Direct Enzymatic Route for the Preparation of Novel Enantiomerically Enriched Hydroxylated β-Amino Ester Stereoisomers


Abstract
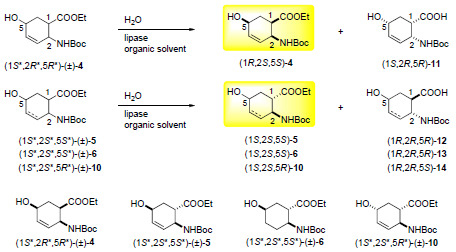
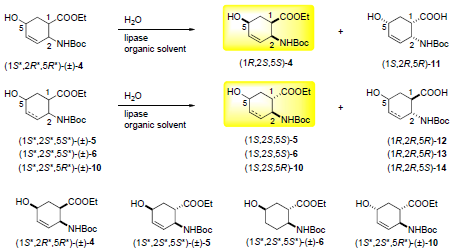
:
1. Introduction
2. Results and Discussion
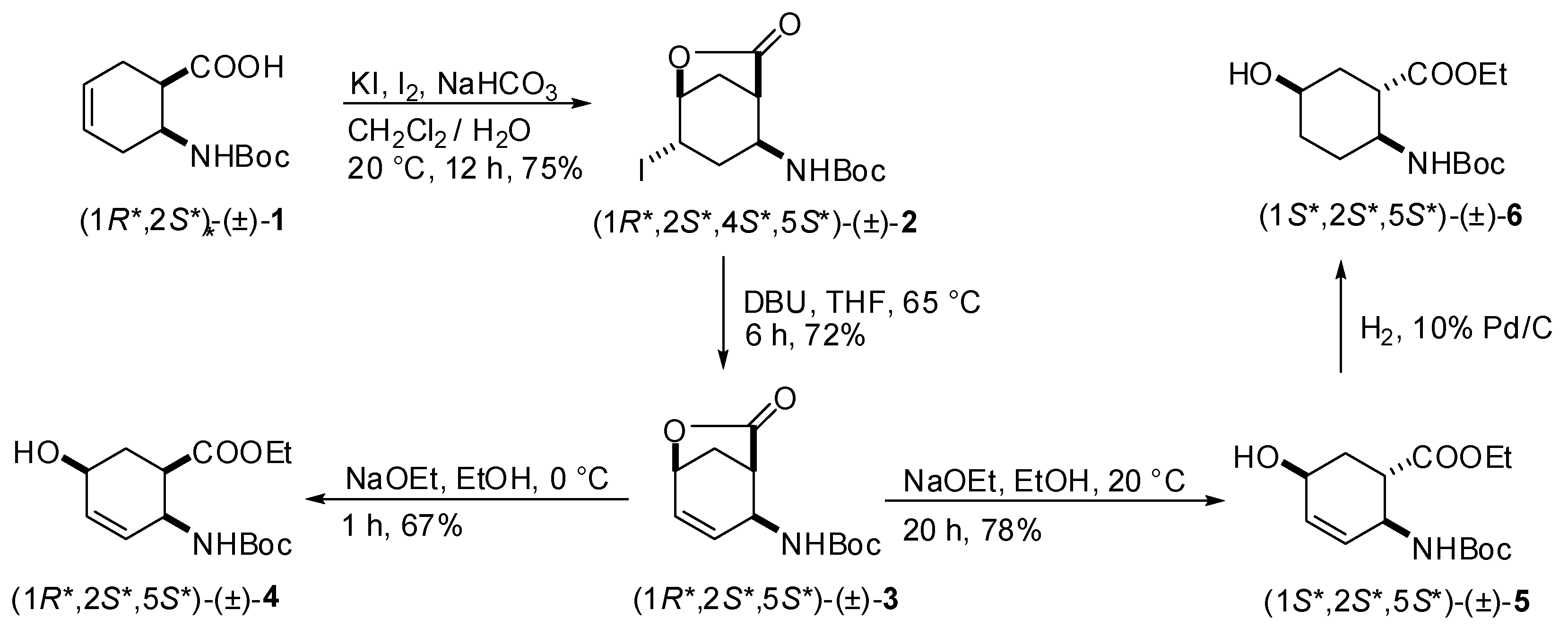
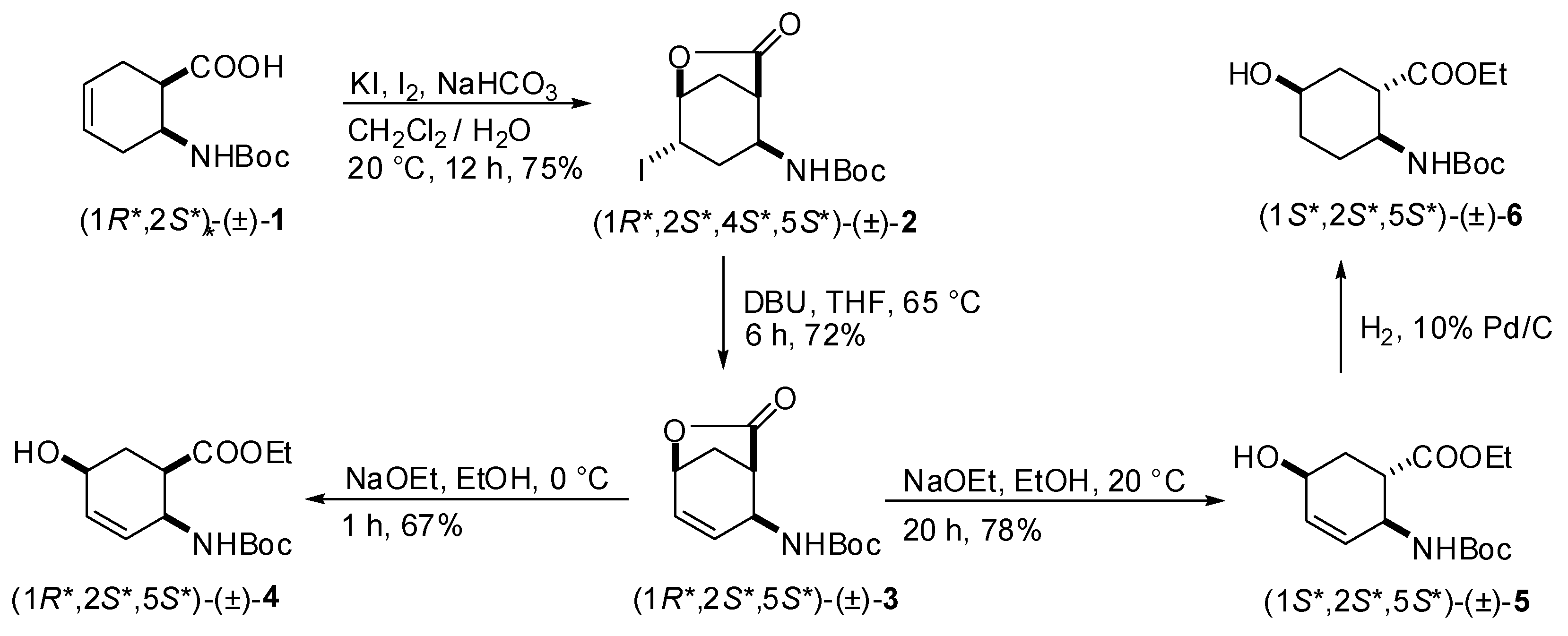
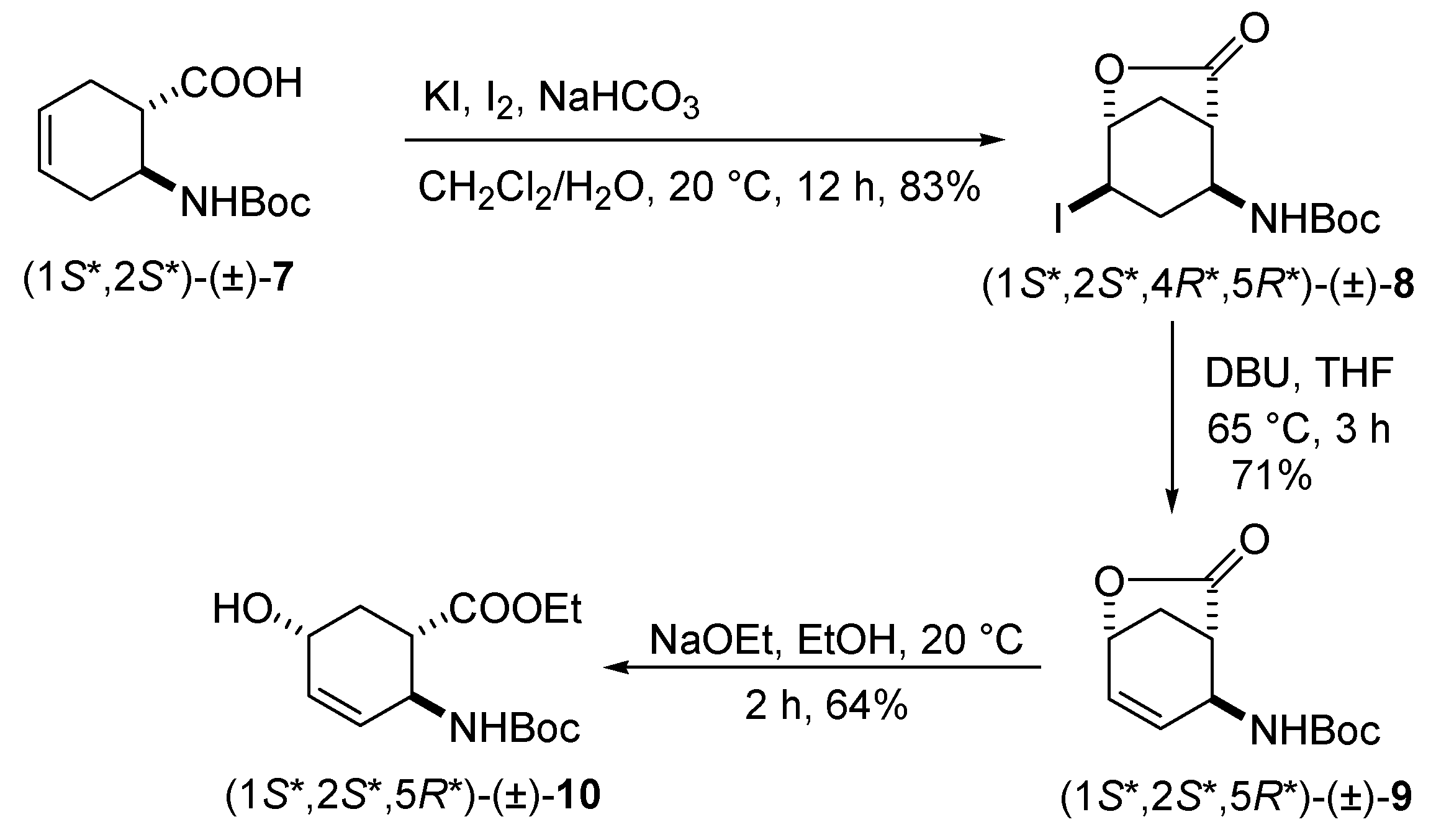
2.1. Synthesis of hydroxy-substituted racemic β-amino esters (1R*,2S*,5S*)-(±)-4, (1S*,2S*,5S*)-(±)-5, (1S*,2S*,5S*)-(±)-6 and (1S*,2S*,5R*)-(±)-10

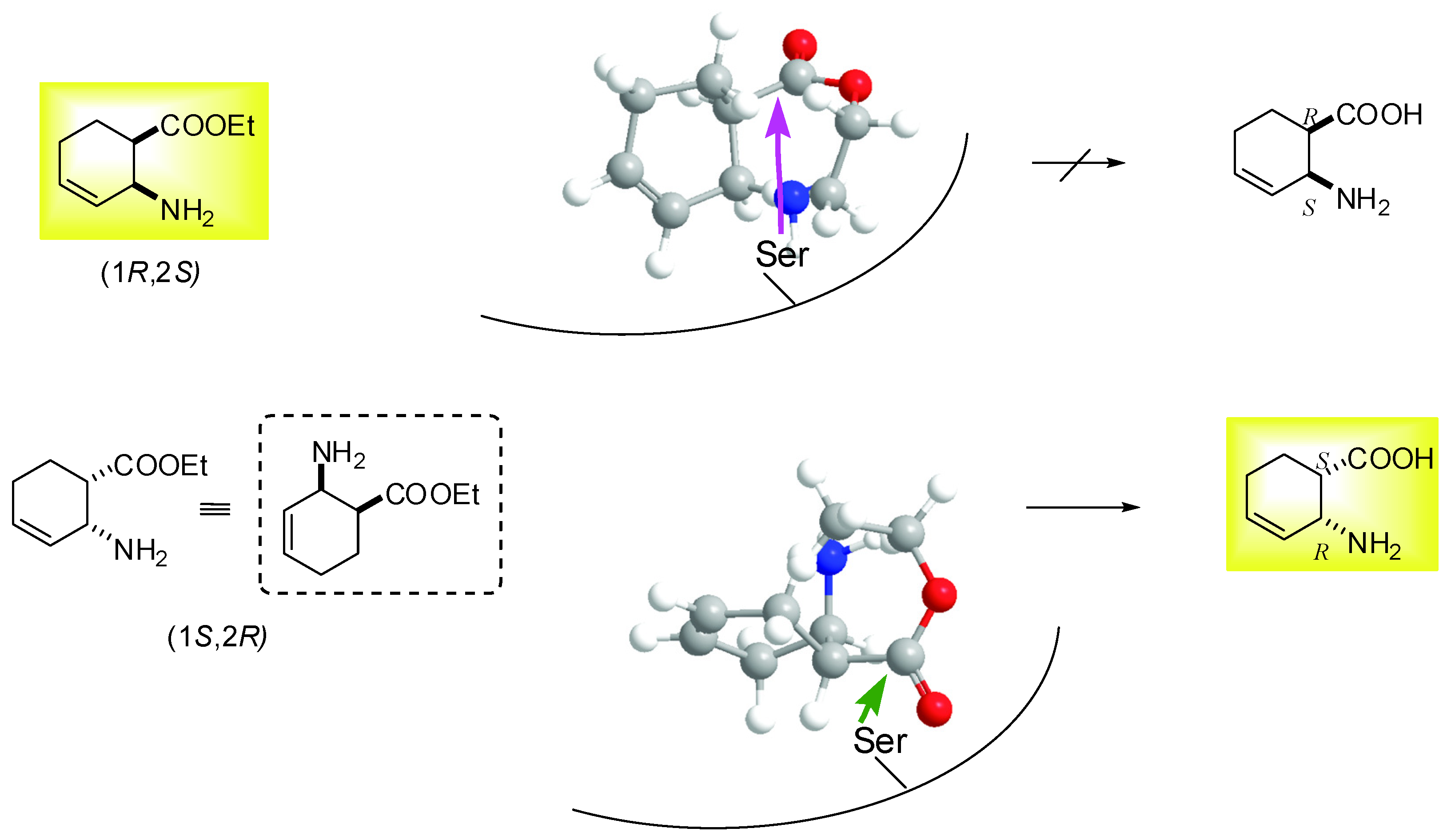
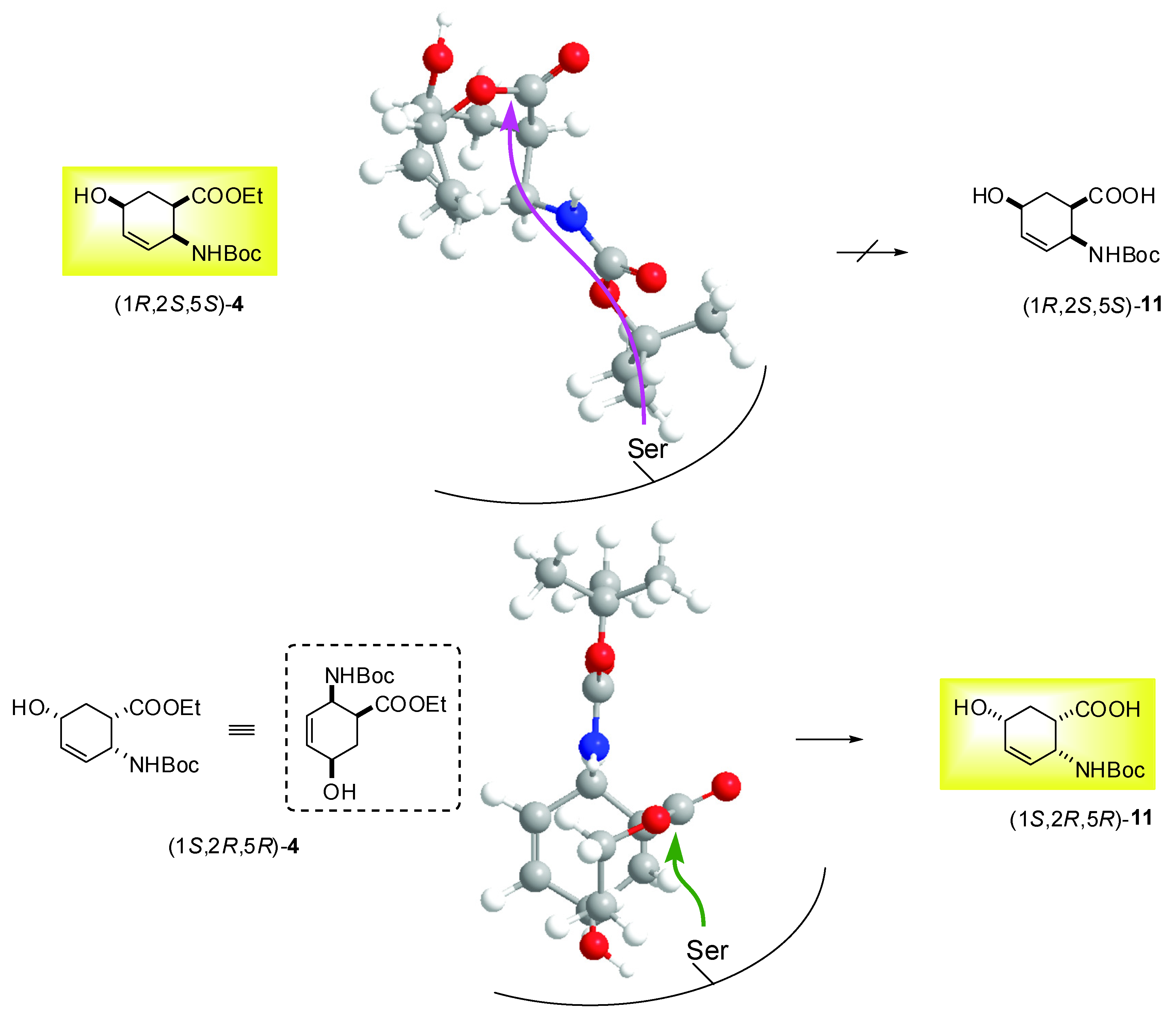
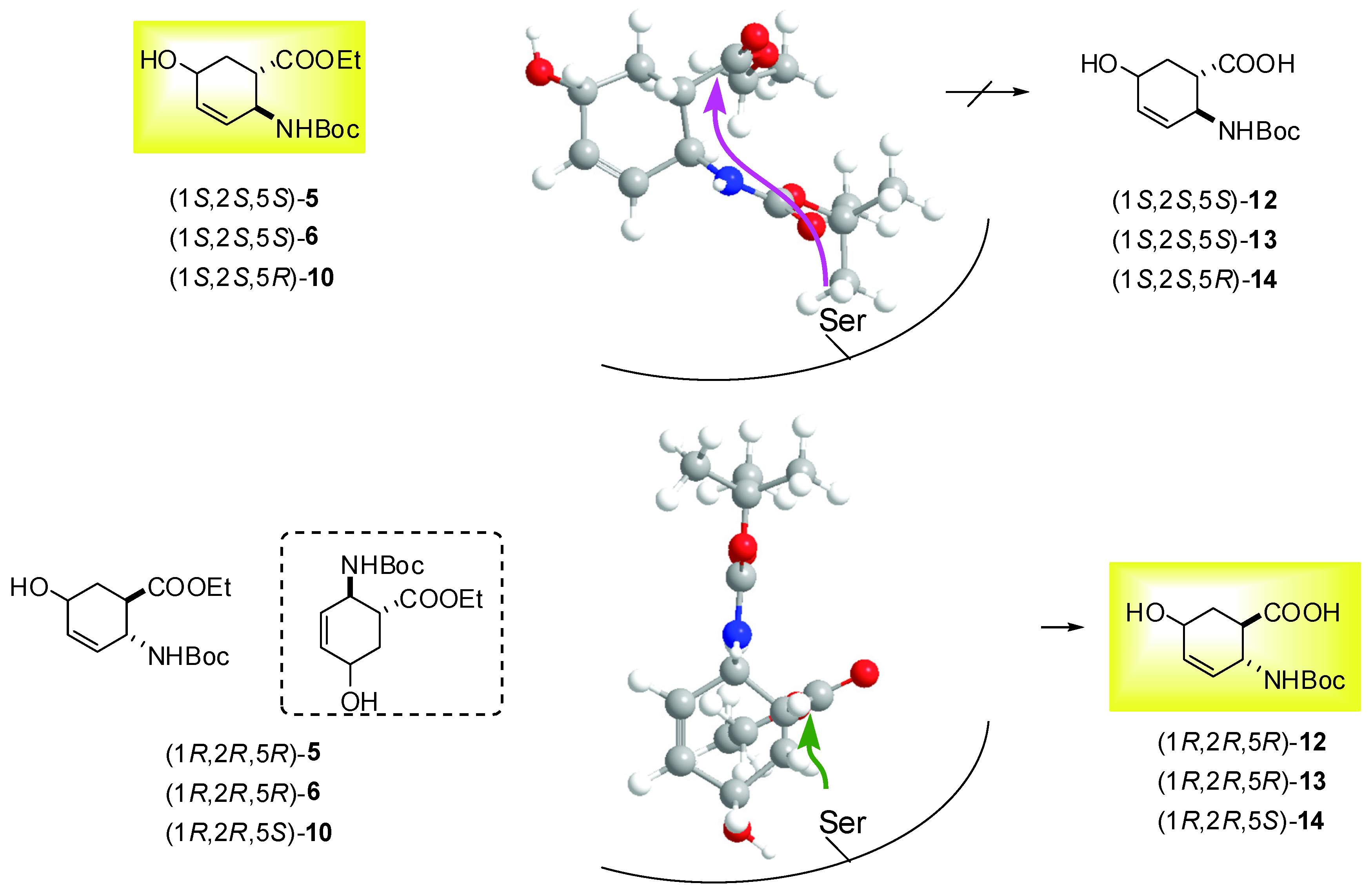
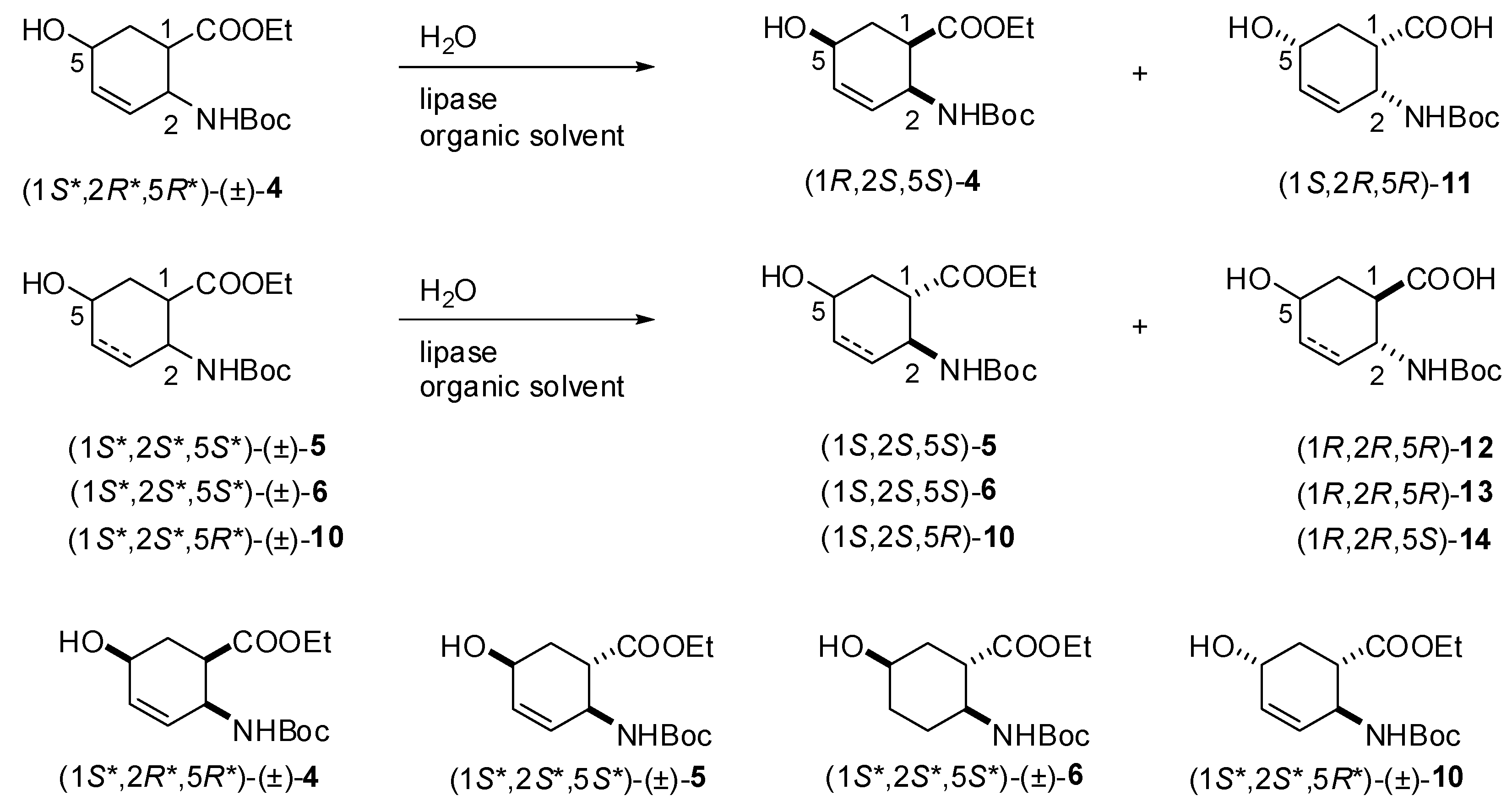
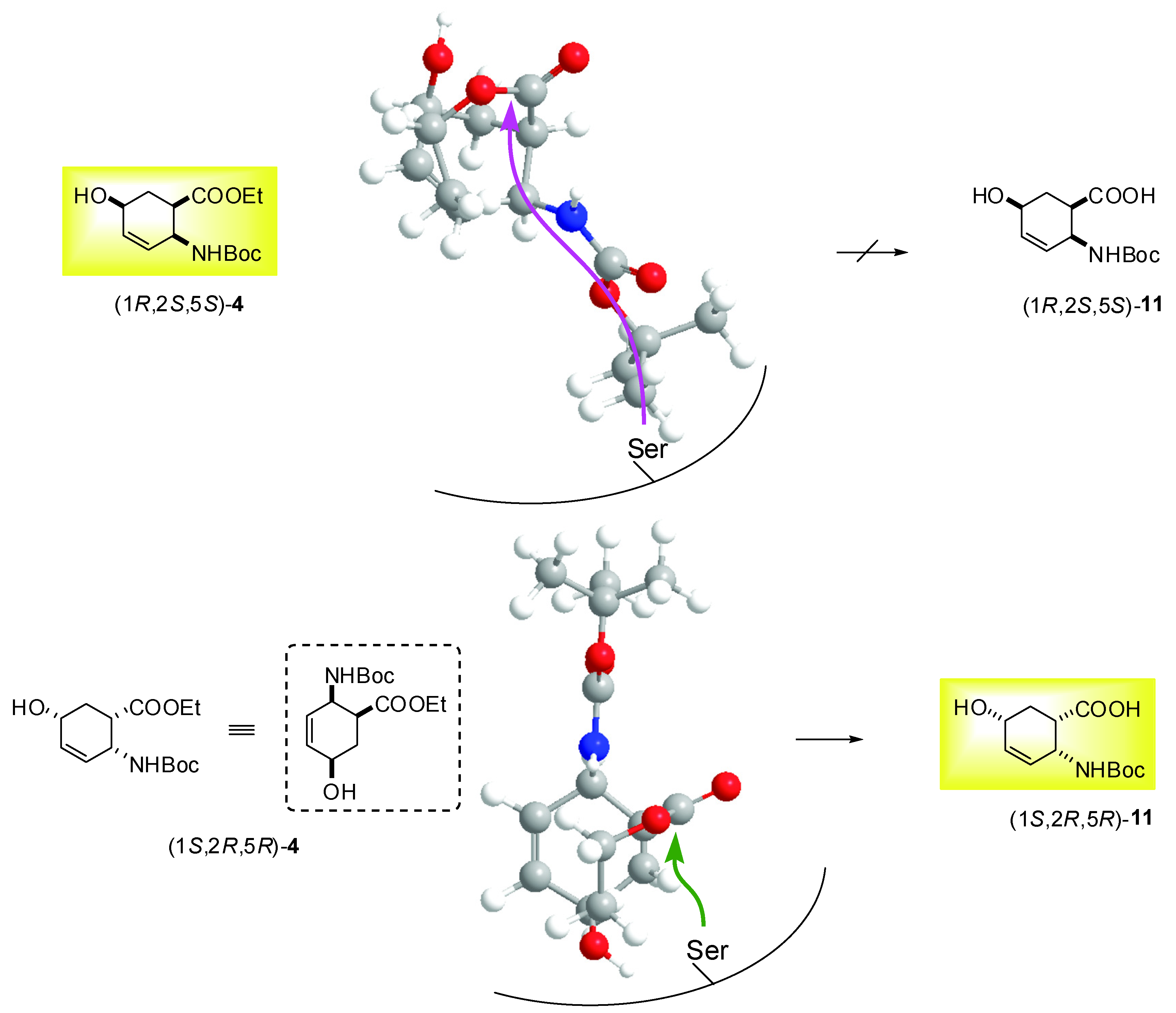
2.2. Enzymatic hydrolysis of hydroxy-substituted β-amino esters (1S*,2R*,5R*)-(±)-4, (1S*,2S*,5S*)-(±)-5, (1S*,2S*,5S*)-(±)-6 and (1S*,2S*,5R*)-(±)-10
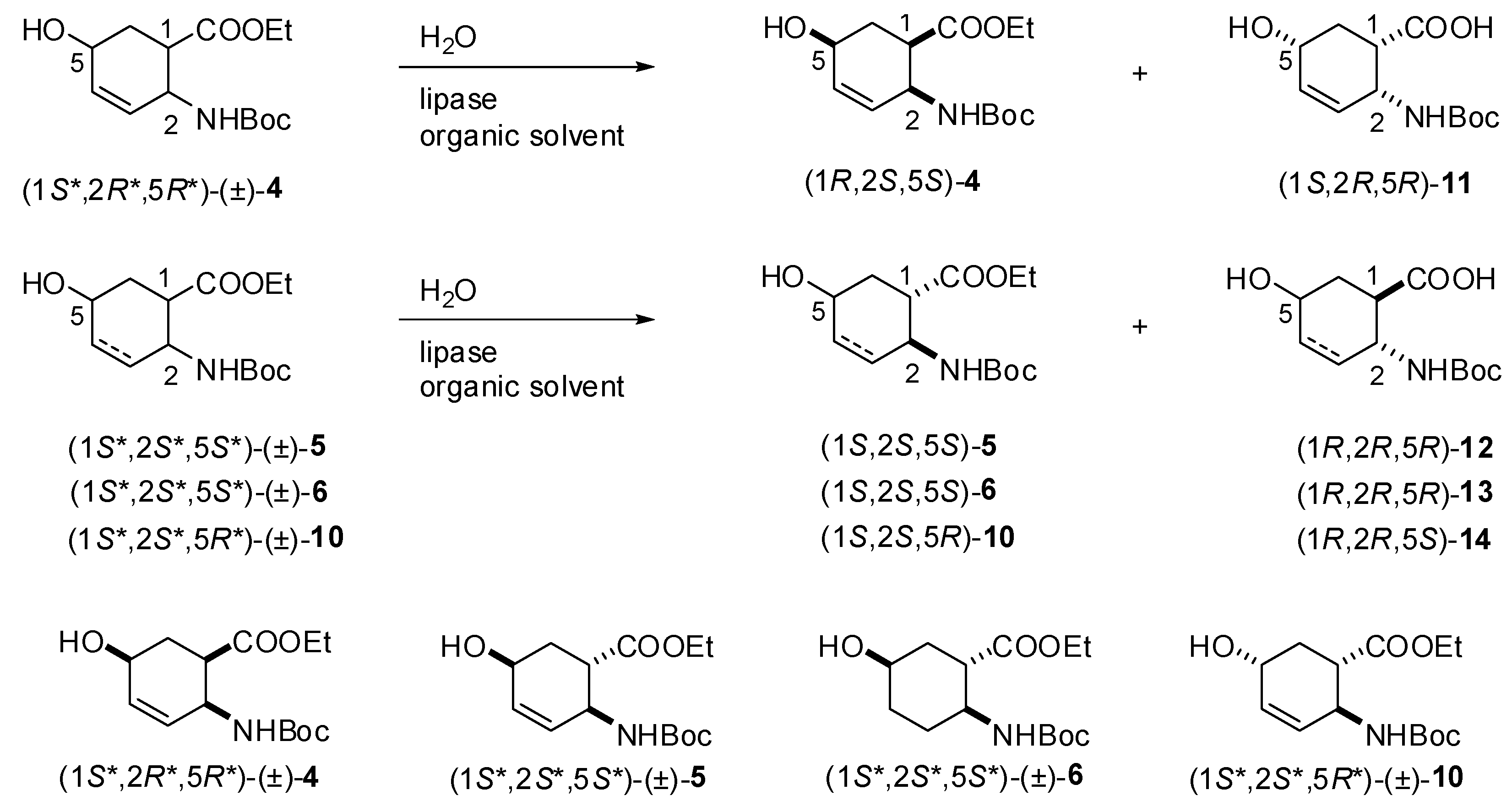

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Enzyme | Quantity of enzyme (mg mL-1) | Temperature (ºC.) | Solvent | Time (h) | Conv.b (%) | eeSc (%) | eePd (%) | Eb |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CAL-B | 30 | 60 | iPr2O | 48 | 37 | 48 | 82 | 16 |
| 2 | CAL-B | 30 | 60 | n-hexane | 48 | 24 | 18 | 58 | 4 |
| 3 | CAL-B | 30 | 60 | toluene | 48 | 35 | 48 | 91 | 34 |
| 4 | CAL-B | 30 | 60 | 1,4-dioxane | 48 | 25 | 16 | 48 | 3 |
| 5 | CAL-B | 30 | 60 | t-BuOMe | 48 | 45 | 75 | 91 | 48 |
