Receptor-Based Virtual Screening of EGFR Kinase Inhibitors from the NCI Diversity Database

Abstract
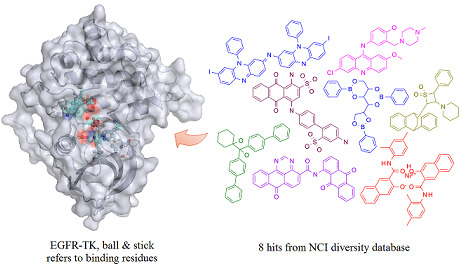
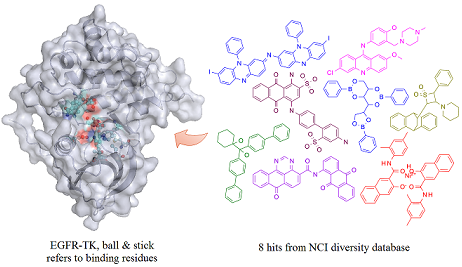
:
1. Introduction

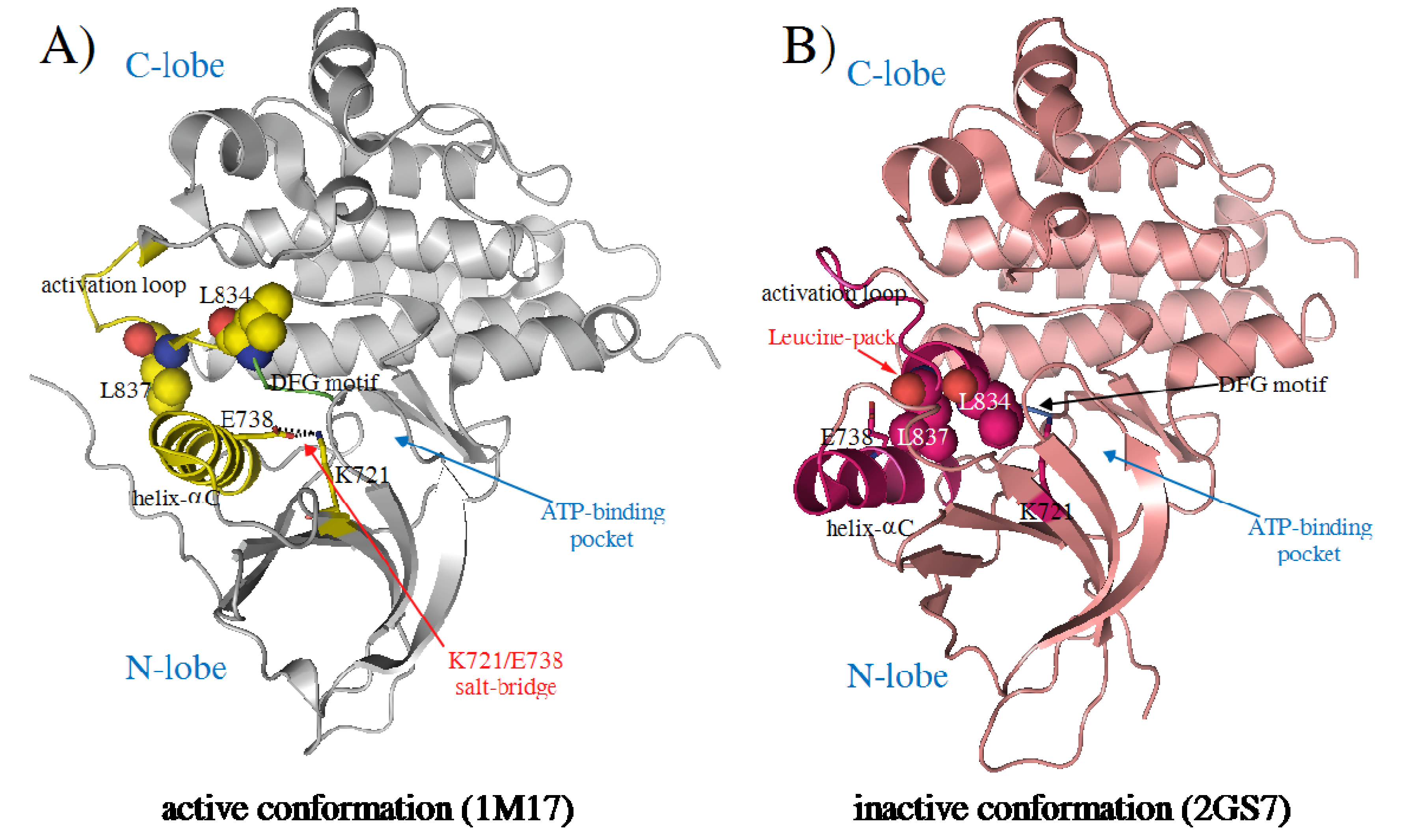
2. Results and Discussion
