Microheterogeneous Catalysis

Department of Physical Chemistry, University of Seville, C/Profesor García González, s/n, 41012, Seville, Spain
*
Author to whom correspondence should be addressed.
Molecules 2010, 15(7), 4815-4874; https://doi.org/10.3390/molecules15074815
Submission received: 1 June 2010
/
Revised: 23 June 2010
/
Accepted: 5 July 2010
/
Published: 9 July 2010
(This article belongs to the Special Issue Heterogeneous Catalysis)
Abstract
:The catalytic effect of micelles, polymers (such as DNA, polypeptides) and nanoparticles, saturable receptors (cyclodextrins and calixarenes) and more complex systems (mixing some of the above mentioned catalysts) have been reviewed. In these microheterogeneous systems the observed changes in the rate constants have been rationalized using the Pseudophase Model. This model produces equations that can be derived from the Brönsted equation, which is the basis for a more general formulation of catalytic effects, including electrocatalysis. When, in the catalyzed reaction one of the reactants is in the excited state, the applicability (at least formally) of the Pseudophase Model occurs only in two limiting situations: the lifetime of the fluorophore and the distributions of the quencher and the probe are the main properties that define the different situations.
1. Introduction
This review deals with catalysis in microheterogeneous systems, that is, in systems in which one (or several) phases are dispersed in another phase (the bulk phase), the dispersed phase being the catalyst. Micellar solutions and microemulsions are typical examples of the systems under consideration.
Generally speaking, the amount of bulk phase in microheterogeneous systems is much greater than the amount of the dispersed phase. This implies that, in order to have effective catalytic activity, the affinity of the dispersed phase by the reactant(s) must be considerably greater than that of the bulk phase, in such a way that a significant part of the reactant(s) is incorporated into the dispersed phase. This affinity results from a variety of interactions such as electrostatic, hydrogen bonding, hydrophobic, π-stacking, etc.
Therefore, if a significant part of the reactant is present in the dispersed phase (catalyst C) the possibility of two reaction paths appears. For the case in which the reaction is slow, compared to the kinetics of the distribution of the reactant(s) between the different phases, one can assume that this distribution is at equilibrium:
![Molecules 15 04815 i001]() in such a way that two populations of reactants, free, Rf, and bound to the catalyst, Rb, are present in the system. These populations react at different rates, so that a catalytic effect appears.
in such a way that two populations of reactants, free, Rf, and bound to the catalyst, Rb, are present in the system. These populations react at different rates, so that a catalytic effect appears.

Several causes can produce an increase in the reaction rate of the bound species. Thus, for bimolecular reactions, if the two reactants are preferentially bound to the dispersed phase, an increase in the local concentrations of these reactants will be observed. This concentration effect will produce an increase in the reaction rate [1]. However, this effect cannot be the cause of the differences of reactivity for the free and bound reactants in the case of unimolecular reactions. For this kind of process, the differences must be related to the properties of the local reaction media that, generally speaking, are quite different from the properties of the bulk phase. So, in the case of solutions containing charged micelles intense local electric fields appear. These fields affect the local properties and, thus, the reaction rate. For example, in the case of electron transfer reactions, the solvent reorganization energy depends on the dielectric characteristic of the surrounding medium [2], and these characteristics are modified by the electric field through solvent saturation effects [3]. On the other hand, the reaction free energy is dependent on the field, because the free energies of the reactant and product states also depend on the dielectric constant of the media. Moreover, the field can change the adiabaticity of the reaction through the polarization of the orbitals of the reactants involved in the electron transfer [4]. The dynamics of the solvent, and thus the pre-exponential term in the rate constant are also changed by the field [5,6]. Indeed, the diffusion coefficients of the intervening species, corresponding to the non homogeneous state (in the presence of the field), are quite different from those of the homogeneous state (without the field) [6]. Therefore, the equilibrium correlations, such as the direct correlation functions, in the presence of a field may also be rather different from those in the absence of the field [7]. Finally, it has been suggested that the fluctuation-dissipation theorem and other important theorems of statistical mechanics may no longer be valid in the presence of a strong field [8,9,10]. All the above mentioned effects can produce dramatic changes in the rate and characteristics of electron transfer reactions in microheterogeneous systems.
Thus, the electron transfer reaction rate within the binuclear pentammineruthenium (III) (μ-cyano)pentacyano-ruthenium (II) complex increases by a factor of about 100 in the presence of hexadecyltrimethylammonium chloride micelles [11]. On the other hand Choudhury et al. [12] claimed that the electron transfer reactions between some coumarin derivatives and amines change from the normal to the inverted regime [13] in micellar solutions.
Of course, electron transfer reactions are neither the only kind of reaction influenced by microheterogeneous catalysis nor the only one that can be influenced by the effects mentioned above. Thus, in the field of Inorganic Chemistry, ligand substitution reactions are also influenced [14,15], as are organic reactions [16] or solvolytic processes [17,18]. Moreover, the influence of microheterogeneous catalysis is not limited to the change of the reaction rate. In some cases a change of the products of a given reaction (which, of course implies a change in the reaction mechanism) has been observed [19].
The effects mentioned above operating in microheterogeneous catalysis indicate clearly the complex character of this type of process. Elucidation of the main factors influencing each particular example is a challenge, which makes the study important from a fundamental point of view.
On the other hand, microheterogeneous catalysis can offer, at least in some cases, advantages in relation to other types of catalysis. Thus, frequently this kind of process can be carried out under average conditions. It is also interesting that this type of reaction does not require, generally speaking, complicated manipulation systems. Moreover, there are many experimental techniques that can provide reliable information on the state of the reactant and the catalyst [17,18]. Finally, an important aspect of microheterogeneous catalysis is its versatility. For example, in systems constituted by surfactants, an external stimulus (light, a variation of pH, etc.) can change the structure of the catalyst drastically: fatty acid soaps aggregate into micelles in a high pH buffer while a simple drop in pH leads to a transformation into bilayer vesicles [20]. In fact, by using this property, the formation of lipid vesicles from micelles and their controlled continuous growth has been achieved with fatty acids [21]. Another example of the versatility of this kind of system comes from the field of protein extraction using microemulsions. In this case, electrostatic interactions compete with biospecific interactions and can disturb the selectivity of the extractive process. However electrostatic interactions can be reduced by doping the interface with non-ionic surfactants [20]. Another way of modulating the balance of electrostatic and other kinds of interactions is to add electrolytes to the systems [22].
The previous paragraphs shows the interest in microheterogeneous catalysis from a basic as well as from an applied point of view. In the following sections of this review this topic will be considered according to the following organization. In Section 2 a common formulation describing homogeneous, heterogeneous and electrocatalysis is presented. Section 3 describes the effects of microheterogeneous catalysts on ground state reactions. These effects, on photochemical reactions, are considered in Section 4. Section 5 gives the general conclusions of this review.
2. A Common Formulation for Homogeneous, Heterogeneous Catalysis and Electrocatalysis
Variations of the rate of a given reaction in the presence of a microcatalyst or a salt, when the concentration of the catalyst or the salt are changed, can be described by equations that are formally identical [23].These variations, in fact, are described by the equation of the Pseudophase Model [24] and the Olson-Simonson equation [25], which are isomorphous.
In both cases, it is supposed that the reactant(s) are present in two states (see Equation 1), which are at equilibrium even if they participate in a chemical reaction, that is, the forward and the reverse processes in Equation 1 are much more rapid than the reactions in which Rf and Rb participates. Under these circumstances, if C (the catalyst) is in excess, the concentrations of free and bound reactants, in the system, are given by [26]:
![Molecules 15 04815 i002]()
![Molecules 15 04815 i003]()


If these two states react at different rates, the observed rate constant, kobs, is given by:
![Molecules 15 04815 i004]() where kf and kb are the rate constants corresponding to the reactions of the free and bound states of R, respectively.
where kf and kb are the rate constants corresponding to the reactions of the free and bound states of R, respectively.