| 6 | CAL-B | 30 | 50 | t-BuOMe | 48 | 35 | 47 | 86 | 21 |
| 7 | CAL-B | 30 | 40 | t-BuOMe | 48 | 36 | 46 | 81 | 15 |
| 8 | CAL-B | 30 | 70 | t-BuOMe | 40 | 35 | 44 | 82 | 16 |
| 9 | CAL-B | 50 | 60 | t-BuOMe | 40 | 40 | 53 | 81 | 16 |
| 10 | CAL-B | 75 | 60 | t-BuOMe | 40 | 42 | 60 | 83 | 20 |
| 11 | Lipase PSe | 30 | 45 | iPr2O | 48 | 22 | 10 | 35 | 2 |
| 12 | CAL-Ae | 30 | 45 | iPr2O | 48 | 44 | 7 | 9 | 1 |
| 13 | Lipase AYe | 30 | 45 | iPr2O | 48 | 11 | 2 | 16 | 1 |
| 14 | Lipase AKe | 30 | 45 | iPr2O | 48 | 25 | 1 | 3 | 1 |
| 15 | PPL | 30 | 45 | iPr2O | 48 | 20 | 5 | 20 | 2 |


| Racemate | Time (days) | Conv.b (%) | Unreacted enantiomer | |||
|---|---|---|---|---|---|---|
| Yield (%) | Isomer | eec (%) | [α]D25 (EtOH) | |||
| (1 S*,2R*,5R*)-(±)-4 | 10 | 43 | 32 | (1R,2S,5S)-4 | 68 | +34 (c 0.19) |
| (1 S*,2S*,5S*)-(±)-5 | 10 | 41 | 34 | (1S,2S,5S)-5 | 78 | +16 (c 0.165) |
| (1 S*,2S*,5S*)-(±)-6 | 10 | 36 | 29 | (1S,2S,5S)-6 | 54 | -4 (c 0.24) |
| (1 S*,2S*,5R*)-(±)-10 | 7 | 43 | 41 | (1S,2S,5R)-10 | 90 | +100 (c 0.155) |
3. Experimental
3.1. Materials and methods
3.2. Typical small-scale enzymatic experiment
3.3. Synthesis of iodolactones tert-butyl (1R*,2S*,4S*,5S*)-4-iodo-7-oxo-6-oxabicyclo[3.2.1]octan-2-ylcarbamate [(1R*,2S*,4S*,5S*)-(±)-2] and tert-butyl (1S*,2S*,4R*,5R*)-4-iodo-7-oxo-6-oxabicyclo-[3.2.1]octan-2-ylcarbamate [(1S*,2S*,4R*,5R*)-(±)-8] [32]