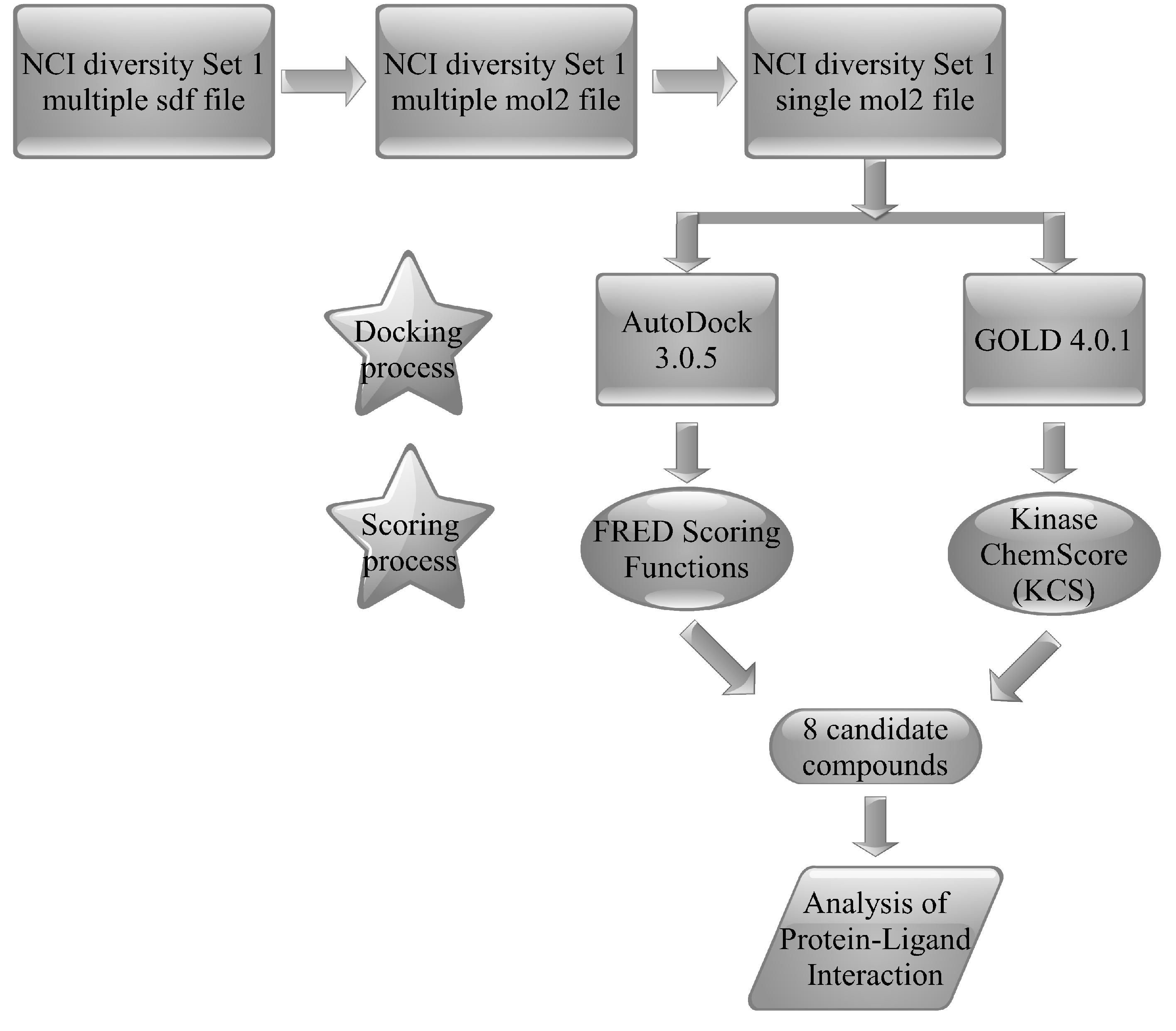

{kind=link}
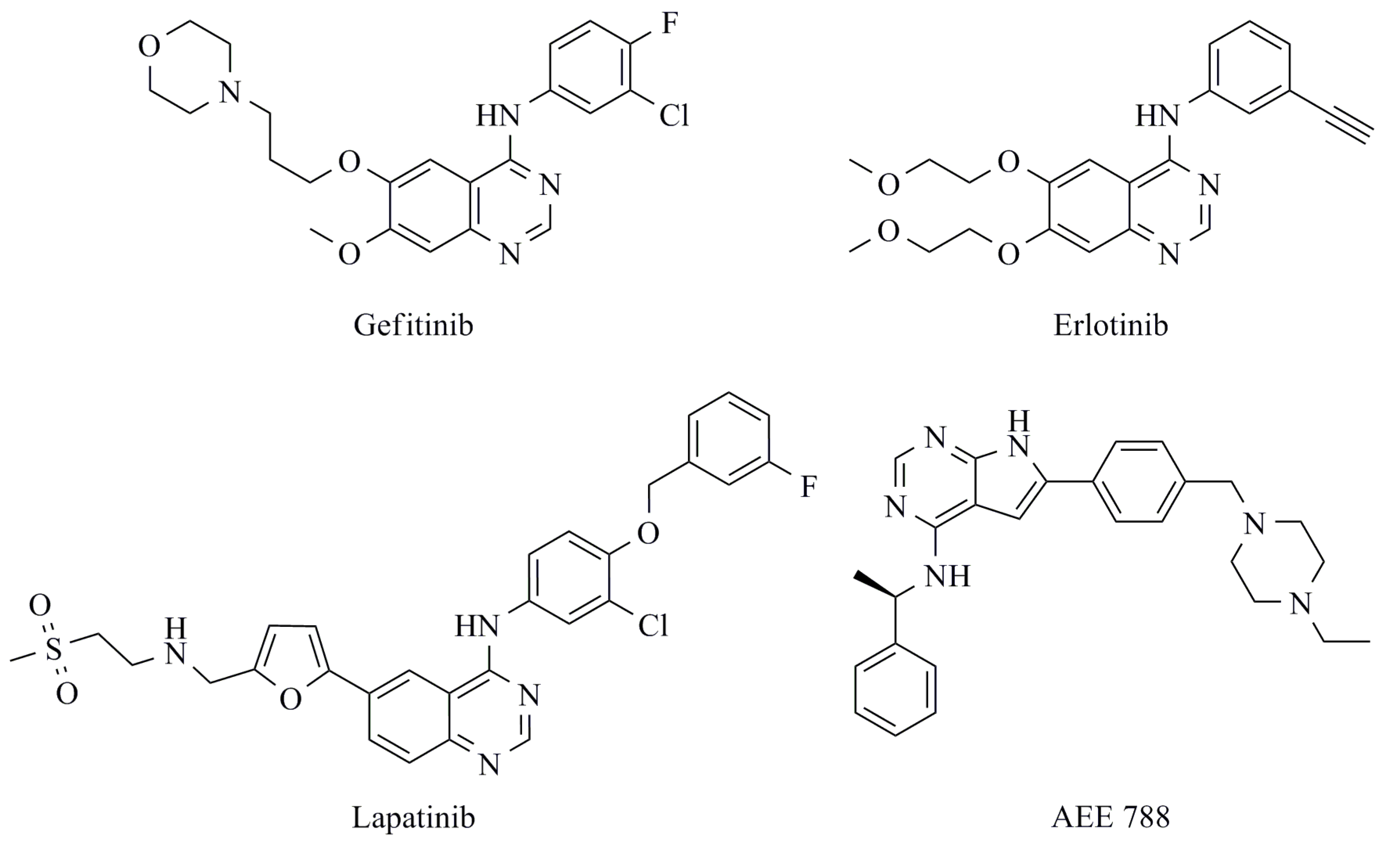{kind=link}
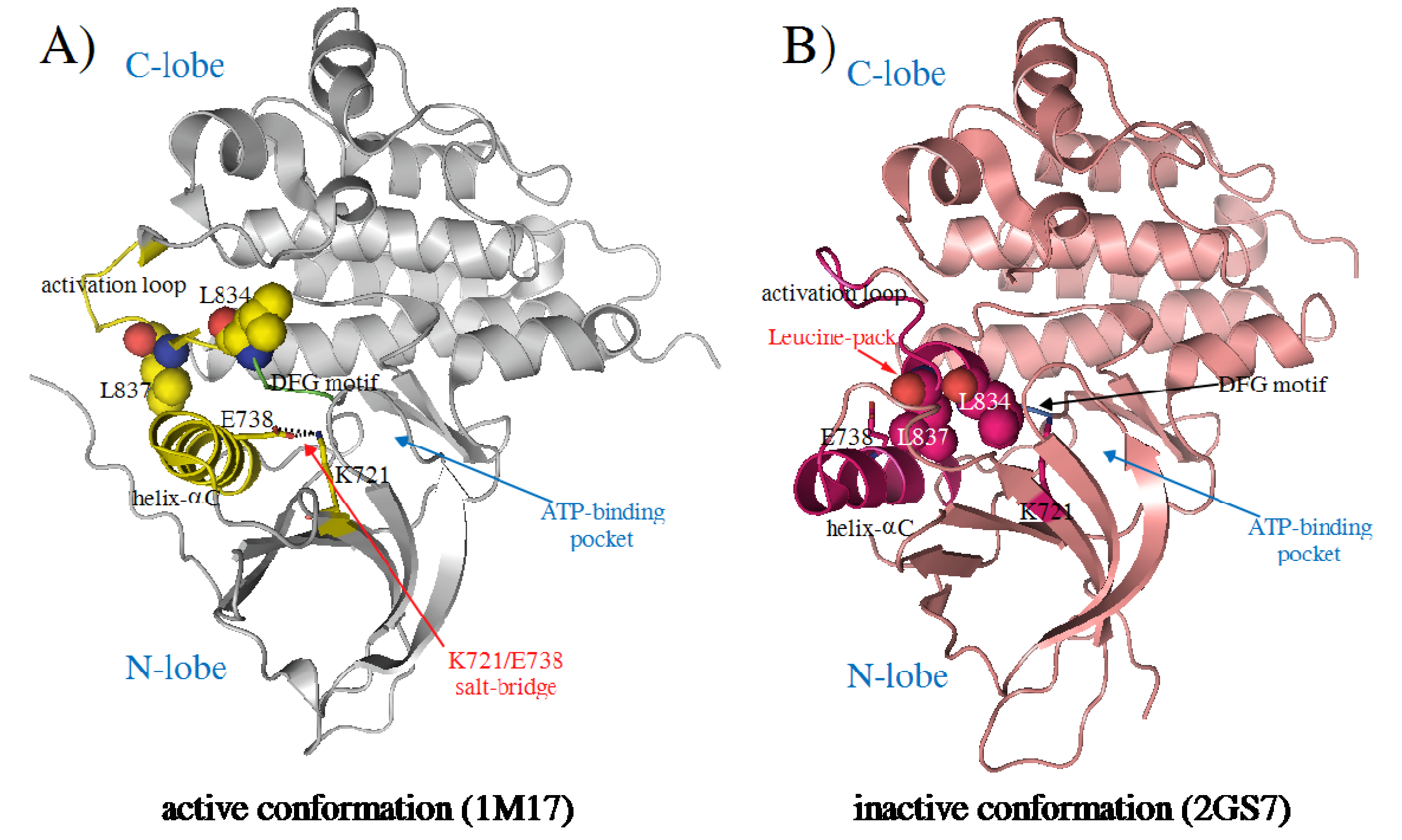{kind=link}
{kind=link}
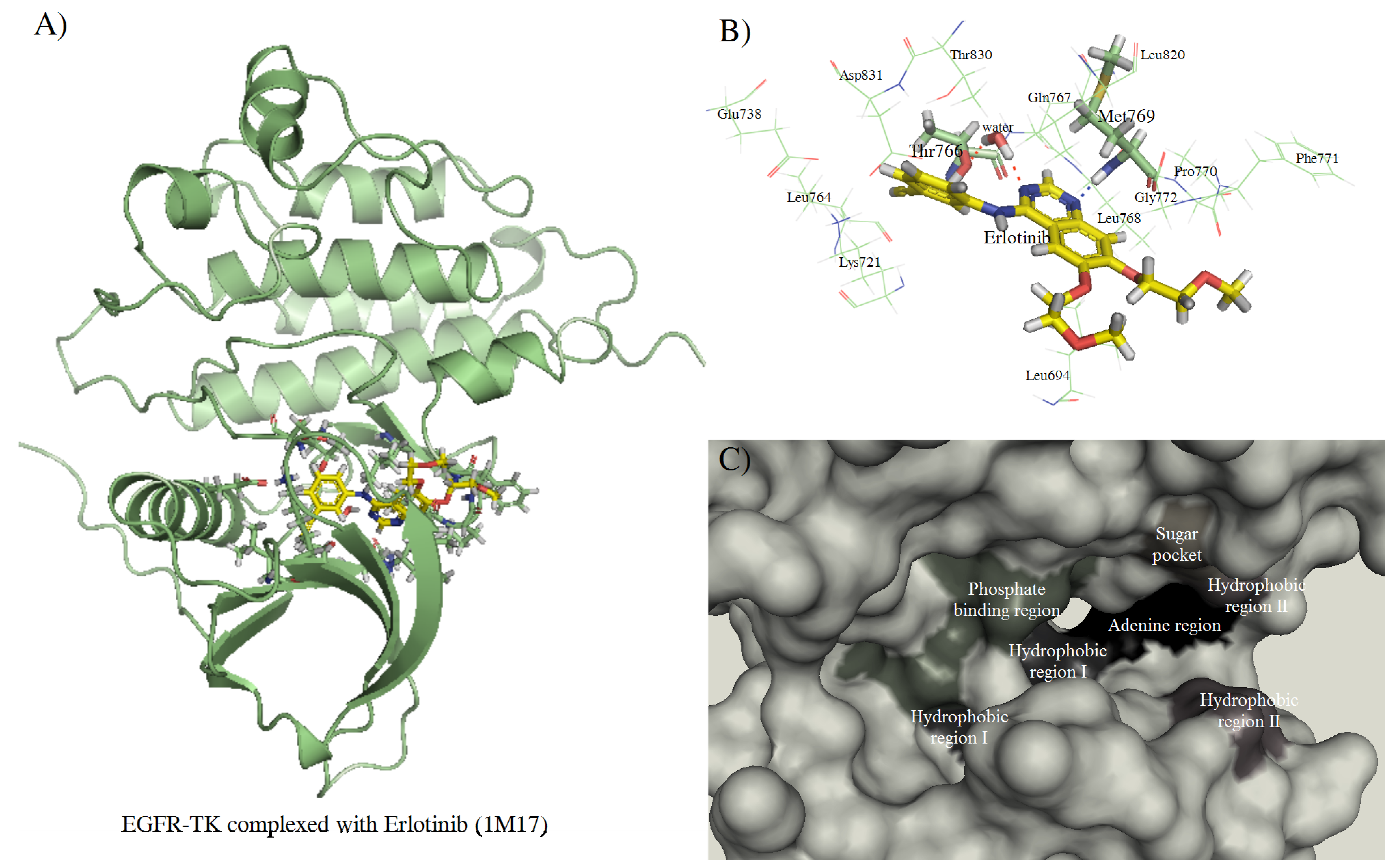{kind=link}
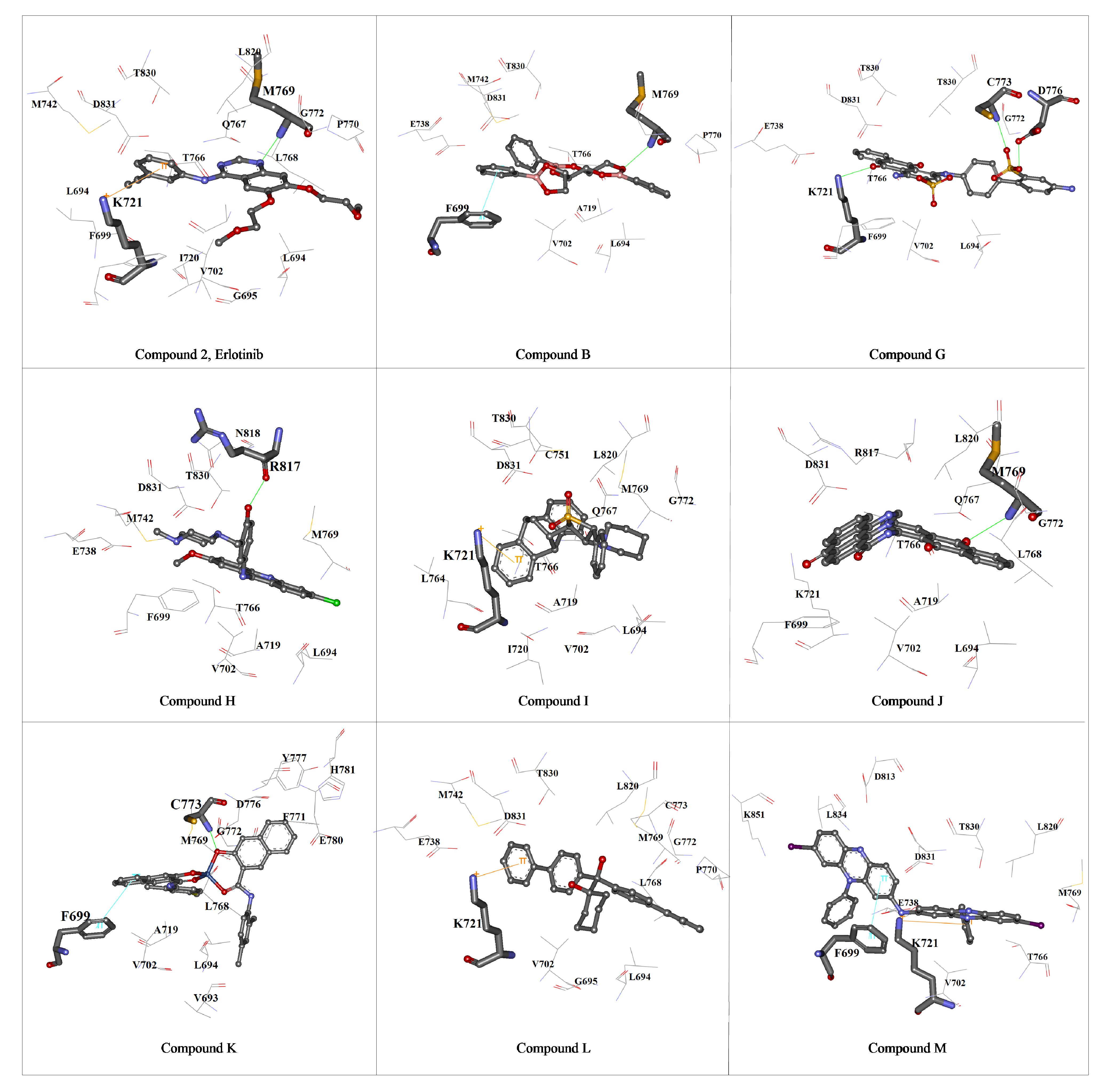{kind=link}
