Regioselective O-Derivatization of Quercetin via Ester Intermediates. An Improved Synthesis of Rhamnetin and Development of a New Mitochondriotropic Derivative

Abstract
:
1. Introduction
2. Results and Discussion
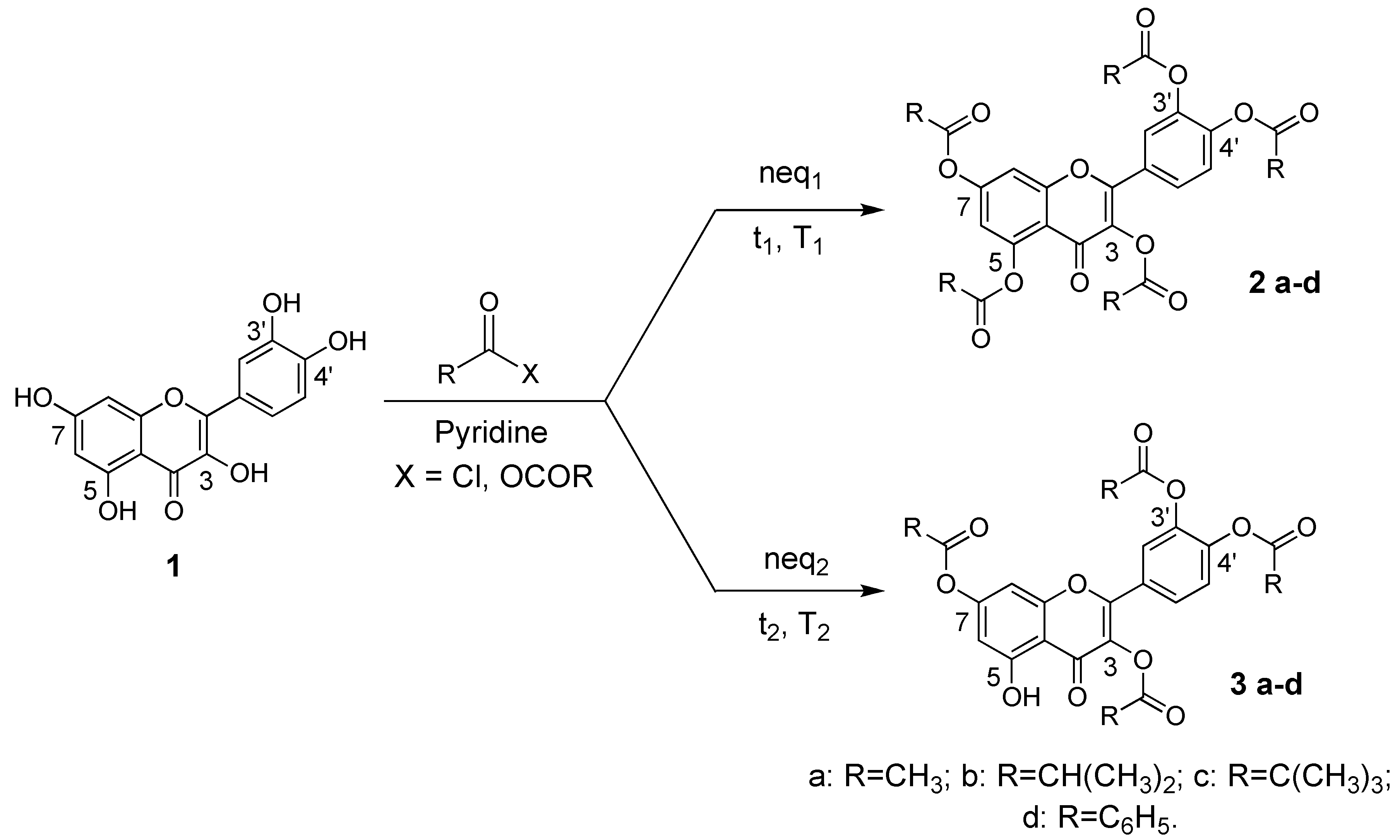
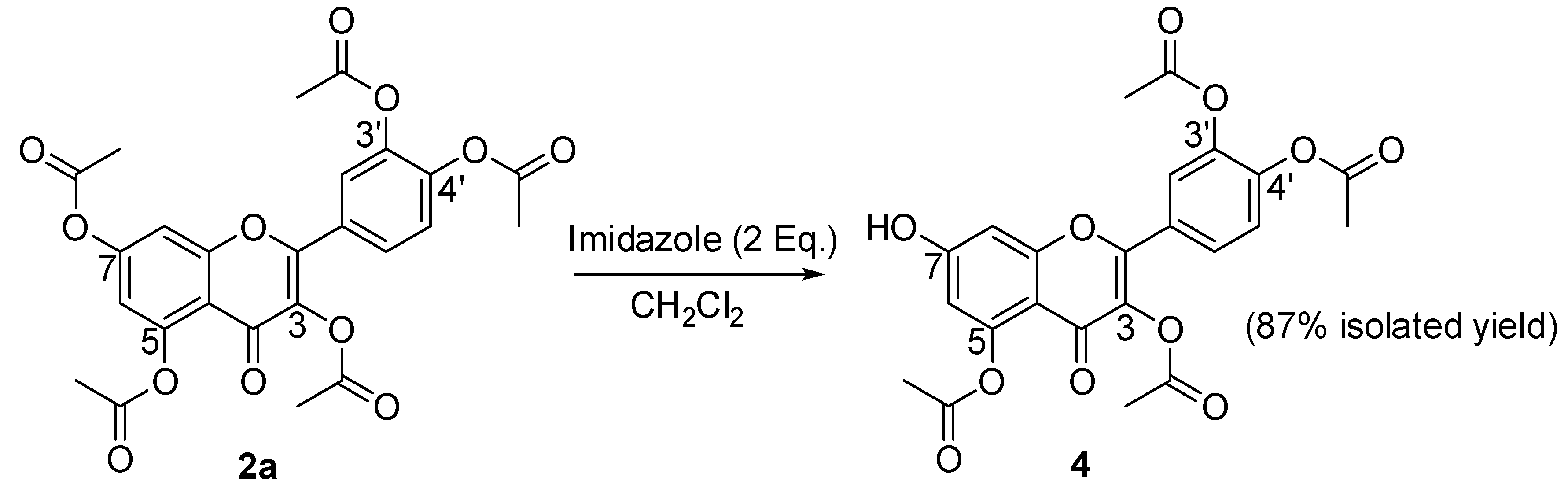
2.1. Synthesis of 3,3’,4’,5,7-pentaacyl quercetins, of 3,3’,4’,7-tetraacyl quercetins and of 3,3’,4’,5-tetraacetyl quercetin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | δ(H-6) | δ(H-8) | δ(H-5’) | δ(H-6’) | δ(H-2’) |
|---|---|---|---|---|---|
| 2a | 6.88 | 7.33 | 7.35 | 7.72 | 7.69 |
| 3a | 6.60 (-0.28) | 6.85 (-0.48) | 7.36 (+0.01) | 7.75 (+0.03) | 7.72 (+0.03) |
| 4 | 6.46 (-0.42) | 6.71 (-0.62) | 7.24 (-0.11) | 7.64 (-0.08) | 7.58 (-0.11) |

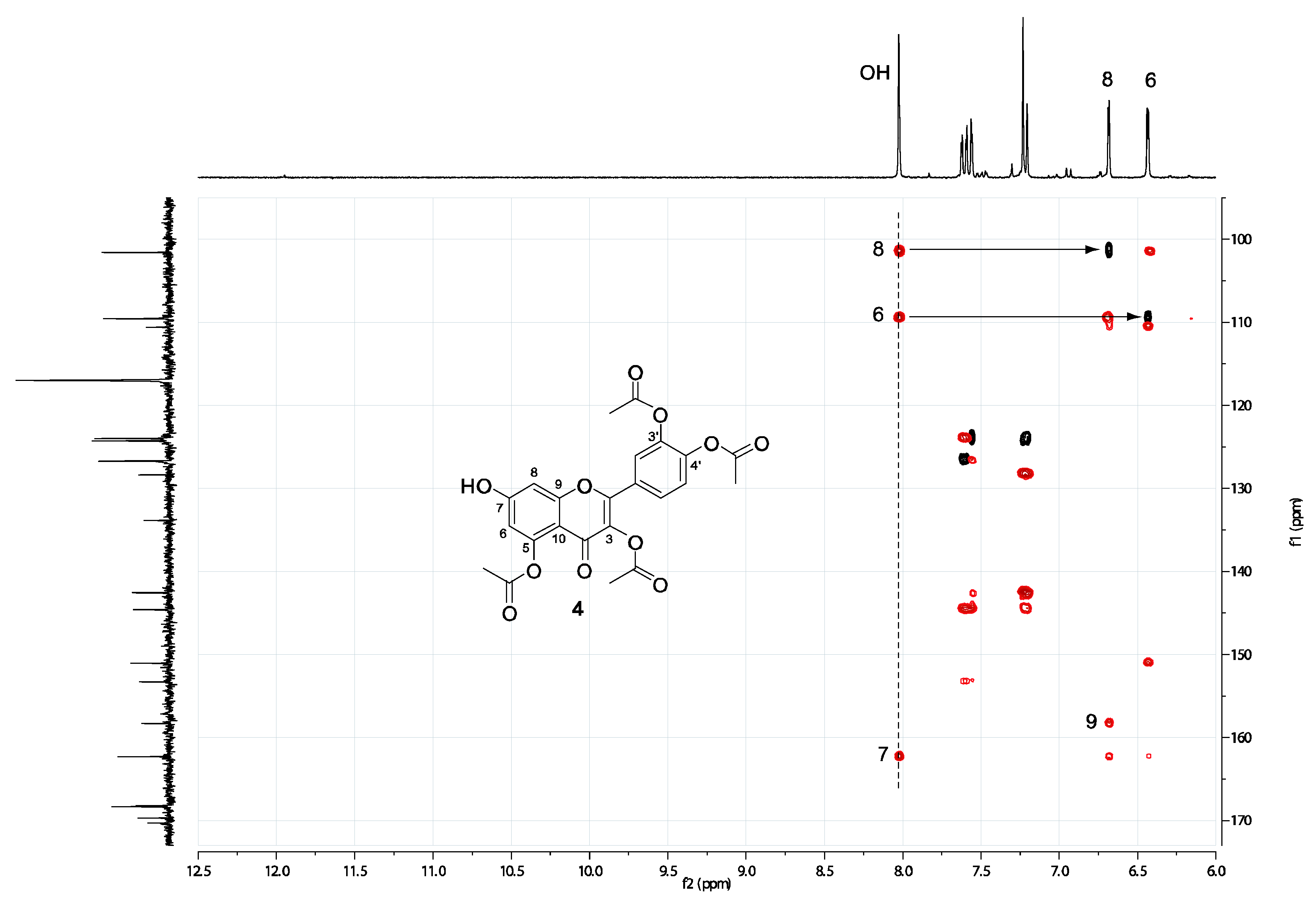
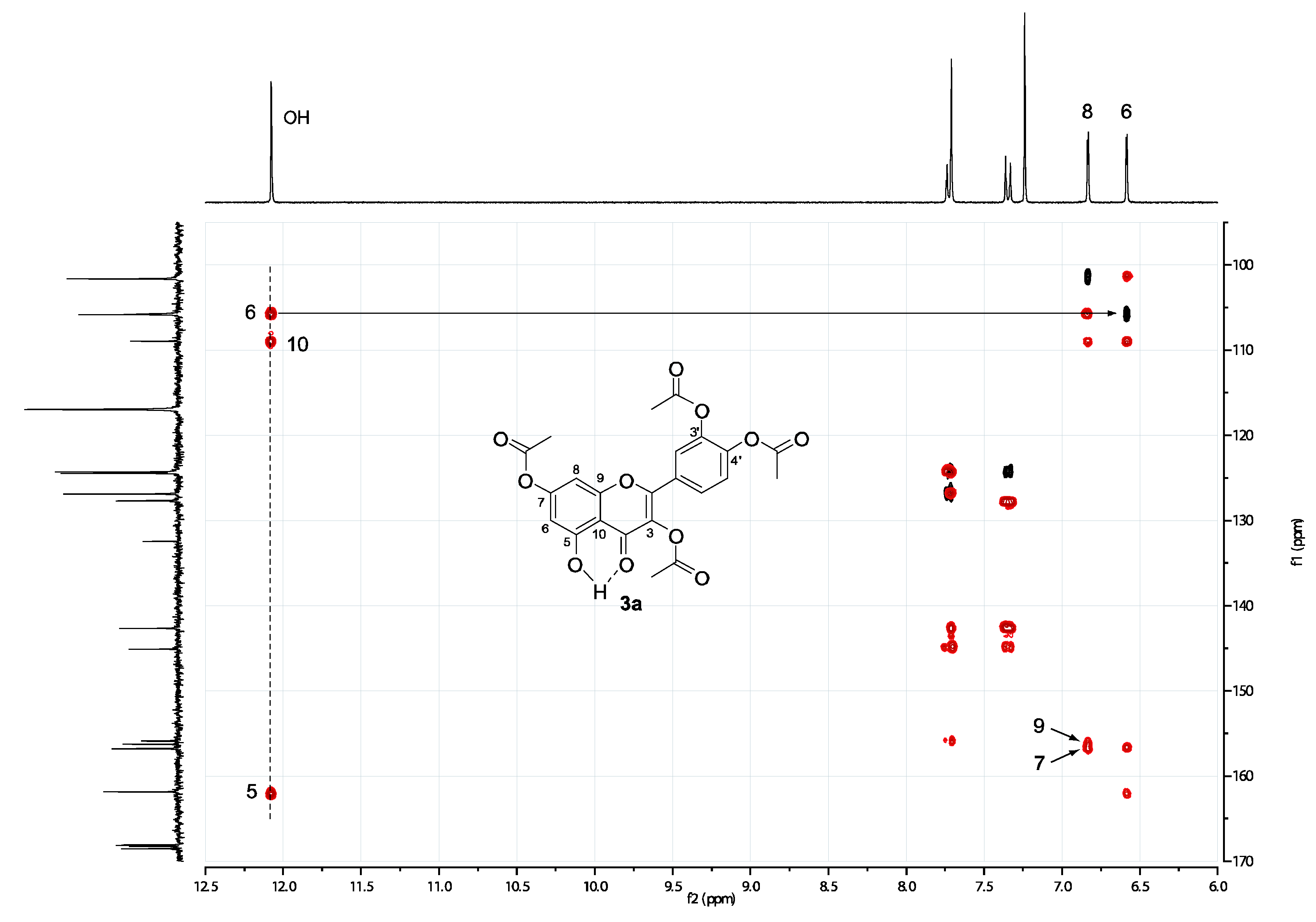
2.2. Spectral assignment of the free hydroxyl position of 3,3’,4’,5-tetraacetylquercetin (4) and 3,3’,4’,7-tetraacetylquercetin (3a)


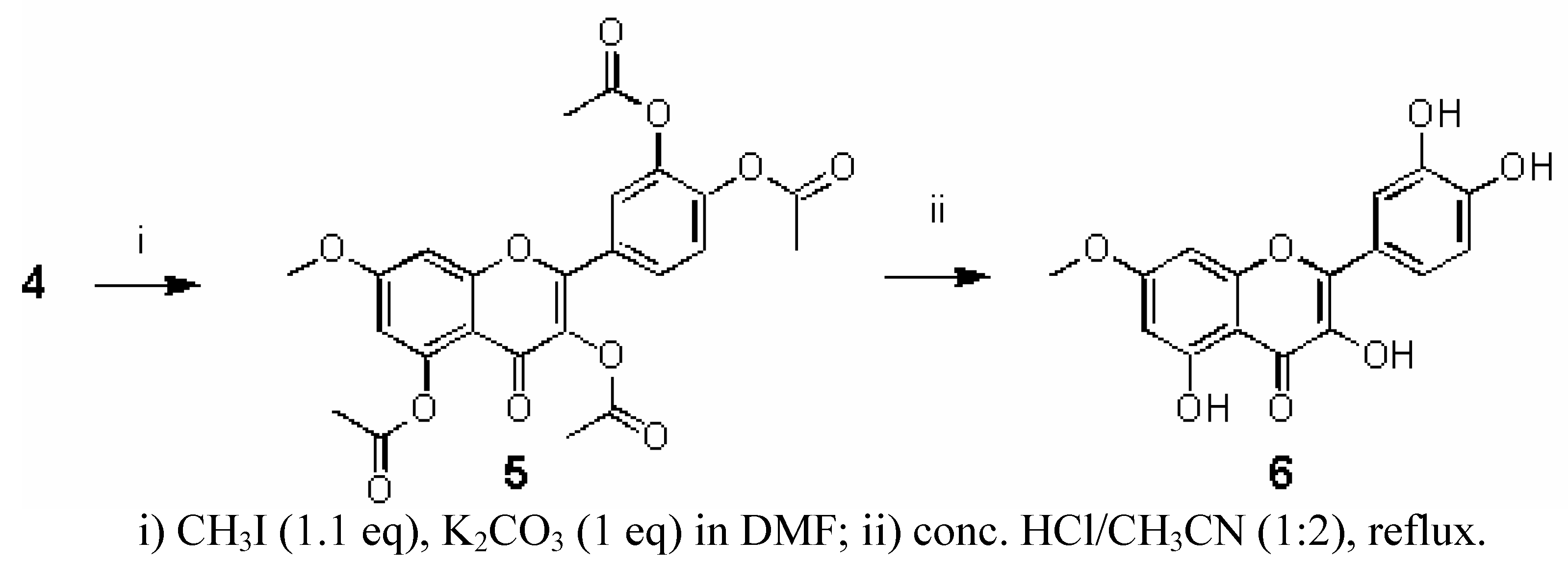
2.3. Synthesis of rhamnetin (7-O-methylquercetin)

2.4. Synthesis of 7-O-(4-triphenylphosphoniumbutyl) quercetin iodide (9)

3. Experimental
3.1. General
3.2. HSQC and HMBC spectra
3.3. Synthesis of 3,3’,4’,5,7-pentaacylquercetins and 3,3’,4’,7-tetraacylquercetins
4. Conclusions