3.4. Dehydroiodination reactions to obtain tert-butyl (1R*,2S*,5S*)-7-oxo-6-oxabicyclo[3.2.1]oct-3-en-2-ylcarbamate [(1R*,2S*,5S*)-(±)-3] and tert-butyl (1S*,2S*,5R*)-7-oxo-6-oxabicyclo[3.2.1]oct-3-en-2-ylcarbamate [(1S*,2S*,5R*)-(±)-9]
3.5. Lactone ring opening reaction to obtain ethyl (1R*,2S*,5S*)-2-(tert-butoxycarbonylamino)-5-hydroxycyclohex-3-enecarboxylate [(1R*,2S*,5S*)-(±)-4], ethyl (1S*,2S*,5S*)-2-(tert-butoxycarbonyl-amino)-5-hydroxycyclohex-3-enecarboxylate [(1S*,2S*,5S*)-(±)-5] and ethyl (1S*,2S*,5R*)-2-(tert-butoxycarbonylamino)-5-hydroxycyclohex-3-enecarboxylate [(1S*,2S*,5R*)-(±)-10]
3.6. Preparation of ethyl (1S*,2S*,5S*)-2-(tert-butoxycarbonylamino)-5-hydroxycyclohexanecarboxylate [(1S*,2S*,5S*)-(±)-6]
3.7. Preparative-scale resolution of (1R*,2S*,5S*)-(±)-4
3.8. Preparative-scale resolution of (1S*,2S*,5S*)-(±)-5
3.9. Preparative-scale resolution of (1S*,2S*,5S*)-(±)-6
3.10. Preparative-scale resolution of (1S*,2S*,5R*)-(±)-10
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors in mg quantities.
References and Notes
- Fülöp, F. The chemistry of 2-aminocycloalkanecarboxylic acids. Chem. Rev. 2001, 101, 2181–2204. [Google Scholar]
- Kiss, L.; Forró, E.; Fülöp, F. Amino Acids,Peptides and Proteins in Organic Chemistry; Hughes, A.B., Ed.; Wiley-VCH: Weinheim, Germany, 2009; Volume 1, pp. 367–409. [Google Scholar]