| Compounds | NSC no. | KCS score fitness value | GOLD Chemscore | ΔG(kJ/mol) | Average GI50† (μg/mL) | ||
|---|---|---|---|---|---|---|---|
| Hbond§ | Lipo‡ | DE Clash٭ | |||||
| M | 402959 | 41.20 | - | 334.07 | 2.18 | -44.57 | 5.50 × 10-5 |
| L | 351123 | 35.05 | - | 308.58 | 0.72 | -36.55 | 9.02 × 10-5 |
| H | 130813 | 34.04 | 0.97 | 257.89 | 0.03 | -34.46 | 1.94 × 10-6 |
| J | 299137 | 32.39 | 0.97 | 212.94 | 0.82 | -34.89 | 7.03 × 10-5 |
| I | 135371 | 32.28 | - | 357.52 | 11.93 | -44.49 | 3.95 × 10-5 |
| B | 48283 | 32.08 | - | 264.37 | 0.30 | -33.85 | 9.76 × 10-5 |
| K | 306698 | 30.24 | 0.78 | 236.62 | 4.21 | -35.78 | 3.25 × 10-6 |
| G | 125910 | 29.20 | 2.86 | 188.34 | 2.34 | -32.77 | 9.86 × 10-5 |
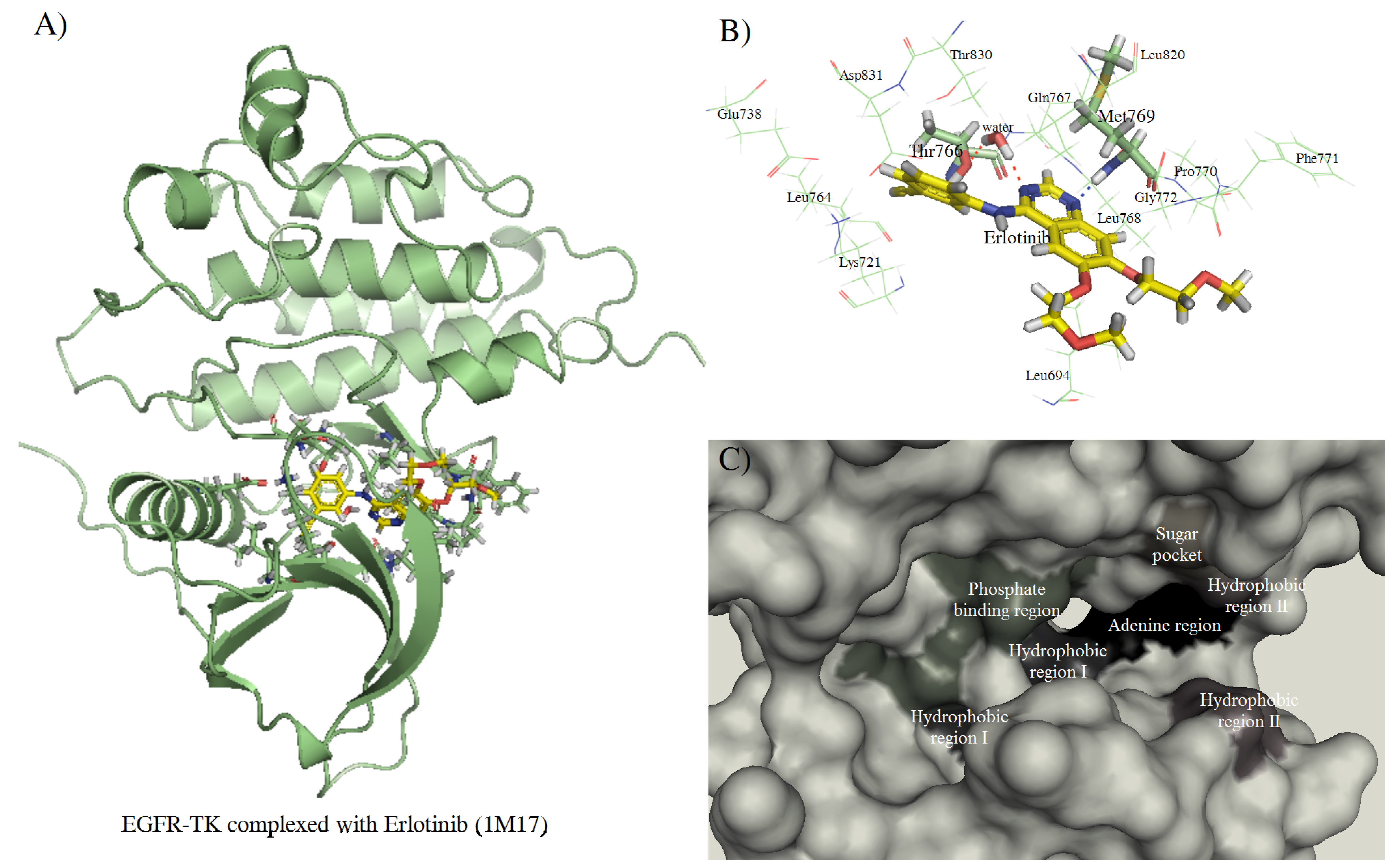
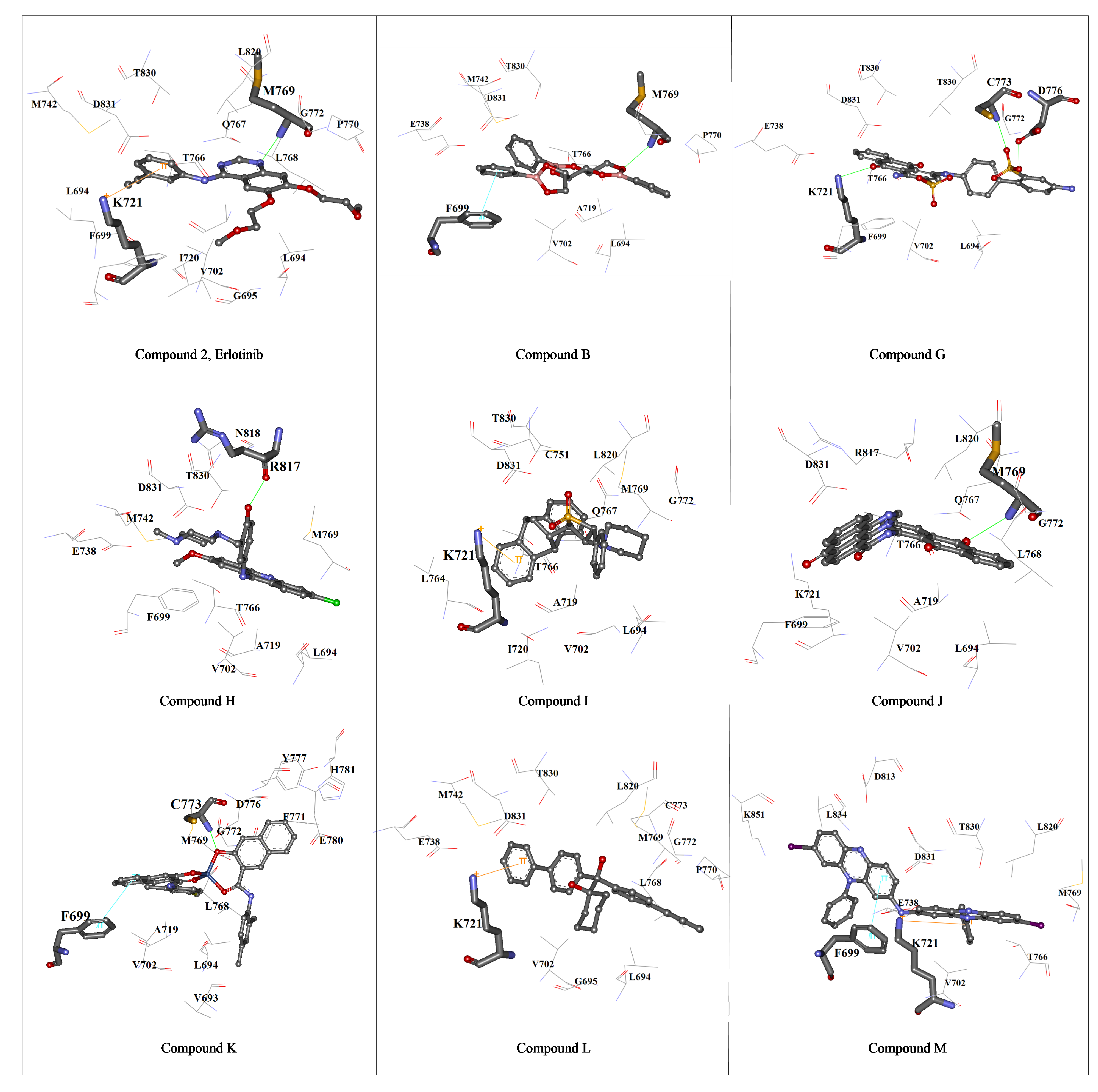
3. Experimental
3.1. Preparation of the protein structure
3.2. Preparation of the ligand structure
3.3. Molecular docking and post-docking analysis
3.3.1. AutoDock 3.0.5
3.3.2. GOLD 4.0.1
3.3.3. Structural analysis and visualization
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgements
- Samples Availability: Samples are not available from the authors.