Equation 3 is the equation of the Pseudophase Model when [C] is the concentration of the dispersed pseudophase (the catalyst), or the Olson-Simonson equation if [C] represents the concentration of the salt influencing the reaction. Of course, this equation also describes the observed behavior of the homogeneous catalysis, provided that the catalyst is in excess over the reactant.
A few years after the publication of Olson and Simonson’s paper, Scatchard [27] showed that Equation 3 can be derived from the Brönsted equation [28]. This equation is:
![Molecules 15 04815 i005]()

or:
![Molecules 15 04815 i006]()

or:
![Molecules 15 04815 i007]()

In this equation (ko)obs is the rate constant in an (arbitrary) reference state and γR and γ‡ are the activity coefficients of the reactant and transition state, corresponding to the selected reference state.
The Brönsted equation is, in some sense an identity because RTlnγi (i = R, ‡) is the change in free energy of i when going from the reference state to the actual state, in which the rate constant is kobs. According to this, Equation 4c establishes that the difference between the free energy of the transition state and the free energy of the reactant, in the actual state, is the same as this difference in the reference state plus the change in the free energy of the transition state minus the free energy of the reactant when going from the reference to the actual state. This statement (see Scheme 1) describes an obvious fact.
Given the identity characteristic of the Brönsted equation, it is clear that it will describe changes in reactivity when going from the reference to the actual state, provided that the Transition State Theory holds. In the case of catalytic reactions an additional condition is that, as mentioned previously, the process in Equation 1 remains at equilibrium in the reacting systems.
Scheme 1.
Schematic Representation of the changes in the activation free energy of a reaction when the system goes from a reference state to the actual state.
Scheme 1.
Schematic Representation of the changes in the activation free energy of a reaction when the system goes from a reference state to the actual state.

Now the relationship between Equation 3 and the Brönsted equation will be established [29]. For this purpose consider a solution of reactant R, and some species, C, able to bind to R (Equation 1). C can be a catalyst or a counterion. The contribution of R to the free energy of the solution is given by:
![Molecules 15 04815 i008]()

or:
![Molecules 15 04815 i009]()

 being the chemical potential of free R and
being the chemical potential of free R and  the chemical potential of R bound to the catalyst (see Equation 1). θ represents the association degree of R to C.
the chemical potential of R bound to the catalyst (see Equation 1). θ represents the association degree of R to C.In Equation 5a, nR is the total number of moles of R in the solution and μR its chemical potential. Equation 5b has been written taking into account the possibility of binding R to C.
It is clear from Equations 5a and 5b that:
![Molecules 15 04815 i010]() but free and bound R are at equilibrium. So their chemical potentials are the same, that is:
but free and bound R are at equilibrium. So their chemical potentials are the same, that is:
![Molecules 15 04815 i011]()


This, taking into account Equation 6, implies that:
![Molecules 15 04815 i012]()

That is:
![Molecules 15 04815 i013]()

On the other hand, it is clear that ![Molecules 15 04815 i237]() and thus:
and thus:
![Molecules 15 04815 i014]()
 and thus:
and thus:

From this equation it follows that:
![Molecules 15 04815 i015]()

Consequently, the experimental activity coefficient γR would be the product of the activity coefficient of free reactants, γRf times the degree of dissociation. If one takes as the reference state the free R (and thus γ'Rf = 1), Equation 11 results in:
![Molecules 15 04815 i016]()

Now consider that the two forms of R (free and bound) can react to give a product. According to the Brönsted equation, the rate constant would be:
![Molecules 15 04815 i017]()

But (k0)obs is just kf, because the reference state is the free ions, so:
![Molecules 15 04815 i018]()

On the other hand, ![Molecules 15 04815 i238]() (see reference 29), consequently Equations 3 and 14 are the same. In the case of homogeneous or microheterogeneous catalysis, the rate of reaction according to previous equations would be given by:
(see reference 29), consequently Equations 3 and 14 are the same. In the case of homogeneous or microheterogeneous catalysis, the rate of reaction according to previous equations would be given by:
![Molecules 15 04815 i019]() where [R] represents the total concentration of the reactant in the system.
where [R] represents the total concentration of the reactant in the system.
 (see reference 29), consequently Equations 3 and 14 are the same. In the case of homogeneous or microheterogeneous catalysis, the rate of reaction according to previous equations would be given by:
(see reference 29), consequently Equations 3 and 14 are the same. In the case of homogeneous or microheterogeneous catalysis, the rate of reaction according to previous equations would be given by:

If the non-catalyzed reaction path is not significant, one can write:

The maximum value of ν corresponds to the case in which the reactant is completely bound to the reactant, in such a way that [R] = [Rb]:
![Molecules 15 04815 i021]()

Writing now ![Molecules 15 04815 i239]() , Equation 16 results in:
, Equation 16 results in:
![Molecules 15 04815 i022]()
 , Equation 16 results in:
, Equation 16 results in:

This equation is similar to the Michaelis-Menten equation, and, as was demonstrated, can be obtained from the Brönsted equation. However, Equation 18 is applicable when C is in excess over R, because this is the condition to apply previous equations and, in particular, Equation 12.
The case of heterogeneous catalysis will be treated now. In particular, the case in which the catalyst is a solid and the reactant is a gas will be considered. In order to use the Brönsted equation, the adsorption/desorption processes will be assumed rapid compared to the reaction step, that is, the equilibrium condition holds.
Under these circumstances, taking as the reference state the free gas, one can write:
![Molecules 15 04815 i023]()
![Molecules 15 04815 i024]()


If KR and ![Molecules 15 04815 i240]() are the equilibrium constants corresponding to the adsorption of the reactant and transition state respectively, it is clear that:
are the equilibrium constants corresponding to the adsorption of the reactant and transition state respectively, it is clear that:
![Molecules 15 04815 i025]() that is:
that is:
![Molecules 15 04815 i026]()
![Molecules 15 04815 i027]()
 are the equilibrium constants corresponding to the adsorption of the reactant and transition state respectively, it is clear that:
are the equilibrium constants corresponding to the adsorption of the reactant and transition state respectively, it is clear that:



From the Brönsted equation, the rate constant corresponding to the reaction of the adsorbed reactant ![Molecules 15 04815 i241]() is given by:
is given by:
![Molecules 15 04815 i028]() where kg is the rate constant for the non-adsorbed reactant, that is, the reactant in the reference state, the gas phase. From Equation 22 it follows:
where kg is the rate constant for the non-adsorbed reactant, that is, the reactant in the reference state, the gas phase. From Equation 22 it follows:
![Molecules 15 04815 i029]()
 is given by:
is given by:


Consider now a system at constant volume and temperature, in such a way that concentration and number of moles of a gas contained in this volume are proportional. This volume contains the gas (the reactant R) that can be adsorbed on a surface contained in the volume (the catalyst) which has N adsorption sites. Under these circumstances, the following process happens:
![Molecules 15 04815 i030]() where ЅF represents a free site on the catalyst. Thus, it can be written:
where ЅF represents a free site on the catalyst. Thus, it can be written:
![Molecules 15 04815 i031]() with the concentrations referred to the (fixed) volume of the system.
with the concentrations referred to the (fixed) volume of the system.


If the gas experiences a reaction (slow enough to maintain the adsorption equilibrium, Equation 24) it is possible to write:
![Molecules 15 04815 i032]()

or :
![Molecules 15 04815 i033]()

Of course:
![Molecules 15 04815 i034]()

From the previous equations it follows that:
![Molecules 15 04815 i035]()
![Molecules 15 04815 i036]() and thus:
and thus:
![Molecules 15 04815 i037]()



So that:
![Molecules 15 04815 i038]()

Equation 30 can be obtained from the Brönsted equation, taking into account that the activity coefficients of the reactant, R, and the transition state, ‡, in the system (not in the adsorbed state, as given by Equations 21) are:
![Molecules 15 04815 i039]()
![Molecules 15 04815 i040]()


Thus:
![Molecules 15 04815 i041]()

But this equation, taking into account Equation 23, is the same as Equation 30. Now consider again Equation 29. Assuming that the rate of the non-catalyzed reaction is negligibly small one can write:
![Molecules 15 04815 i042]() or (see Equation 28b):
or (see Equation 28b):
![Molecules 15 04815 i043]()


Of course, concentration of the adsorbed reactant is the same as concentration of occupied sites, if each reactant molecule adsorbs while occupying only one site. Thus:
![Molecules 15 04815 i044]()

Taking into account that:
![Molecules 15 04815 i045]()

θ being the fraction of the covered surface, it follows:
![Molecules 15 04815 i046]()
![Molecules 15 04815 i047]()


If the adsorption of the gas is of the Langmuir type, θ is given by [30,31]:
![Molecules 15 04815 i048]() and thus:
and thus:
![Molecules 15 04815 i049]() which is the familiar equation for the rate of a catalyzed reaction when the reactant gas is adsorbed on the catalyst with a Langmuir type adsorption isotherm.
which is the familiar equation for the rate of a catalyzed reaction when the reactant gas is adsorbed on the catalyst with a Langmuir type adsorption isotherm.