- Zegarac, M.; Mestrovic, E.; Hulita, N.K.; Filic, D.; Dumic, M.; Grunenberg, A.; Keil, B.; Ceric, H. Solid state forms of (-)-(1R,2S)-2-amino-4-methylenecyclopentanecarboxylic acid. PCT Int. Appl. WO 2005100302 A1.
- Hameršak, Z.; Roje, M.; Avdagić, A. Quinine-mediated parallel kinetic resolution of racemic cyclic anhydride: Stereoselective synthesis, relative and absolute configuration of novel alicyclic β-amino acids. Tetrahedron Asymmetr. 2007, 18, 635–644. [Google Scholar] [CrossRef]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.I. β-Amino Acids. Versatile peptidomimetics. Curr. Med. Chem. 2002, 9, 811–822. [Google Scholar] [CrossRef]
- Martinek, T.A.; Mándity, I.M.; Fülöp, L.; Tóth, G.K.; Vass, E.; Hollósi, M.; Forró, E.; Fülöp, F. Effects of the alternating backbone configuration on the secondary structure and self-assembly of β-peptides. J. Am. Chem. Soc. 2006, 128, 13539–13544. [Google Scholar]
- Price, J.L.; Horne, W.S.; Gellman, S.H. Discrete heterogeneous quaternary structure formed by α/β-peptide foldamers and α-peptides. J. Am. Chem. Soc. 2007, 129, 6376–6377. [Google Scholar]
- Mándity, I.M.; Wéber, E.; Martinek, T.A.; Olajos, G.; Tóth, G.K.; Vass, E.; Fülöp, F. Design of peptidic foldamer helices: A stereochemical patterning approach. Angew. Chem. Int. Ed. 2009, 48, 2171–2175. [Google Scholar]
- Fernández, D.; Torres, E.; Avilés, F.X.; Ortuňo, R.M.; Vendrell, J. Cyclobutane-containing peptides: Evaluation as novel metallocarboxypeptidase inhibitors and modelling of their mode of action. Bioorg. Med. Chem. 2009, 17, 3824–3828. [Google Scholar] [CrossRef]