References
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217. [Google Scholar] [CrossRef]
- Choowongkomon, K.; Carlin, C.R.; Sonnichsen, F.D. A structural model for the membrane-bound form of the juxtamembrane domain of the epidermal growth factor receptor. J. Biol. Chem. 2005, 280, 24043–24052. [Google Scholar]
- Walton, G.M.; Chen, W.S.; Rosenfeld, M.G.; Gill, G.N. Analysis of deletions of the carboxyl terminus of the epidermal growth factor receptor reveals self-phosphorylation at tyrosine 992 and enhanced in vivo tyrosine phosphorylation of cell substrates. J. Biol. Chem. 1990, 265, 1750–1754. [Google Scholar]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef]
- Woodburn, J.R. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol. Ther. 1999, 82, 241–250. [Google Scholar] [CrossRef]
- Rowinsky, E.K. The erbB family: Targets for therapeutic development against cancer and therapeutic strategies using monoclonal antibodies and tyrosine kinase inhibitors. Annu. Rev. Med. 2004, 55, 433–457. [Google Scholar] [CrossRef]
- Baselga, J.; Cortes, J. Epidermal growth factor receptor pathway inhibitors. Cancer Chemother. Biol. Response Modif. 2005, 22, 205–223. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef]
- Kawamoto, T.; Sato, J.D.; Le, A.; Polikoff, J.; Sato, G.H.; Mendelsohn, J. Growth stimulation of A431 cells by epidermal growth factor: identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc. Natl. Acad. Sci. USA 1983, 80, 1337–1341. [Google Scholar] [CrossRef]
- Mendelsohn, J. Antibody-mediated EGF receptor blockade as an anticancer therapy: From the laboratory to the clinic. Cancer Immunol. Immunother. 2003, 52, 342–346. [Google Scholar]
- Rubenstein, M.; Slobodskoy, L.; Mirochnik, Y.; Guinan, P. Backbone modification alters the efficacy of antisense oligonucleotides directed against mRNA encoding either TGF-alpha or EGFR in the treatment of prostate cancer cell lines. Methods Find. Exp. Clin. Pharmacol. 2002, 24, 649–652. [Google Scholar] [CrossRef]
- Dean, G.S.; Pusztai, L.; Xu, F.J.; O'Briant, K.; DeSombre, K.; Conaway, M.; Boyer, C.M.; Mendelsohn, J.; Bast, R.C., Jr. Cell surface density of p185(c-erbB-2) determines susceptibility to anti-p185(c-erbB-2)-ricin A chain (RTA) immunotoxin therapy alone and in combination with anti-p170(EGFR)-RTA in ovarian cancer cells. Clin. Cancer Res. 1998, 4, 2545–2550. [Google Scholar]
- Bruell, D.; Bruns, C.J.; Yezhelyev, M.; Huhn, M.; Muller, J.; Ischenko, I.; Fischer, R.; Finnern, R.; Jauch, K.W.; Barth, S. Recombinant anti-EGFR immunotoxin 425(scFv)-ETA' demonstrates anti-tumor activity against disseminated human pancreatic cancer in nude mice. Int. J. Mol. Med. 2005, 15, 305–313. [Google Scholar]
- Ghosh, S.; Liu, X.P.; Zheng, Y.; Uckun, F.M. Rational design of potent and selective EGFR tyrosine kinase inhibitors as anticancer agents. Curr. Cancer Drug Targets 2001, 1, 129–140. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; Alligood, K.J.; Rusnak, D.W.; Gilmer, T.M.; Shewchuk, L. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cells 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Blair, J.A.; Rauh, D.; Kung, C.; Yun, C.H.; Fan, Q.W.; Rode, H.; Zhang, C.; Eck, M.J.; Weiss, W.A.; Shokat, K.M. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat. Chem. Biol. 2007, 3, 229–238. [Google Scholar] [CrossRef]
- Zhang, X.; Pickin, K.A.; Bose, R.; Jura, N.; Cole, P.A.; Kuriyan, J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 2007, 450, 741–744. [Google Scholar]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Lengauer, T.; Lemmen, C.; Rarey, M.; Zimmermann, M. Novel technologies for virtual screening. Drug Discov. Today 2004, 9, 27–34. [Google Scholar] [CrossRef]
- Peng, H.; Huang, N.; Qi, J.; Xie, P.; Xu, C.; Wang, J.; Yang, C. Identification of novel inhibitors of BCR-ABL tyrosine kinase via virtual screening. Bioorg. Med. Chem. Lett. 2003, 13, 3693–3699. [Google Scholar]
- Foloppe, N.; Fisher, L.M.; Howes, R.; Kierstan, P.; Potter, A.; Robertson, A.G.; Surgenor, A.E. Structure-based design of novel Chk1 inhibitors: Insights into hydrogen bonding and protein-ligand affinity. J. Med. Chem. 2005, 48, 4332–4345. [Google Scholar] [CrossRef]
- Burkhard, P.; Hommel, U.; Sanner, M.; Walkinshaw, M.D. The discovery of steroids and other novel FKBP inhibitors using a molecular docking program. J. Mol. Biol. 1999, 287, 853–858. [Google Scholar] [CrossRef]