Acknowledgements
- Sample Availability: Not available.
References and Notes
- Bischoff, S.C. Quercetin: Potentials in the prevention and therapy of disease. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 733–740. [Google Scholar] [CrossRef]
- Murakami, A.; Ashida, H.; Terao, J. Multitargeted cancer prevention by quercetin. Cancer Lett. 2008, 269, 315–325. [Google Scholar] [CrossRef]
- Boots, A.W.; Haenen, G.R.; Bast, A. Health effects of quercetin: From antioxidant to nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar] [CrossRef]
- Hirpara, K.V.; Aggarwal, P.; Mukherjee, J.; Joshi, N.; Burman, A.C. Quercetin and its derivatives: Synthesis, pharmacological uses with special emphasis on anti-tumor properties and prodrug with enhanced bio-availability. Anticancer Agents Med. Chem. 2009, 9, 138–161. [Google Scholar]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81 Suppl. 1, 230S–242S. [Google Scholar]
- Williamson, G.; Manach, C. Bioavailability and bioefficacy of polyphenols in humans. II. Review of 93 intervention studies. Am. J. Clin. Nutr. 2005, 81 Suppl. 1, 243S–255S. [Google Scholar]
- Silberberg, M.; Morand, C.; Mathevon, T.; Besson, C.; Manach, C.; Scalbert, A.; Rémésy, C. The bioavailability of polyphenols is highly governed by the capacity of the intestine and of the liver to secrete conjugated metabolites. Eur. J. Nutr. 2006, 45, 88–96. [Google Scholar] [CrossRef]
