalysis in nanopores of mesoporous materials. Chem. Commun. 2007, 547–558. [Google Scholar]
- Song, C.E.; Lee, S.-G. Supported chiral catalysts on inorganic materials. Chem. Rev. 2002, 102, 3495–3524. [Google Scholar] [CrossRef]
- Thomas, J.M.; Raja, R. Exploiting nanospace for asymmetric catalysis: Confinement of immobilized, single-site chiral catalysts enhances enantioselectivity. Acc. Chem. Res. 2007, 41, 708–720. [Google Scholar] [CrossRef]
- Fraile, J.M.; Garcia, J.I.; Mayoral, J.A. Noncovalent immobilization of eneatioselective catalysts. Chem. Rev. 2009, 109, 360–417. [Google Scholar] [CrossRef]
- Bigi, F.; Franca, B.; Moroni, L.; Maggi, R.; Sartori, G. Heterogeneous enantioselective epoxidation of olefins catalysed by unsymmetrical (salen)Mn(III) complexes supported on amorphous or MCM-41 silica through a new triazine-based linker. Chem. Comm. 2002, 716–717. [Google Scholar]
- Xiang, S.; Zhang, Y.L.; Xin, Q.; Li, C. Enantioselective epoxidation of olefins catalyzed by Mn (salen)/MCM-41 synthesized with a new anchoring method. Chem Commun. 2002, 2696–2697. [Google Scholar]
- Bianchini, C.; Barbaro, P. Recent aspects of asymmetric catalysis by immobilized chiral metal catalysts. Top. Catal. 2002, 19, 17–32. [Google Scholar]
- Sabater, M.J.; Corma, A.; Domenech, A.; Fornes, V.; Garcia, H. Chiral salen manganese complex encapsulated within zeolite Y: A heterogeneous enantioselective catalyst for the epoxidation of alkenes. Chem. Commun. 1997, 1285–1286. [Google Scholar]
- Zhang, R.; Yu, W.Y.; Wong, K.Y.; Che, C.M. Highly efficient asymmetric epoxidation of alkenes with a D4-symmetric chiral dichlororuthenium(IV) porphyrin catalyst. J. Org. Chem. 2001, 66, 8145–8153. [Google Scholar] [CrossRef]
- Bianchini, C.; Burnaby, D.G.; Evans, J.; Frediani, P.; Meli, A.; Oberhauser, W.; Psaro, R.; Sordelli, L.; Vizza, F. Preparation, characterization, and performance of tripodal polyphosphine rhodium catalysts immobilized on silica via hydrogen bonding. J. Am. Chem. Soc. 1999, 121, 5961–5971. [Google Scholar]
- de Rege, F.M.; Morita, D.K.; Ott, K.C.; Tumas, W.; Broene, R.D. Non-covalent immobilization of homogeneous cationic chiral rhodium–phosphine catalysts on silica surfaces. Chem. Commun. 2000, 1797–1798. [Google Scholar]
- Bianchini, C.; Barbaro, P.; Dal Santo, V.; Gobetto, R.; Meli, A.; Oberhauser, W.; Psaro, R.; Vizza, F. Immobilization of optically active rhodium-diphosphine complexes on porous silica via hydrogen bonding. Adv. Synth. Catal. 2001, 343, 41–45. [Google Scholar] [CrossRef]
- Thomas, J.M.; Maschmeyer, T.; Johnson, B.F.G.; Shephard, D.S. Constrained chiral catalysts. J. Mol. Catal. A Chem. 1999, 141, 139–144. [Google Scholar] [CrossRef]
- Raynor, S.A.; Thomas, J.M.; Raja, R.; Johnson, B.F.G.; Bell, R.G.; Mantle, M.D. A one-step, enantioselective reduction of ethyl nicotinate to ethyl nipecotinate using a constrained, chiral, heterogeneous catalyst. Chem. Commun. 2000, 1925–1926. [Google Scholar]
- Goettmann, F.; Sanchez, C. How does confinement affect the catalytic activity of mesoporous materials? J. Mater. Chem. 2007, 17, 24–30. [Google Scholar] [CrossRef]
- Jones, M.D.; Raja, R.; Thomas, J.M.; Johnson, B.F.G.; Lewis, D.W.; Rouzaud, J.; Harris, K.D.M. Enhancing the enantioselectivity of novel homogeneous organometallic hydrogenation catalysts. Angew. Chem. Int. Ed. 2003, 42, 4326–4331. [Google Scholar]
- Zhang, H.; Zhang, Y.; Li, c. Enantioselective epoxidation of unfunctionalized olefins catalyzed by the Mn(salen) catalysts immobilized in the nanopores of mesoporous materials. J. Catal. 2006, 238, 369–381. [Google Scholar] [CrossRef]
- Barbosa, E.F.G.; Sprio, M. Joint homogeneous and heterogeneous catalysis: A new synergic effect. J. Chem. Soc. Chem. Commun. 1977, 423–424. [Google Scholar] [CrossRef]
- Gao, H.; Angelici, R.J. Combination catalysts consisting of a homogeneous catalyst tethered to silica-supported palladium heterogeneous catalyst: Arene hydrogenation. J. Am. Chem. Soc. 1997, 119, 6937–6938. [Google Scholar] [CrossRef]
- Gao, H.; Angelici, R.J. Rhodium amine complexes tethered on silica-supported metal catalysts. Highly active catalysts for the hydrogenation of arenes. New J. Chem. 1999, 23, 633–640. [Google Scholar] [CrossRef]