- Kapferer, P.; Vasella, A. Electrophilic bromination of N-acylated cyclohex-3-en-1-amines: Synthesis of 7-azanorbornanes. Helv. Chim. Acta 2004, 87, 2764–2789. [Google Scholar] [CrossRef]
- Carter, P.H. Process for preparation of diprotected 3-(4-fluorobenzyl)-1-(2,5-diaminocyclo-hexyl)piperidine via reductive amination. U.S. Pat. Appl. 2005/277,666, 2005. [Chem. Abstr. 2005, 144, 51452]. [Google Scholar]
- Fülöp, F.; Miklós, F.; Forró, E. Diexo-3-aminonorbornane-2-carboxylic acid as highly applicable chiral source for the enantioselective synthesis of heterocycles. Synlett 2008, 1687–1689. [Google Scholar]
- Kiss, L.; Nonn, M.; Forró, E.; Sillanpää, R.; Fülöp, F. Synthesis of novel isoxazoline-fused cispentacin stereoisomers. Tetrahedron Lett. 2009, 50, 2605–2608. [Google Scholar]
- Gedey, S.; Van der Eycken, J.; Fülöp, F. Synthesis of alicyclic β-lactams via the Ugi reaction on a solid support. Lett. Org. Chem. 2004, 1, 215–217. [Google Scholar]
- Kanizsai, I.; Gyónfalvi, S.; Szakonyi, Z.; Sillanpää, R.; Fülöp, F. Synthesis of bi- and tricyclic β-lactam libraries in aqueous medium. Green Chem. 2007, 9, 357–360. [Google Scholar]
- Kiss, L.; Forró, E.; Sillanpää, R.; Fülöp, F. Diastereo- and enantioselective synthesis of orthogonally protected 2,4-diaminocyclopentanecarboxylates: a flip from β-amino- to β,γ-diaminocarboxylates. J. Org. Chem. 2007, 72, 8786–8790. [Google Scholar]