Finally, the case of electrocatalysis will be considered. This is the term frequently used by electrochemists to describe the effect of the electrode potential on the kinetics of electrode reactions [32]. The rate of these reactions is measured as the current density, that is, the current per unit area of the electrode.
In the following paragraphs, the fundamental equation of the electrocatalysis, that is, the Butler-Volmer equation will be derived from the Brönsted equation. For this purpose, consider an electrode process in which an electron acceptor A is reduced at the electrode:
![Molecules 15 04815 i050]()

D represents the reduced A, that is, an electron donor. For simplicity, the charges on A and D are not written. Of course, they have a unit difference in their charges.
The net rate of reaction expressed as the current density is:
![Molecules 15 04815 i051]() where F is the Faraday constant and the anodic current is considered to be positive.
where F is the Faraday constant and the anodic current is considered to be positive.

Taking as the reference states for D and A the states of these species in the actual solution when the electrode potential difference is zero, that is, when the potential of the metal (electrode) and the solution are the same, the chemical potentials of A and D when the solution changes its potential (with respect to the metal) are given by:
![Molecules 15 04815 i052]()
![Molecules 15 04815 i053]()


Assuming that the transition state bears a charge ![Molecules 15 04815 i241]() , its chemical potential will be:
, its chemical potential will be:
![Molecules 15 04815 i054]()
, its chemical potential will be:

It is clear that the activity coefficients of D, A and transition state, with respect to the selected reference state, are given by:
![Molecules 15 04815 i055]()

According to this, the variation of φs will imply a change in the activation free energy, and thus, a change in the rate constant. According to the Brönsted equation, the changes are given by:
![Molecules 15 04815 i056]()
![Molecules 15 04815 i057]()


 y
y  being the rate constants at the reference state, that is when φS = φM.
being the rate constants at the reference state, that is when φS = φM.From Equations 42 to 45 it follows that:
![Molecules 15 04815 i058]()
![Molecules 15 04815 i059]()


In this way Equation 41 becomes:
![Molecules 15 04815 i060]()

Eliminating the condition of constant φM, this equation would be written, in terms of ∆φ′ = φs −φM as:
![Molecules 15 04815 i061]() or, as is customary in the electrochemical context:
or, as is customary in the electrochemical context:
![Molecules 15 04815 i062]() with ∆φ = −∆φ′ = φM −φs.
with ∆φ = −∆φ′ = φM −φs.


When there is no current at the electrode, ∆φ reaches its equilibrium value, ∆φe. In this case:
![Molecules 15 04815 i063]()

In terms of i0 and the overpotential, η = ∆φ − ∆φe, Equation 49 reads:
![Molecules 15 04815 i064]()

This is the Butler-Volmer equation [33,34], the fundamental equation of electrocatalysis. On the other hand, using Equation 50 it is possible to express ∆φe, that is the equilibrium electrode potential, as given by the Nernst equation, in terms of ![Molecules 15 04815 i244]() and
and ![Molecules 15 04815 i245]() :
:
![Molecules 15 04815 i065]()
 and
and  :
:

Moreover, as established above, ![Molecules 15 04815 i244]() and
and ![Molecules 15 04815 i245]() correspond to a reference state (the actual solution when ∆φ = 0) different from the customary one. These rate constants can be written as:
correspond to a reference state (the actual solution when ∆φ = 0) different from the customary one. These rate constants can be written as:
![Molecules 15 04815 i066]()
![Molecules 15 04815 i067]()
and correspond to a reference state (the actual solution when ∆φ = 0) different from the customary one. These rate constants can be written as:


In terms of the rate constant, ![Molecules 15 04815 i246]() and
and ![Molecules 15 04815 i247]() , and the activity coefficients, referred to the customary reference state (of infinite dilution). In this way:
, and the activity coefficients, referred to the customary reference state (of infinite dilution). In this way:
![Molecules 15 04815 i068]() or:
or:
![Molecules 15 04815 i069]()
 and
and  , and the activity coefficients, referred to the customary reference state (of infinite dilution). In this way:
, and the activity coefficients, referred to the customary reference state (of infinite dilution). In this way:


In Equation 55, ![Molecules 15 04815 i248]() and αA and αD are the activities of the acceptor and donor, respectively.
and αA and αD are the activities of the acceptor and donor, respectively.
 and αA and αD are the activities of the acceptor and donor, respectively.
and αA and αD are the activities of the acceptor and donor, respectively.Of course, in the application of the Nernst equation, ∆φe is measured with respect to the potential of a reference electrode, ∆φref, in such a way that it produces:
![Molecules 15 04815 i070]()

E being ∆φ − ∆φref and E° being ![Molecules 15 04815 i249]() .
.
 .
.Thus it has been demonstrated that both the Butler-Volmer equation as well as the Nernst equation can be deduced from the Brönsted equation. In fact, the Nernst equation offers the possibility of checking the treatment developed in this Section and in particular Equation 12 in which this treatment is based. Thus, Equation 56 can be written as:
![Molecules 15 04815 i071]() where E°′ is the so-called standard formal redox potential and is given by:
where E°′ is the so-called standard formal redox potential and is given by:
![Molecules 15 04815 i072]()


This equation, using Equation 12, becomes:
![Molecules 15 04815 i073]()

Equation 59 has been checked recently by one of us through the measurements of the standard formal redox potentials of the redox couple [Fe(CN)5(4-CNpy)]2−/3− (4-CNpy = 4-cyanopyridine) in the presence of micelles of hexadecyltrimethylammonium chloride (CTAC) [35]. From these results it is clear that Equation 59 describes well the changes in the standard formal redox potential when the concentration of micelles is varied. It is interesting to note that Equation 59 permits us to calculate the binding constant of reactants to a microcatalyst (receptor) through measurements of standard formal redox potentials, as Almgren et al. pointed out [36]. On the other hand, changes in the half wave potentials of redox couples in micellar solutions are in agreement with previous equations [37], as are the changes in the equilibrium constants for the reactions of ferrocene, Fe(Cp)2, with a series of cobalt complexes in micellar solutions, when the concentration of micelles is changed [38].
Thus, it is possible to conclude that the previous treatment has been checked employing kinetic and thermodynamic results. This treatment reveals the possibility of obtaining the equations describing the different types of catalysis (homogeneous, microheterogeneous, heterogeneous and electrocatalysis) from a common starting point, the Brönsted equation. This circumstance indicates that the distinction between these different types of catalysis, although useful, is not a radical question.
3. Microheterogeneous Catalysis with Participation of Ground-State Reactants
3.1. General
In this section representative examples of microheterogeneous catalysis will be considered. Here we limit ourselves to the cases in which reactants are in their ground states. The case in which excited states of the reactants participates will be considered in the next section of the review.
The cause for this separation relies in the fact that application of treatments developed in Section 2, implies that the binding of the reactant and catalyst (Equation 1) must be at equilibrium. This requirement is generally accomplished by thermal (ground-state) processes, but in the case of photochemical (excited state) reactions, the possibility of reaching the equilibrium in Equation 1 depends on the lifetime of the excited state and on the rates of the forward and reverse processes in this equation, in such a way that the equilibrium condition does not always hold.
This section has been divided into four subsections corresponding to the cases, in which the catalysts are: (i) micelles (both direct and reverse); (ii) polymers; (iii) saturable receptors, such as cyclodextrins and related systems and (iv) complex systems. Although there is no essential differences between receptors, in the sense that the treatment developed in the previous section can be applied in all of these cases, they have characteristics that can produce some differences. Thus, in the case of saturable receptors, the number of guests bound to a given microcatalyst is generally small and well defined (one or two in some cases). This produces some differences from micelles because, although these kinds of receptors are definitively saturable, given the small concentrations of reactants generally employed in relation to the number of binding sites on the micelles, they behave as practically unsaturable receptors.
Moreover, reactions between two reactants bound to different cyclodextrins cannot be excluded. On the contrary, the reaction between two reactants bound to different charged micelles is impeded by the repulsive micelle-micelle (or polymer-polymer) interaction. According to this, bound reactants must be on the same micelle. This implies that, for a bimolecular reaction in the presence of micelles there is only two possible reaction paths:
![Molecules 15 04815 i074]()
![Molecules 15 04815 i075]() whereas in the case of cyclodextrins there are four possible reaction paths:
whereas in the case of cyclodextrins there are four possible reaction paths:
![Molecules 15 04815 i076]()
![Molecules 15 04815 i077]()
![Molecules 15 04815 i078]()
![Molecules 15 04815 i079]()