- Biasutto, L.; Marotta, E.; De Marchi, U.; Zoratti, M.; Paradisi, C. Ester-based precursors to increase the bioavailability of quercetin. J. Med. Chem. 2007, 50, 241–253. [Google Scholar]
- Biasutto, L.; Marotta, E.; Mattarei, A.; Beltramello, S.; Caliceti, P.; Salmaso, S.; Bernkop-Schnürch, A.; Garbisa, S.; Zoratti, M.; Paradisi, C. Absorption and metabolism of resveratrol carboxyesters and methanesulfonate by explanted rat intestinal segments. Cell. Physiol. Biochem. 2009, 24, 557–566. [Google Scholar] [CrossRef]
- Mattarei, A.; Biasutto, L.; Marotta, E.; De Marchi, U.; Sassi, N.; Garbisa, S.; Zoratti, M.; Paradisi, C. A mitochondriotropic derivative of quercetin: A strategy to increase the effectiveness of polyphenols. ChemBioChem 2008, 16, 2633–2642. [Google Scholar]
- Biasutto, L.; Mattarei, A.; Marotta, E.; Bradaschia, A.; Sassi, N.; Garbisa, S.; Zoratti, M.; Paradisi, C. Development of mitochondria-targeted derivatives of resveratrol. Bioorg. Med. Chem. Lett. 2008, 18, 5594–5597. [Google Scholar] [CrossRef]