- Juaristi, E.; Soloshonok, V.A. Enantioselective synthesis of β-amino acids, 2nd ed; Wiley-Interscience: New York, NY, USA, 2005. [Google Scholar]
- Mowery, B.P.; Lee, S.E.; Kissounko, D.A.; Epand, R.F.; Epand, R.M.; Weisblum, B.; Stahl, S.S.; Gellman, S.H. Mimicry of antimicrobial host-defense peptides by random copolymers. J. Am. Chem. Soc. 2007, 129, 15474–15476. [Google Scholar]
- Juaristi, E.; Lopez-Ruiz, H. Recent advances in the enantioselective synthesis of beta-amino acids. Curr. Med. Chem. 1999, 6, 983–1004. [Google Scholar]
- Palkó, M.; Kiss, L.; Fülöp, F. Syntheses of hydroxylated cyclic β-amino acids and derivatives. Curr. Med. Chem. 2005, 12, 3063–3083. [Google Scholar] [CrossRef]
- Kingston, D.G.I.; Newman, D.J. Taxoids: Cancer-fighting compounds from nature. Curr. Opin. Drug Disc. Dev. 2007, 10, 130–144. [Google Scholar]
- Bunnage, M.E.; Ganesh, T.; Masesane, I.B.; Orton, D.; Steel, P.G. Asymmetric Synthesis of the Putative Structure of (−)-Oryzoxymycin. Org. Lett. 2003, 5, 239–242. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ohbayashi, M.; Hirata, K.; Nishizawa, R.; Isobe, M. Synthesis of Blastidic acid and Cytosinine, two components of Blasticidin S. Synlett 2001, 1763–1766. [Google Scholar]
- Kiss, L.; Forró, E.; Fülöp, F. A new strategy for the regio- and stereoselective hydroxylation of trans-2-aminocyclohexenecarboxylic acid. Tetrahedron Lett. 2006, 47, 2855–2858. [Google Scholar] [CrossRef]