This, of course, produces different equations giving the experimental rate constants, kobs: from the arguments given in Section 2 it is possible to show that, in the micellar case, kobs is given by [39]:
![Molecules 15 04815 i080]() where kf is the rate constant for reaction 60a and
where kf is the rate constant for reaction 60a and ![Molecules 15 04815 i250]() the rate constant for the unimolecular [40,41] process 60b. K1 and K2 represents the binding constants of the reactants to the micelles.
the rate constant for the unimolecular [40,41] process 60b. K1 and K2 represents the binding constants of the reactants to the micelles.

 the rate constant for the unimolecular [40,41] process 60b. K1 and K2 represents the binding constants of the reactants to the micelles.
the rate constant for the unimolecular [40,41] process 60b. K1 and K2 represents the binding constants of the reactants to the micelles.In the case of cyclodextrins (and related systems) Equations 61a–d produce the following equation for kobs [39]:
![Molecules 15 04815 i081]()

Moreover, in the case of saturable receptors, the binding constant is a true constant, that is, independent of the concentrations of receptors and reactants. On the contrary, the binding constants can change, in the case of micelles and polyelectrolytes, due to the condensation of counter-ions [42] or cooperative effects [43]. Of course, the treatment developed in Section 2 is still valid, provided that the influence of these effects is incorporated into the treatment [44].
On the other hand, micelles and polyelectrolytes present some differences. Thus, in the case of micelles constituted by a unique surfactant all the sites at the micellar surfaces are equivalent. On the contrary, polymeric materials are frequently heterogeneous in the sense that they show different kinds of binding sites. For example, in the case of DNA it is known that ligands can bind to the polymer through electrostatic interactions with phosphate groups, or in the major or minor grooves, as well as through intercalative binding mode [45,46]. It is clear that the reactivity of ligands that depends on the binding will be different in these different sites. Indeed, as the proportion of occupied sites of each class will be dependent on the relative concentrations of ligand and polymer, the (macroscopic) measured binding constant will be dependent on this proportion. In this regard, the polymer behaves as a mixture of receptors.
3.2. Micellar Solutions
Reactivity in normal and reverse micelles has been the objective of a great deal of research activity. Thus, many kinds of reactions such as, oxidation, hydroformylation, carbonylation, dehalogenation, free radical polymerization, enzymatic reactions [47], ligand substitution reactions [48], electron transfer reactions [49] etc., have been studied in these kinds of systems.
This interest arises from several reasons such as the possibility of localization of reactants in suitable microenvironments, which enables catalyzing and control of a wide variety of reactions [50]. Thus, for example, microemulsions have been used to induce regioselectivity in organic reactions [47]. On the other hand it is possible to solubilize in micellar solutions reagents that cannot be dissolved in the same bulk solvent. In other cases, interest arises because simple microheterogeneous systems mimic some of the most important characteristics of biological systems. Some studies on reactivity in micellar systems are aimed at learning about the structure of these systems [51]. In relation to the latter kind of study, however, it is important to realize that a rate constant is a macroscopic magnitude (in some sense, if one accepts Transition State Theory, a thermodynamic magnitude). As such, it represents an average value and, consequently, cannot give structural information directly. However, thermodynamic and kinetic data are useful in order to check the structural information obtained from other techniques.
Many (but not all) of the reactions in micellar systems happen, at least partially at the interfaces, in this case, at liquid interfaces. An important aspect of these interfaces is the fact that their properties are not uniform through them. This implies that reactivity at different points of the interfaces can be different. Moreover, this inhomogeneous character implies that the total intermolecular force on a given molecule at the interfacial regions is strongly anisotropic. As a result, certain molecular orientations and localizations may be preferred at the interface [52]. Of course, localization and orientation are specific, that is, they depend on the characteristics of the molecule that is being considered. This gives rise to some problems when one is considering some properties of the interfaces. For example, dielectric permittivity at the micellar interfaces has been deduced sometimes employing some probes and measuring properties of the probes related to the dielectric polarity of their environments [53]. These dielectric data are then used to explain reactivities of reactants different from the probes. However, this would not be correct unless one is secure about the fact that probes and reagents are localized in the same region of the interfaces. This caution must also be considered when one is dealing with electric potentials at the interfaces.
These potentials are determined by using suitable indicators [54]. In the best of cases, this procedure would give the electric potential at the points where the indicators are placed, which are not necessarily the same as the points where other reactants are localized. In fact, it has been suggested that ions of different charges, but with the same charge sign, are localized on different regions of the DNA/water interface. This different localization arises as a consequence of dielectric saturation effects caused by the high electric field at the interface. This effect would push the ions towards the aqueous phase, but more so to the ion with the higher charge. In other words, the ions will feel different potentials according to their different charges [22].
Coming back to consideration of the reactivity in micellar (direct micelles) solutions, the micellar effect has been interpreted generally using the Pseudophase [24] and related models, such as the Pseudophase Ion Exchange Model [55]. The Pseudophase Model of Menger and Portnoy produces the same equations derived in Section 2 from the Brönsted equation. Thus for a true unimolecular reaction, as established in Section 2, Equation 3 is obtained. This equation still is valid for bimolecular reactions provided that one of the reactants remains mainly in the aqueous pseudophase [56]. Thus, for a reaction:
![Molecules 15 04815 i082]()

if only the reactant B is partitioned between the micelles and the bulk pseudophase, taking into account the volume of this pseudophase has practically the same value as the total volume of the system, one can write:
![Molecules 15 04815 i083]() where:
where:
![Molecules 15 04815 i084]()
![Molecules 15 04815 i085]()
![Molecules 15 04815 i086]()




In this way, kf ≈ kf where kf has the same meaning as in Equation 3 and kf is the rate constant in the aqueous pseudophase corresponding to the concentrations of the reactants in this pseudophase. On the other hand, kb is related to the true (second order) rate constant of the catalyzed reaction, kb, by:
![Molecules 15 04815 i087]() (notice that according to Equation 65 [A]b << [A]f and [Ab] << [Af]). Equation 67 can be written as:
(notice that according to Equation 65 [A]b << [A]f and [Ab] << [Af]). Equation 67 can be written as:
![Molecules 15 04815 i088]() where:
where:
![Molecules 15 04815 i089]()



Of course, κA has a different meaning from KA (see Equation 1), but they are related:
![Molecules 15 04815 i090]()

φ being:
![Molecules 15 04815 i091]()

In other words, for bimolecular reactions, if only one of the reactants is appreciably partitioned between the micellar and aqueous pseudophases, Equation 3 is formally valid. In this equation kf has the same meaning as that of unimolecular reactions, but the meaning of kb is somewhat different, as given by Equation 68.
In the case of bimolecular reactions, when the two reactants are partitioned between the micellar and aqueous pseudophases, Equation 3 cannot be applied. Instead Equation 62 must be used. This equation reveals that, in this case, the effect of the catalyst comes, at least in part, from a concentration effect, because local concentration of the reactants is increased. On the other hand, if the intrinsic reactivity at the micellar pseudophase is greater than in the aqueous (or bulk) pseudophase, an additional increase in rate will be observed. Equation 62 implies that kobs will go through a maximum when [C] is changed. Thus, for small concentrations of C, in such a way that K1[C] and K2[C] << 1, kobs will depend linearly on this concentration:
![Molecules 15 04815 i092]()

However, at big enough concentrations of C, the quadratic term in Equation 62 will dominate, thus producing a diminution of kobs. This behavior reflects the fact that when [C] is small there is a high probability of finding two reactants on the same micelle, in such a way that the concentration effect is operative. On the other hand, when the concentration of the catalyst is big enough, the probability of finding two reactants on the same micelle is low, and the concentration effect disappears.
It is interesting to note the this behavior was predicted theoretically by Scatchard [27], for salt effects on reactions between ions of the same charge sign, starting from the Brönsted equation and employing the Mayer’s treatment [57] of ionic solutions for the calculation of the activity coefficient of reactants and the transition state. This fact reveals that there are no essential differences between the catalytic effects of salts and micelles on ionic reactions [23].
Equations 3 and 62 have been checked many times in studies on reactivity in micellar solutions. However, several causes can produce deviations from the behavior predicted by these equations. In particular, for charged reactants, the binding constants (Equation 1) change when the concentration of charged micelles does so. This is a consequence of the fact that when the concentration of micelles increases, the association degree of the counterions (of the micelles) does so. Obviously, this produces a diminution of the charge of the micelles that, in turn, reduces the electrostatic component of the free energy of binding.
The effects of counterion association (with the micelles) can be incorporated into previous equations in a simple way [42]. Thus, the free energy corresponding to K (Equation 1) can be written, without any lost of generality, as the sum of two contributions: a nonelectrostatic contribution, ∆Gnel, and an electrostatic contribution ∆Gel, dependent on the electrostatic field at the interface, that is, on the degree of association with the counterions:
![Molecules 15 04815 i093]()