- Biasutto, L.; Sassi, N.; Mattarei, A.; Marotta, E.; Cattelan, P.; Toninello, A.; Garbisa, S.; Zoratti, M.; Paradisi, C. Impact of mitochondriotropic quercetin derivatives on mitochondria. Biochim. Biophys. Acta 2010, 1797, 189–196. [Google Scholar]
- Hoye, A.T.; Davoren, J.E.; Wipf, P.; Fink, M.P.; Kagan, V.E. Targeting mitochondria. Acc. Chem. Res. 2008, 41, 87–97. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar]
- Murphy, M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta 2008, 1777, 1028–1031. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Anisimov, V.N.; Antonenko, Y.N.; Bakeeva, L.E.; Chernyak, B.V.; Erichev, V.P.; Filenko, O.F.; Kalinina, N.I.; Kapelko, V.I.; Kolosova, N.G.; Kopnin, B.P.; Korshunova, G.A.; Lichinitser, M.R.; Obukhova, L.A.; Pasyukova, E.G.; Pisarenko, O.I.; Roginsky, V.A.; Ruuge, E.K.; Senin, I.I.; Severina, I.I.; Skulachev, M.V.; Spivak, I.M.; Tashlitsky, V.N.; Tkachuk, V.A.; Vyssokikh, M.Y.; Yaguzhinsky, L.S.; Zorov, D.B. An attempt to prevent senescence: A mitochondrial approach. Biochim. Biophys. Acta 2009, 1787, 437–461. [Google Scholar] [CrossRef]
- Metodiewa, D.; Jaiswal, A.K.; Cenas, N.; Dickancaité, E. Quercetin may act as a cytotoxic prooxidant afyer its metabolic activation to semiquinone and quinoidal product. Free Radic. Biol. Med. 1999, 26, 107–116. [Google Scholar] [CrossRef]