- Kiss, L.; Forró, E.; Sillanpää, R.; Fülöp, F. Novel functionalized cispentacin derivatives. Synthesis of 1,2,3-triazole-substituted 2-aminocyclopentanecarboxylate stereoisomers. Tetrahedron Asymmetr. 2008, 19, 2856–2860. [Google Scholar] [CrossRef]
- Benedek, G.; Palkó, M.; Wéber, E.; Martinek, T.A.; Forró, E.; Fülöp, F. Efficient synthesis of 3,4- and 4,5-dihydroxy-2-aminocyclohexanecarboxylic acid enantiomers. Tetrahedron Asymmetr. 2009, 20, 2220–2225. [Google Scholar] [CrossRef]
- Kiss, L.; Fülöp, F. Selective Syntheses of Functionalized Cyclic β-Amino Acids via Transformation of the Ring C-C Double Bonds. Synlett 2010, 1302–1314. [Google Scholar]
- Forró, E.; Fülöp, F. The first direct enzymatic hydrolysis of alicyclic β-amino esters: -a route to enantiopure cis and trans β-amino acids. Chem. Eur. J. 2007, 13, 6397–6401. [Google Scholar] [CrossRef]
- Tasnádi, G.; Forró, E.; Fülöp, F. An efficient new enzymatic method for the preparation of β-aryl-β-amino acid enantiomers. Tetrahedron Asymmetr. 2008, 19, 2072–2077. [Google Scholar]
- Tasnádi, G.; Forró, E.; Fülöp, F. Burkholderia cepacia lipase is an excellent enzyme for the enantioselective hydrolysis of β-heteroaryl-β-amino esters. Tetrahedron Asymmetr. 2009, 15, 1771–1777. [Google Scholar]