The electrostatic contribution can be expressed as:
![Molecules 15 04815 i094]() where ZR is the charge of the reactant and Ψ the electrostatic potential measured with some probe, generally an indicator. Parameter α takes into account that the locations of the indicator and the reactant can be different, as previously mentioned. If one defines:
where ZR is the charge of the reactant and Ψ the electrostatic potential measured with some probe, generally an indicator. Parameter α takes into account that the locations of the indicator and the reactant can be different, as previously mentioned. If one defines:
![Molecules 15 04815 i095]()


It is obvious that:
![Molecules 15 04815 i096]()

This procedure for the incorporation of the effects that counterion binding has on the changes in reactivity, has been checked several times by us [42,44,58].
Separation of the electrostatic and non-electrostatic components of the free energy of binding can be accomplished using de Lippard’s equation [59]. According to this equation, in the presence of an added salt, log Kel is proportional to - log[X], X being the counterion of the micelles. Thus:
![Molecules 15 04815 i097]()

Consequently, a plot of log K vs. log[X] gives, as the intercep, log Knel. Once Knel is known, Kel, for each salt concentration can be obtained as:
![Molecules 15 04815 i098]()

In this way, we have determined the electrostatic and non-electrostatic component of the free energy of binding of [Ru(NH3)5Pz]2+ to gold nanoparticles [60] and DNA [22,61]. The effects of the changes in the composition of the interface have also been focused from other points of view. Thus as mentioned previously Quina and Chaimovich [55] developed the Ion Exchange Pseudophase Model by applying to the micellar catalysis a treatment analogous to the classical treatment of ion-exchange resins. They considered the case of a micellar solution formed from the detergent DY where a salt BX of the reactive ion, X−, is present. According to the authors, X− occupies a site at the interface through an ion exchange process:
![Molecules 15 04815 i099]() where:
where:
![Molecules 15 04815 i100]()


They assumed that: (1) The distribution of aggregate sizes can be represented in terms of the most probable aggregation number, ![Molecules 15 04815 i251]() ; (2) The ion-ion and ion head-group interaction are non-cooperative, in such a way that ion exchange rates are dependent only on the number of ions in a given aggregate and the concentrations of free ions in the bulk; (3) The degrees of ionization α of the individual micellar species MYiXi are the same independently of the i and j values. If m is the total number of occupied sites at the micellar surface:
; (2) The ion-ion and ion head-group interaction are non-cooperative, in such a way that ion exchange rates are dependent only on the number of ions in a given aggregate and the concentrations of free ions in the bulk; (3) The degrees of ionization α of the individual micellar species MYiXi are the same independently of the i and j values. If m is the total number of occupied sites at the micellar surface:
![Molecules 15 04815 i101]()
 ; (2) The ion-ion and ion head-group interaction are non-cooperative, in such a way that ion exchange rates are dependent only on the number of ions in a given aggregate and the concentrations of free ions in the bulk; (3) The degrees of ionization α of the individual micellar species MYiXi are the same independently of the i and j values. If m is the total number of occupied sites at the micellar surface:
; (2) The ion-ion and ion head-group interaction are non-cooperative, in such a way that ion exchange rates are dependent only on the number of ions in a given aggregate and the concentrations of free ions in the bulk; (3) The degrees of ionization α of the individual micellar species MYiXi are the same independently of the i and j values. If m is the total number of occupied sites at the micellar surface:

(4) The ion-exchange processes are rapid relative to the lifetime of micelles; (5) The activities of the different micellar and free species present are treatable in terms of their analytical concentrations. An additional assumption is that KX/Y, α and the cmc are independent of the total detergent concentration [55]. Under these assumptions the following equations hold for the case in which BY is added to the solution:
![Molecules 15 04815 i102]()
![Molecules 15 04815 i103]()
![Molecules 15 04815 i104]()
![Molecules 15 04815 i105]()
![Molecules 15 04815 i106]()





From these equations and Equation 80 it follows that:
![Molecules 15 04815 i107]()

This equation may be resolved for [Xb] to give:
![Molecules 15 04815 i108]()
![Molecules 15 04815 i109]()


Once [Xb] is known, [Xf] can be calculated from:
![Molecules 15 04815 i110]() in such a way that the observed rate constant would be given by:
in such a way that the observed rate constant would be given by:
![Molecules 15 04815 i111]()


The authors also considered more complicated situations such as the effect of the addition of a salt ZB, the case in which the micellar system remains buffered ,as well as the reaction of X with an uncharged substrate, S, (when [X] >> [S]) whose binding to the micelle is described by a binding constant Ks.
The problem that we are considering has been also envisaged by Rodenas et al. [62,63]. They employed a treatment based on a cell model of the solution. In this model the micellar solution is divided into cells, each of them containing a micelle and the amount of water and electrolyte given by the whole concentration of the particular systems. The cells and the micelles are considered to be spherical, with radius rc and rm, respectively. The micellar charge is considered to be uniformly distributed over the micellar surface. The distribution of ions around the micelles is calculated from the non-linearized Poisson-Boltzmann equation. This equation under spherical symmetry conditions is given by:
![Molecules 15 04815 i112]() where ε represents the permittivity of the solution, and zi and ci the charges and concentrations of the ions present in the solution. By integration of this equation one can obtain the concentration of the reactive ion(s) close to the micellar surface for different concentrations of supporting electrolytes.
where ε represents the permittivity of the solution, and zi and ci the charges and concentrations of the ions present in the solution. By integration of this equation one can obtain the concentration of the reactive ion(s) close to the micellar surface for different concentrations of supporting electrolytes.

In the previous paragraphs we have considered changes in the binding constants of the reactants to the micelles caused by counterion condensation. There are other possible causes for a change in K. Thus, this parameter can change as a consequence of a change in the micellar shape. Obviously a change in the shape of the micelles implies a change in the surface curvature and thus in the density of charge in the micelles. This would imply a change in the electrostatic component of the free energy of binding of charged substrates (see Equations 73-76). This possibility of change of the binding constant has kinetic consequences, as shown by Moya et al. [64,65]. These authors showed that the change in the micellar shape implies changes in the dissociation degree of the counterions, aggregation number, water content in the interfacial region and local polarity [66]. These changes, as previously mentioned, influence the binding constant, as well as local reactivity. Thus, according to the authors, and due to these changes, an increase in the reactivity is observed, for the reactions of methyl 4-nitrobenzene-sulfonate and methyl naphtalene-2-sulfonate with bromide ion, as a consequence of the sphere to rod transition in solution containing gemini surfactants [64], as well as in solution containing mixed single chain and gemini surfactants.
Another cause of failure of the simple equation derived from the Brönsted equation or from the Pseudophase Model has been observed for reactions between ions of opposite charge sign in micellar solutions [67]. In this case, only one of the reactant ions will be associated to the micelles. The other reactant will remain in the bulk pseudophase. If the charge of the reactant located in the bulk pseudophase is high and/or its concentration (relatively) high and/or the micellar concentration is low, this reactant will compete with the micelles for capturing the other reactant. This will produce a change in K (Equation 1) that will depend on the concentration of the reactant non-associated to the micelle. Thus, consider the reaction between two oppositely charged reactants A and B, and suppose that only A is associated with the micelles, according to Equation 1. Now suppose that A and B can also be associated forming ion pairs A/B. Reactant A can now be in three states: free, bound to the micelles, AB, and bound to B, A/B. Thus, if one defines:
![Molecules 15 04815 i113]()
![Molecules 15 04815 i114]()
![Molecules 15 04815 i115]() where A′f represents the total concentration of A which is not bound to the micelles, that is:
where A′f represents the total concentration of A which is not bound to the micelles, that is:
![Molecules 15 04815 i116]()




The apparent binding constant, Kapp, will be given by:
![Molecules 15 04815 i117]() or:
or:
![Molecules 15 04815 i118]()


That is:
![Molecules 15 04815 i119]()