- Sarno, S.; Moro, S.; Meggio, F.; Zagotto, G.; Dal Ben, D.; Ghibellini, P.; Battistutta, R.; Zanotti, G.; Pinna, L.A. Toward the rational design of protein kinase casein kinase-2 inhibitors. Pharmacol. Ther. 2002, 93, 159–168. [Google Scholar] [CrossRef]
- van Dijk, C.; Driessen, A.; Recourt, K. The uncoupling efficiency and affinity of flavonoids for vesicles. Biochem. Pharmacol. 2000, 60, 1593–1600. [Google Scholar] [CrossRef]
- Montenegro, L.; Carbone, C.; Maniscalco, C.; Lambusta, D.; Nicolosi, G.; Ventura, C.A.; Puglisi, G. In vitro evaluation of quercetin-3-O-acyl esters as topical drugs. Int. J. Pharm. 2007, 336, 257–262. [Google Scholar] [CrossRef]
- Bouktaib, M.; Lebrun, S.; Atmani, A.; Rolando, C. Hemisynthesis of all the O-monomethylated analogues of quercetin including the major metabolites, through selective protection of phenolic functions. Tetrahedron 2002, 58, 10001–10009. [Google Scholar] [CrossRef]
- Hodnick, W.F.; Duval, D.L.; Pardini, R.S. Inhibition of mitochondrial respiration and cyanide-stimulated generation of reactive oxygen species by selected flavonoids. Biochem. Pharmacol. 1994, 47, 573–580. [Google Scholar] [CrossRef]
- Picq, M.; Prigent, A.F.; Némoz, G.; André, A.C.; Pacheco, H. Pentasubstituted quercetin analogues as selective inhibitors of particulate 3’:5’-cyclic-AMP phosphodiesterase from rat brain. J. Med. Chem. 1982, 25, 1192–1198. [Google Scholar] [CrossRef]