- Tasnádi, G.; Forró, E.; Fülöp, F. An improved enzymatic method for the preparation of valuable β-arylalkyl-substituted β-amino acid enantiomers. Org. Biomol. Chem. 2010, 8, 886–895. [Google Scholar]
- Fülöp, F.; Palkó, M.; Forró, E.; Dervarics, M.; Martinek, T.A.; Sillanpää, R. Facile regio- and diastereoselective syntheses of hydroxylated 2-aminocyclohexanecarboxylic acids. Eur. J.Org. Chem. 2005, 3214–3220. [Google Scholar]
- Forró, E. New gas chromatographic method for the enantioseparation of β-amino acids by a quick double-derivatization technique. J. Chromatogr. A 2009, 1216, 1025–1029. [Google Scholar]
© 2010 by the authors;
Share and Cite
Forró, E.; Schönstein, L.; Kiss, L.; Vega-Peñaloza, A.; Juaristi, E.; Fülöp, F. Direct Enzymatic Route for the Preparation of Novel Enantiomerically Enriched Hydroxylated β-Amino Ester Stereoisomers. Molecules 2010, 15, 3998-4010. https://doi.org/10.3390/molecules15063998
Forró E, Schönstein L, Kiss L, Vega-Peñaloza A, Juaristi E, Fülöp F. Direct Enzymatic Route for the Preparation of Novel Enantiomerically Enriched Hydroxylated β-Amino Ester Stereoisomers. Molecules. 2010; 15(6):3998-4010. https://doi.org/10.3390/molecules15063998
Chicago/Turabian StyleForró, Enikő, László Schönstein, Loránd Kiss, Alberto Vega-Peñaloza, Eusebio Juaristi, and Ferenc Fülöp. 2010. "Direct Enzymatic Route for the Preparation of Novel Enantiomerically Enriched Hydroxylated β-Amino Ester Stereoisomers" Molecules 15, no. 6: 3998-4010. https://doi.org/10.3390/molecules15063998