According to this equation, the value of the effective binding constant to the micelles to be used in Equation 3 will be Kapp, that is, dependent on the concentration of B. Notice that if the concentration of B is not in great excess over the concentration of A, its value will change along the reaction time, this producing a deviation of the simple behavior predicted by Equation 3. On the other hand, the binding constant for reactant A to a given kind of micelle, determined from kinetic data corresponding to reactions with two different reactants B and C, having different values of K′, will be different [67].
An interesting point of view in relation to micellar catalysis is due to Piszkiewicz [68,69,70]. The starting point for Piszkiewicz was the fact that micellar catalysis has been considered frequently as a model for enzyme catalyzed processes. This fact comes from the circumstance that there are important similarities between enzymes and micelles: their structures are similar in that they have hydrophobic cores with polar groups on their surfaces. On the other hand, the structures of micelles are disrupted by common protein denaturing agents such as urea and guanidinium salts. Moreover, both catalytic micelles and enzymes bind substrates in a noncovalent manner and, in some cases, the kinetic of micellar catalysis may be saturated by the substrate, as is the case in enzymatic reactions [69]. Finally, the kinetics of micelle catalyzed reactions shows, sometimes, similarities to the kinetics of processes catalyzed by many regulatory enzymes in the sense that they present positive homotropic interactions (positive cooperativity) [68].
Taking into account these facts, Piszkiewicz proposed a model of micellar catalysis different from the classical Pseudophase Model. In this model it is postulated that the reactant, R, and surfactant molecules, T, aggregate to form supramolecular TnR complexes, according to:
![Molecules 15 04815 i120]()
![Molecules 15 04815 i121]()
![Molecules 15 04815 i122]()



According to this, kobs is given by:
![Molecules 15 04815 i123]()

The reaction mechanism in Equation 93 is similar to the mechanism proposed by Hill [71] to describe the sigmoidal dependence of rate on substrate concentration in enzymatic reactions:
![Molecules 15 04815 i124]()
![Molecules 15 04815 i125]()


According to this mechanism, the reaction rate, v, would be given by:
![Molecules 15 04815 i126]()
![Molecules 15 04815 i127]()


Equations 94 and 96a can be rearranged to give, respectively:
![Molecules 15 04815 i128]() and:
and:
![Molecules 15 04815 i129]()


If in Equation 97a kf << kobs (that is if the non-catalyzed reaction is not significant) it results in:
![Molecules 15 04815 i130]() which is formally identical to Equation 97b.
which is formally identical to Equation 97b.

Previous equations for micellar catalysis describe the sigmoidal variations of kobs vs. [T] observed at low concentrations of surfactants in some micelle-catalyzed reactions [68,69,70]. An interesting point in this treatment is that n is much lower than the aggregation number normally found for micelles. This has been considered as a manifestation of catalysis by pre-micellar structures. This possibility has been treated by us [72,73].
On the other hand, Biresaw and Bunton [74] have also considered the catalytic effect of premicellar aggregates. Their model also explains the sigmoidal dependence of the observed rate constant on [T], at low values of this concentration (see also reference [73]). This treatment is based on the following assumptions: (1) the model is applied to short chain surfactants that forms polydisperse aggregates (instead of micelles) in such a way that they do not exhibit a critical micelle concentration; (2) This aggregation may be assisted by cooperative interaction with the reactant (which experiences the catalyzed reaction), as in Equation 95a; (3) The aggregates are formed (at least formally) via a stepwise-self-association process, consistent with the absence of a critical micellar concentration; (4) The distribution of the reactant between the continuous phase (water) and the aggregate with q monomers, Tα, for q ≥ 2 is given by an equilibrium constant K′α; (5) The rate constant for the reactant bound to Tα is k′α. Two simplifying assumptions are: (6) k′α = kb, independently of q and (7) K′α = qKs, Ks being the intrinsic substrate binding constant expressed per monomer concentration. According to these assumptions:
![Molecules 15 04815 i131]() where Rq represents the reactant bound to Tq. The concentration of Rq, according to previous assumptions, is:
where Rq represents the reactant bound to Tq. The concentration of Rq, according to previous assumptions, is:
![Molecules 15 04815 i132]() and thus:
and thus:
![Molecules 15 04815 i133]()



If [Rb] is the total concentration of the bound reactant,
![Molecules 15 04815 i134]()

So:
![Molecules 15 04815 i135]() and consequently:
and consequently:
![Molecules 15 04815 i136]()


This equation can be simplified assuming that stepwise association constants corresponding to processes:
![Molecules 15 04815 i137]() are independent of the value of i, that is:
are independent of the value of i, that is:
![Molecules 15 04815 i138]()


Under this circumstance, results for kobs:
![Molecules 15 04815 i139]()
![Molecules 15 04815 i140]()


This equation describes well the catalytic effect of the aggregates on unimolecular reactions, of the type described by the Piszkiewicz model, with reasonable values of the parameters appearing in the equations [74]. As, in the Piszkiewicz model, a sigmoidal dependence of kobs of [T] is predicted by this treatment. However, maximum values of q are greater than the values of n found in the Piszkiewicz treatment.
All the previous considerations and treatments can be applied to both, direct and reverse micelles. The latter, however, have some special characteristics that produce some differences with the direct micelles. Thus, in reverse micelles most of the water present in the system is confined water, particularly, at low values of the molar ratio w = [H2O]/[surfactant] [75]. This water has very different properties from bulk water (see Introduction section) which influences reactivity. The unique feature of the entrapped water can be exploited fruitfully in various applications in biotechnology [17,18]. It is interesting to note that in the last few years the interest of the studies (including reactivity studies) in reverse micelles is more to learn about the characteristics of confined water than to learn about the catalytic properties of reverse micelles. However, at least in some cases, reverse micellar systems can become more complex that systems containing direct micelles. Solutions of direct micelles are considered biphasic (or pseudobiphasic) systems. In the case of reverse micelles one can conceive the possibility of reactions that happen simultaneously at the interface (the surfactant region) the confined water region and the oil or continuous pseudophase. In this way, the system becomes a pseudo-triphasic system.
For these reasons, some specific treatments have been developed for reactions in reverse micelles. Thus, in reference [44] the oxidation of [Ru(NH3)Pz]2+ by S2O82− (Pz = pyrazine) in reverse micelles was studied. The studies were conducted at different w values and at constant concentrations of the reactants in the aqueous pseudophase. Given the ionic character of the reactants it was considered that they were absent in the oil pseudophase. In order to explain the reactivity trends, it was supposed that one of the reactants (the ruthenium complex) was adsorbed at the interface, the other remaining mainly in the aqueous pseudophase. For a given value of the molar ratio, w, assuming that all the surfactant and water molecules are incorporated into the micelles, if V is the volume of the water pool it is possible to write:
![Molecules 15 04815 i141]()

Mw and ρw being the molar mass and the density of water. From the value of the molar ratio it follows that:
![Molecules 15 04815 i142]()

This number is equal (or proportional) to the number of binding sites in a droplet. Thus:
![Molecules 15 04815 i143]() where β is a proportionality constant, which takes into account the possibility of a substrate binding to more than one polar head of surfactant.
where β is a proportionality constant, which takes into account the possibility of a substrate binding to more than one polar head of surfactant.

Considering the microheterogeneous equilibrium:
![Molecules 15 04815 i144]()

If one assumes a Langmuir type of the adsorption process, one can write:
![Molecules 15 04815 i145]()

θ being the fraction of occupied sites
On the other hand:
![Molecules 15 04815 i146]()

and:
![Molecules 15 04815 i147]() and, evidently:
and, evidently:
![Molecules 15 04815 i148]()


Under the working conditions in this work, θ << 1 (even if the ruthenium complex is totally adsorbed). Thus:
![Molecules 15 04815 i149]()

in such a way that:
![Molecules 15 04815 i150]()

From this equation,
![Molecules 15 04815 i151]()
![Molecules 15 04815 i152]() and using Equation 34 one finds:
and using Equation 34 one finds:
![Molecules 15 04815 i153]()



From Equations 118a and 119 it follows that the observed (second order) rate constant is given by:
![Molecules 15 04815 i154]()

ks being the rate constant for the surface reaction, and kf the corresponding parameter for the water pool.
As mentioned previously, microemulsions can behave in some cases as (pseudo) triphasic systems. For these cases, the most direct way to find the equation giving the observed rate constant is to employ the formulation developed in Section 2, that is, the formulation based on the Brönsted equation. Using this approach it can be shown [76], for the general case of a system containing a continuous phase, I, and n-1 dispersed phases in which a reaction is possible, that the observed rate constant, if one takes phase I as reference, is given by:
![Molecules 15 04815 i155]()

This equation for the particular case of a microemulsion oil/water/surfactant, if one takes as reference the oil phase gives:
![Molecules 15 04815 i156]() where Kwater and Ksurfactant are defined as in Equation 1.
where Kwater and Ksurfactant are defined as in Equation 1.