- Op de Beck, P.; Dijoux, M.G.; Cartier, G.; Mariotte, A.-M. Quercitrin-3’-sulphate from leaves of Leea guinensis. Phytochemistry 1998, 47, 1171–1173. [Google Scholar]
- Rietjens, I.M.; Awad, H.M.; Boersma, M.G.; van Iersel, M.L.; Vervoort, J.; Van Bladeren, P.J. Structure activity relationship for the chemical behaviour and toxicity of electrophilic quinones/quinone methides. Adv. Exp. Med. Biol. 2001, 500, 11–21. [Google Scholar]
- Needs, P.W.; Williamson, G. Syntheses of daidzein-7-yl beta-D-glucopyranosiduronic acid and daidzein-4’,7-yl di-beta-D-glucopyranosiduronic acid. Carbohydr. Res. 2001, 330, 511–515. [Google Scholar] [CrossRef]
- Shin, J.S.; Kim, K.S.; Kim, M.B.; Jeong, J.H.; Kim, B.K. Synthesis and hypoglycemic effect of chrysin derivatives. Bioorg. Med. Chem. Lett. 1999, 9, 869–874. [Google Scholar] [CrossRef]
- Li, M.; Han, X.; Yu, B. Facile synthesis of flavonoid 7-O-glycosides. J. Org. Chem. 2003, 17, 6842–6845. [Google Scholar]
- Musialik, M.; Kuzmicz, R.; Pawlowski, T.S.; Litwinienko, G. Acidity of hydroxyl groups: an overlooked influence on antiradical properties of flavonoids. J. Org. Chem. 2009, 74, 2699–2709. [Google Scholar] [CrossRef]