- Bianchini, C.; Dal Santo, V.; Meli, A.; Oberhauser, W.; Psaro, R.; Vizza, F. Preparation, Characterization, and Performance of the Supported Hydrogen-Bonded Ruthenium Catalyst [(sulphos)Ru(NCMe)3](OSO2CF3)/SiO2. Comparisons with Analogous Homogeneous and Aqueous-Biphase Catalytic Systems in the Hydrogenation of Benzylideneacetone and Benzonitrile. Organometallics 2000, 19, 2433. [Google Scholar] [CrossRef]
- Bianchini, C.; Dal Santo, V.; Meli, A.; Moreno, M.; Psaro, R.; Sordelli, L.; Vizza, F. Hydrogenation of Arenes over Heterogeneous Catalysts that Combine a Metal Phase and a Grafted Metal Complex: Role of the Single-Site Catalyst. Angew. Chem. Int. Ed. 2003, 42, 2636–2639. [Google Scholar] [CrossRef]
- Barbaro, P.; Bianchini, C.; Dal Santo, V.; Meli, A.; Moneti, S.; Psaro, R.; Scaffidi, A.; Sordelli, L.; Vizza, F. Hydrogenation of Arenes over Silica-Supported Catalysts that Combine a Grafted Rhodium Complex and Palladium Nanoparticles: Evidence for Substrate Activation on Rhsingle-site-Pdmetal Moieties. J. Am. Chem. Soc. 2006, 128, 7065–7076. [Google Scholar]
- Barbaro, P.; Bianchini, C.; Dal Santo, V.; Meli, A.; Moneti, S.; Pirovano, C.; Psaro, R.; Sordelli, L.; Vizza, F. Benzene Hydrogenation by Silica-Supported Catalysts Made of Palladium Nanoparticles and Electrostatically Immobilized Rhodium Single Sites. Organometallics 2008, 27, 2809–2824. [Google Scholar] [CrossRef]
- Yang, H.; Gao, H.; Angelici, R.J. Hydrogenation of arenes under mild conditions using rhodium pyridylphosphine and bipyridyl complexes tethered to a silica-supported palladium heterogeneous catalyst. Organometallics 2000, 19, 622–629. [Google Scholar]
- Yang, H.; Gao, H.; Angelici, R.J. Hydrodefluorination of fluorobenzene and 1,2-difluorobenzene under mild conditions over rhodium pyridylphosphine and bipyridyl complexes tethered on a silica-supported palladium catalyst. Organometallics 1999, 18, 2285–2287. [Google Scholar] [CrossRef]
- Abu-Reziq, R.; Avnir, D.; Miloslavski, I.; Schumann, H.; Blum, J. Entrapment of metallic palladium and a rhodium(I) complex in a silica sol-gel matrix - Formation of a highly active recyclable arene hydrogenation catalyst. J. Mol. Catal. A Chem. 2002, 185, 179–185. [Google Scholar] [CrossRef]
- Abu-Reziq, R.; Avnir, D.; Blum, J. Catalytic hydrogenolysis of aromatic ketones by a sol-gel entrapped combined Pd-[Rh(cod)Cl]2 catalyst. J. Mol. Catal. A Chem. 2002, 187, 277–281. [Google Scholar] [CrossRef]
- Pugin, B.; Müller, M. Heterogeneous Catalystsand Fine Chemical, 3rd; Guisnet, M., Barbier, J., Barrault, J., Bouchoule, C., Duprez, D., Perot, G., Montassier, C., Eds.; Elsevier: Amsterdam, The Netherlands, 1993; Volume 78, p. 107. [Google Scholar]
- Pugin, B. Immobilized catalysts for enantioselective hydrogenation: The effect of site-isolation. J. Mol. Catal. A Chem. 1996, 107, 273–279. [Google Scholar] [CrossRef]
- Stanger, K.J.; Wiench, J.W.; Pruski, M.; Angelici, R.J. 31P NMR and IR characterization of enantioselective olefin and arene hydrogenation catalysts containing a rhodium-chiral phosphine complex tethered on silica. J. Mol. Catal. A Chem. 2003, 195, 63–82. [Google Scholar] [CrossRef]
- Abu-Reziq, R.; Wang, D.; Post, M.L.; Alper, H. Separable catalysts in one-pot syntheses for greener chemistry. Chem. Mater. 2008, 20, 2544–2550. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2010 by the authors;
Share and Cite
Dal Santo, V.; Liguori, F.; Pirovano, C.; Guidotti, M. Design and Use of Nanostructured Single-Site Heterogeneous Catalysts for the Selective Transformation of Fine Chemicals. Molecules 2010, 15, 3829-3856. https://doi.org/10.3390/molecules15063829
Dal Santo V, Liguori F, Pirovano C, Guidotti M. Design and Use of Nanostructured Single-Site Heterogeneous Catalysts for the Selective Transformation of Fine Chemicals. Molecules. 2010; 15(6):3829-3856. https://doi.org/10.3390/molecules15063829
Chicago/Turabian StyleDal Santo, Vladimiro, Francesca Liguori, Claudio Pirovano, and Matteo Guidotti. 2010. "Design and Use of Nanostructured Single-Site Heterogeneous Catalysts for the Selective Transformation of Fine Chemicals" Molecules 15, no. 6: 3829-3856. https://doi.org/10.3390/molecules15063829