Other treatments for particular cases have been developed. Thus, Garcia-Rios et al. considered the cases of thermodynamics and kinetics of complexation of Ni2+ and Co2+ by PADA (pyridine-2-azo-p-dimethylaniline) in AOT/isooctane/water microemulsions [77,78]. The authors considered microemulsions to be made up of three different microenvironments: a continuous phase formed by the alkane, a disperse phase formed by the aqueous micro-droplets and a surfactant film that separates the aqueous phase from the alkane phase. The authors, from the variation of the absorption spectrum of the ligand (PADA), concluded that it is distributed among the three pseudophases. The distribution equilibrium constants ![Molecules 15 04815 i252]() (oi = oil/interface) and
(oi = oil/interface) and ![Molecules 15 04815 i253]() (wi = water/interface) are defined as:
(wi = water/interface) are defined as:
![Molecules 15 04815 i157]()
![Molecules 15 04815 i158]() where w has its habitual meaning and z is the molar ratio [isooctane] /[AOT]. In fact
where w has its habitual meaning and z is the molar ratio [isooctane] /[AOT]. In fact ![Molecules 15 04815 i253]() is high, in such a way that the ligand PADA can be considered distributed only between the oil and surfactant pseudophases. On the other hand, the Ni2+ and Co2+ ions are present only in the aqueous pseudophase and at the interface. This distribution is viewed as an ion exchange process between the counterions of the surfactant, Na+, and the M2+ cations, and quantitatively described by an equilibrium constant
is high, in such a way that the ligand PADA can be considered distributed only between the oil and surfactant pseudophases. On the other hand, the Ni2+ and Co2+ ions are present only in the aqueous pseudophase and at the interface. This distribution is viewed as an ion exchange process between the counterions of the surfactant, Na+, and the M2+ cations, and quantitatively described by an equilibrium constant ![Molecules 15 04815 i254]() given by:
given by:
![Molecules 15 04815 i159]()
 (oi = oil/interface) and
(oi = oil/interface) and  (wi = water/interface) are defined as:
(wi = water/interface) are defined as:

 is high, in such a way that the ligand PADA can be considered distributed only between the oil and surfactant pseudophases. On the other hand, the Ni2+ and Co2+ ions are present only in the aqueous pseudophase and at the interface. This distribution is viewed as an ion exchange process between the counterions of the surfactant, Na+, and the M2+ cations, and quantitatively described by an equilibrium constant
is high, in such a way that the ligand PADA can be considered distributed only between the oil and surfactant pseudophases. On the other hand, the Ni2+ and Co2+ ions are present only in the aqueous pseudophase and at the interface. This distribution is viewed as an ion exchange process between the counterions of the surfactant, Na+, and the M2+ cations, and quantitatively described by an equilibrium constant  given by:
given by:

From previous considerations it is obvious that the complexation reaction can happens only at the interface, according to:
![Molecules 15 04815 i160]()

K in previous equation is related to the observed equilibrium constant Kobs (referred to the concentrations of the reactant in the system) by:
![Molecules 15 04815 i161]()

According to this equation, the plot of ![Molecules 15 04815 i255]() vs. z should be linear. From the slope and intercept of this plot the authors were able to obtain K and
vs. z should be linear. From the slope and intercept of this plot the authors were able to obtain K and ![Molecules 15 04815 i252]() . An interesting result is that K depends on w, that is, the structure of the interface depends on the water contents of the system [77].
. An interesting result is that K depends on w, that is, the structure of the interface depends on the water contents of the system [77].
 vs. z should be linear. From the slope and intercept of this plot the authors were able to obtain K and . An interesting result is that K depends on w, that is, the structure of the interface depends on the water contents of the system [77].
vs. z should be linear. From the slope and intercept of this plot the authors were able to obtain K and . An interesting result is that K depends on w, that is, the structure of the interface depends on the water contents of the system [77].Using the same reaction scheme, the authors were able to relate the observed rate constant, kobs, to the rate constant for the formation/dissociation rate constants at the interface:
![Molecules 15 04815 i162]()
![Molecules 15 04815 i163]()


From this equation the authors derived the values of ![Molecules 15 04815 i256]() and
and ![Molecules 15 04815 i257]() . These values, as K, depend on the water contents of the microemulsion. In fact
. These values, as K, depend on the water contents of the microemulsion. In fact ![Molecules 15 04815 i256]() changes much more than
changes much more than ![Molecules 15 04815 i257]() in such a way that variations of
in such a way that variations of ![Molecules 15 04815 i256]() control the changes in the equilibrium constant.
control the changes in the equilibrium constant.
 and
and  . These values, as K, depend on the water contents of the microemulsion. In fact changes much more than in such a way that variations of control the changes in the equilibrium constant.
. These values, as K, depend on the water contents of the microemulsion. In fact changes much more than in such a way that variations of control the changes in the equilibrium constant.To conclude this section one can say that two state models describe well the variations in reactivity in micellar systems. These models, in fact, are particular cases of the Brönsted formulation: they can be deduced from it. This equation, indeed, permits generalization to multiphasic systems. It is important to realize that final equations, in any case, depend on parameters defined as equilibrium or distribution constant. However, these parameters are not true constants, in the sense that they depend on the structure of the system (micelles) that can change if one works in a wide range of conditions. Several examples of the causes that produce changes in these “constants” have been considered, as well as the procedures to take them into account.
3.3. Polymers and Related Microcatalysts
Polymers can act as receptors with some similarities to micelles. Thus, the reactivity in the presence of DNA, a negatively charged natural polymer, can be modelled according to the Pseudophase Model. In fact, changes in kobs for reactions in the presence of DNA are given by Equation 3 (when only one of the reactants participating in the reaction is bound to polymer), or Equation 62 (if the two reactants are bound to DNA) [79]. However, there are some differences between polymer effects and micellar effects. Thus, polymers are, generally speaking, heterogeneous materials in the sense that they show different kind of binding sites. In the case of DNA, for example, some ligands bind to DNA through electrostatic interactions with phosphate groups, whereas others prefer major or minor grooves to bind. Finally, other ligands are intercalated between the base pairs [45,46]. Moreover, polyelectrolytes are more flexible than micelles and, according to the properties of the surrounding medium can suffer conformational changes that in turn produce changes in the binding constants of the ligands (reactants). For example, changes in the ionic strength will produce, changes in the binding constants for reactants having charges of opposite signs to those of the polymer [80,81]. In part, this effect can be ascribed to the screening effect of the ionic clouds of the polymer and ligand. However, this screening effect is not the unique effect of increasing ionic strength. This is clearly demonstrated by the fact that the binding constant of the neutral ligand of pyrene-1-carboxyaldehyde to DNA decreases when the ionic strength of the surrounding medium increases [82].This points to the idea that the conformation of DNA is altered by the salt (probably by the counterions of the salt). In fact, electrolytes can produce conformational changes in DNA dispersed in water by going from a random coil to a helix structure [83]. Neutral cosolutes, like alcohols, also produce changes in the binding constants [82] of ligands to DNA, as do surfactants [84]. In this case, this change is due to the conformal change of DNA from the random-coil to globular structure induced by the surfactant [85]. Obviously, the structural changes can be induced by the ligand (reactant) itself, which would produce a change in the binding constant of the ligand. This circumstance would give rise, at least formally, to cooperative effects [86]. Fortunately, equations of the Pseudophase Model are flexible enough to incorporate the above mentioned effect. It suffices to consider that the binding constants, in this case, depend on the concentration of the species in the reaction media producing the alteration on the DNA structure. In fact, the reactant(s) can induce a change in the DNA structure. In this case, the binding constant will be dependent on the adsorption degree of the reactant(s), that, in turn, will be dependent on the relation of the concentrations of the catalyst (DNA) and the reactant. If, as generally happens, the concentration of the reactant is fixed and the DNA concentration is changed, one will observe a dependence of the binding constant on the concentration of the catalyst. If the binding constant decreases when the concentration of DNA increases, this will be the consequence of a positive cooperativity. On the contrary, if the binding constant increases when the DNA concentration does so, the system shows a negative cooperativity [45].
Unless for the existence of cooperativity, the behavior of DNA as catalyst is quite similar to the effect of charged micelles, as are the effects of nanoparticles [60] and charged dendrimers [44], that is, changes in kobs can be described by Equation 3 or 62 with the binding constant appearing in these equations being true constants or parameters which depend on the concentration of the catalyst.
As mentioned previously, changes in the ionic strength in the solutions containing DNA influence the strength of binding of charged solutes. These changes follow the Lippard’s equation [59]. In the presence of a sodium salt this equation is:
![Molecules 15 04815 i164]()

In this equation Knel represents the non-electrostatic part of K and β is a constant dependent on the charge of the ligand. Knel can be obtained from the (linear) plot of log k vs. log[Nα+] and, once the non-electrostatic part of the binding is established, the electrostatic part can be determined (see Equation 78). In turn, one can write:
![Molecules 15 04815 i165]() in such a way that one can determine the value of the difference in the electrostatic potential between the solution and the region close to DNA where the ligand binds to it [22]. Following this approach, the values of ∆Ψ have been estimated using as probes [Ru(NH3)5Pz]2+ and [Co(NH3)5Pz]3+. The values of ∆Ψ for these probes are consistent, which gives support to the procedure.
in such a way that one can determine the value of the difference in the electrostatic potential between the solution and the region close to DNA where the ligand binds to it [22]. Following this approach, the values of ∆Ψ have been estimated using as probes [Ru(NH3)5Pz]2+ and [Co(NH3)5Pz]3+. The values of ∆Ψ for these probes are consistent, which gives support to the procedure.