- Sarmiento, M.; Wu, L.; Keng, Y.F.; Song, L.; Luo, Z.; Huang, Z.; Wu, G.Z.; Yuan, A.K.; Zhang, Z.Y. Structure-based discovery of small molecule inhibitors targeted to protein tyrosine phosphatase 1B. J. Med. Chem. 2000, 43, 146–155. [Google Scholar] [CrossRef]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Ortiz, M.A.; Abagyan, R.A.; Piedrafita, F.J. In silico identification of novel EGFR inhibitors with antiproliferative activity against cancer cells. Bioorg. Med. Chem. Lett. 2006, 16, 1969–1974. [Google Scholar] [CrossRef]
- Assefa, H.; Kamath, S.; Buolamwini, J.K. 3D-QSAR and docking studies on 4-anilinoquinazoline and 4-anilinoquinoline epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. J. Comput. Aided Mol. Des. 2003, 17, 475–493. [Google Scholar] [CrossRef]
- Aparna, V.; Rambabu, G.; Panigrahi, S.K.; Sarma, J.A.; Desiraju, G.R. Virtual screening of 4-anilinoquinazoline analogues as EGFR kinase inhibitors: importance of hydrogen bonds in the evaluation of poses and scoring functions. J. Chem. Inf. Model. 2005, 45, 725–738. [Google Scholar] [CrossRef]
- Gundla, R.; Kazemi, R.; Sanam, R.; Muttineni, R.; Sarma, J.A.; Dayam, R.; Neamati, N. Discovery of novel small-molecule inhibitors of human epidermal growth factor receptor-2: Combined ligand and target-based approach. J. Med. Chem. 2008, 51, 3367–3377. [Google Scholar] [CrossRef]
- La Motta, C.; Sartini, S.; Tuccinardi, T.; Nerini, E.; Da Settimo, F.; Martinelli, A. Computational studies of epidermal growth factor receptor: Docking reliability, three-dimensional quantitative structure-activity relationship analysis, and virtual screening studies. J. Med. Chem. 2009, 52, 964–975. [Google Scholar]
- Janmaat, M.L.; Giaccone, G. Small-molecule epidermal growth factor receptor tyrosine kinase inhibitors. Oncologist 2003, 8, 576–586. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Plants as a source of anti-cancer agents. J. Ethnopharmacol. 2005, 100, 72–79. [Google Scholar] [CrossRef]
- Riely, G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, S146–S149. [Google Scholar] [CrossRef]
- Balius, T.E.; Rizzo, R.C. Quantitative prediction of fold resistance for inhibitors of EGFR. Biochemistry 2009, 48, 8435–8448. [Google Scholar] [CrossRef]
- Johnson, L.N. Protein kinase inhibitors: Contributions from structure to clinical compounds. Q. Rev. Biophys. 2009, 42, 1–40. [Google Scholar] [CrossRef]
- Liu, B.; Bernard, B.; Wu, J.H. Impact of EGFR point mutations on the sensitivity to gefitinib: Insights from comparative structural analyses and molecular dynamics simulations. Proteins 2006, 65, 331–346. [Google Scholar] [CrossRef]
- Tripos Sybyl7.3.; Tripos Associates: St. Louis, MO, USA.
- PMOL2Q. Available online: http://bioinfo.tau.ac.il/man/pmol2q/README.txt accessed on 19 December 2007.
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- NCI/DTP. Available online: http://dtp.nci.nih.gov/branches/dscb/repo_open.html/. accessed on 23 May 2010.
- openbabelGUI. Available online: http://openbabel.sourceforge.net/wiki/Main_Page/. accessed on 20 February 2008.
- splitmol2. Available online: http://shoichetlab.compbio.ucsf.edu/pipermail/zinc-fans/2005-February/000006.html/. accessed on 1 December 2007.
- FRED program; OpenEye Scientific Software, Inc.: Santa Fe, NM, USA. Available online: www.eyesopen.com/ accessed on 1 December 2007.
- Clark, A.M.; Labute, P. 2D depiction of protein-ligand complexes. J. Chem. Inf. Model. 2007, 47, 1933–1944. [Google Scholar] [CrossRef]
- VIDA 3.0.0 program; OpenEye Scientific Software, Inc.: Santa Fe, NM, USA. Available online: www.eyesopen.com/. assessed on 4 June 2010.
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: : Palo Alto, CA, USA, 2002. Available online: http://www.pymol.org/. assessed on 4 June 2010.
© 2010 by the authors;
Share and Cite
Choowongkomon, K.; Sawatdichaikul, O.; Songtawee, N.; Limtrakul, J. Receptor-Based Virtual Screening of EGFR Kinase Inhibitors from the NCI Diversity Database. Molecules 2010, 15, 4041-4054. https://doi.org/10.3390/molecules15064041
Choowongkomon K, Sawatdichaikul O, Songtawee N, Limtrakul J. Receptor-Based Virtual Screening of EGFR Kinase Inhibitors from the NCI Diversity Database. Molecules. 2010; 15(6):4041-4054. https://doi.org/10.3390/molecules15064041
Chicago/Turabian StyleChoowongkomon, Kiattawee, Orathai Sawatdichaikul, Napat Songtawee, and Jumras Limtrakul. 2010. "Receptor-Based Virtual Screening of EGFR Kinase Inhibitors from the NCI Diversity Database" Molecules 15, no. 6: 4041-4054. https://doi.org/10.3390/molecules15064041