- Piotto, M.; Bourdonneau, M.; Elbayed, K.; Wieruzeski, J.M.; Lippens, G. New DEFT sequences for the acquisition of one-dimensional carbon NMR spectra of small unlabelled molecules. Magn. Reson. Chem. 2006, 44, 943–947. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mattarei, A.; Biasutto, L.; Rastrelli, F.; Garbisa, S.; Marotta, E.; Zoratti, M.; Paradisi, C. Regioselective O-Derivatization of Quercetin via Ester Intermediates. An Improved Synthesis of Rhamnetin and Development of a New Mitochondriotropic Derivative. Molecules 2010, 15, 4722-4736. https://doi.org/10.3390/molecules15074722
Mattarei A, Biasutto L, Rastrelli F, Garbisa S, Marotta E, Zoratti M, Paradisi C. Regioselective O-Derivatization of Quercetin via Ester Intermediates. An Improved Synthesis of Rhamnetin and Development of a New Mitochondriotropic Derivative. Molecules. 2010; 15(7):4722-4736. https://doi.org/10.3390/molecules15074722
Chicago/Turabian StyleMattarei, Andrea, Lucia Biasutto, Federico Rastrelli, Spiridione Garbisa, Ester Marotta, Mario Zoratti, and Cristina Paradisi. 2010. "Regioselective O-Derivatization of Quercetin via Ester Intermediates. An Improved Synthesis of Rhamnetin and Development of a New Mitochondriotropic Derivative" Molecules 15, no. 7: 4722-4736. https://doi.org/10.3390/molecules15074722