Another kind of polymer with similar behavior to DNA are polypeptides [87,88]. Sanchez et al. employed lysine/glycine polypeptides with different degrees of polymerization. The different peptides produce a decrease in the rate of the reaction between [Co(C2O4)3]3- and [Ru(NH3)5Pz]2+. From these rate changes, the binding constant of the cobalt complex can be assessed. An interesting result from these studies is that the binding free energy, per monomer increases with the degree of polymerization. That is, the aggregation of monomers increases the free energy of binding. This increase in the negative free energy of binding represents a driving force for the polymerization process. Thus for n (number of monomer) 7, 14 and 21 the differences in the free energies of polymerization in the presence and in the absence of the cobalt complex are respectively. −9.5, −16.7 and −18.5 kJ/mol.
As to the effects of dendrimers on chemical reactivity, they behave as do DNA and other polymers such as polypeptides. Thus, in the presence of negatively charged dendrimers, the changes in the rate constant of the oxidation of [Ru(NH3)5Pz]2+ by ![Molecules 15 04815 i258]() (only the ruthenium complex binds to dendrimers) follow the expected trend according to the Pseudophase Model [44]. The same behavior is found when the oxidant is [Co(C2O4)3]3-, thus confirming that only the ruthenium complex binds to the dendrimers [89].
(only the ruthenium complex binds to dendrimers) follow the expected trend according to the Pseudophase Model [44]. The same behavior is found when the oxidant is [Co(C2O4)3]3-, thus confirming that only the ruthenium complex binds to the dendrimers [89].
 (only the ruthenium complex binds to dendrimers) follow the expected trend according to the Pseudophase Model [44]. The same behavior is found when the oxidant is [Co(C2O4)3]3-, thus confirming that only the ruthenium complex binds to the dendrimers [89].
(only the ruthenium complex binds to dendrimers) follow the expected trend according to the Pseudophase Model [44]. The same behavior is found when the oxidant is [Co(C2O4)3]3-, thus confirming that only the ruthenium complex binds to the dendrimers [89]. The effects of dendrimers on ligand substitution reaction have also been studied [15,90]. As in other kinds of reactions, the Pseudophase Model also holds for this type of process. It is interesting to note that in reference [90] the authors studied the influence of the dendrimers at several values of the ionic strength. By following the same procedure as in the case of DNA, the electrostatic and nonelectrostatic components of the binding constant were separated.
Nanoparticles exhibit similar behavior to polymers and dendrimers. Thus the kinetics of the oxidation of [Ru(NH3)5Pz]2+ by ![Molecules 15 04815 i257]() has been studied in the presence of gold nanoparticles caped with N-(2-mercaptopropyonyl)glycine [60]. The kinetics, when the concentration of nanoparticles is changed, follows the trend corresponding to the equation of the Pseudophase Model with a high binding constant for the union of the ruthenium complex to the nanoparticles. According to the authors there is some anticooperative character in this union. However, this anticooperativity can be only formal in the sense that it could be due to saturation effects at the surface of the catalyst. The authors worked at different ionic strength in such a way that they were able to separate the electrostatic and non-electrostatic components of the binding free energy. According to their results, the electrostatic component is the main driving force for the union.
has been studied in the presence of gold nanoparticles caped with N-(2-mercaptopropyonyl)glycine [60]. The kinetics, when the concentration of nanoparticles is changed, follows the trend corresponding to the equation of the Pseudophase Model with a high binding constant for the union of the ruthenium complex to the nanoparticles. According to the authors there is some anticooperative character in this union. However, this anticooperativity can be only formal in the sense that it could be due to saturation effects at the surface of the catalyst. The authors worked at different ionic strength in such a way that they were able to separate the electrostatic and non-electrostatic components of the binding free energy. According to their results, the electrostatic component is the main driving force for the union.
has been studied in the presence of gold nanoparticles caped with N-(2-mercaptopropyonyl)glycine [60]. The kinetics, when the concentration of nanoparticles is changed, follows the trend corresponding to the equation of the Pseudophase Model with a high binding constant for the union of the ruthenium complex to the nanoparticles. According to the authors there is some anticooperative character in this union. However, this anticooperativity can be only formal in the sense that it could be due to saturation effects at the surface of the catalyst. The authors worked at different ionic strength in such a way that they were able to separate the electrostatic and non-electrostatic components of the binding free energy. According to their results, the electrostatic component is the main driving force for the union.The effects of AlOOH nanoparticles as microcatalyst have been studied by Sánchez et al. [91]. The study was performed at different ionic strength using as a probe the kinetics of the oxidation reaction of [Ru(NH3)5Pz]2+ by ![Molecules 15 04815 i258]() at pH = 5.4. In this case, it is the anion the ligand that binds to the nanoparticle. The rate of reaction changes by a factor of about ten in the presence of nanoparticles; this change decreasing as the ionic strength increases. Again, as in the case of gold nanoparticles, the main component of the free energy of binding is the electrostatic part.
at pH = 5.4. In this case, it is the anion the ligand that binds to the nanoparticle. The rate of reaction changes by a factor of about ten in the presence of nanoparticles; this change decreasing as the ionic strength increases. Again, as in the case of gold nanoparticles, the main component of the free energy of binding is the electrostatic part.
at pH = 5.4. In this case, it is the anion the ligand that binds to the nanoparticle. The rate of reaction changes by a factor of about ten in the presence of nanoparticles; this change decreasing as the ionic strength increases. Again, as in the case of gold nanoparticles, the main component of the free energy of binding is the electrostatic part.So, it can be said that polymers and related receptors behave in a similar way to that of micelles, although in some cases they can exhibit a somewhat more complex behavior due to the conformational changes that the process of reactant binding can induce in the polymer. But even in these cases, the Pseudophase Model can still be applied, although the binding “constant” would be variable, that is, it would dependent on the [receptor]/[ligand] ratio.
3.4. Solutions Containing Saturable Receptors
Now the case of microheterogeneous catalysis produced by receptors such as cyclodextrins and similar receptors will be considered. First of all, it is important to realize that there are no significant differences between these kinds of receptors and, for example, micelles. Thus, the Pseudophase Model can be applied to the interpretation of data in, for example, cyclodextrins solutions [44]. However, we have considered pertinent the inclusion of a separate section for these kinds of receptors, that is, of receptors in which the number of guest molecules is small and well defined (one, or two, in some cases) [92]. Moreover, as mentioned previously, reactions between two reactants bound to different receptors are possible, in such a way that Equation 63 would apply.
However, for low concentrations of the receptor and/or big binding constant to saturable receptor, application of equations such as Equations 3 or 63 must be done with caution: strictly speaking, in these equations [C] represents the concentration of free catalyst. When this concentration is big enough, it can be taken as the total concentration of C. However, some saturable receptors (as for example β-CD) present a low solubility in such a way that the concentration of a free catalyst can be quite different from the total concentration. Under these circumstances, if one uses equation 3 with [C] = total concentration of the catalyst, a variable (apparent) binding constant will be obtained, and, for a fixed ligand concentration this constant will increase when the concentration of C does so, in such a way that the system would behave, formally, as a system in which the ligand/catalyst binding was anti-cooperative. Thus, it can easily be shown that the apparent binding constant Kapp defined as:
![Molecules 15 04815 i166]() is related to the true binding constant, K, by:
is related to the true binding constant, K, by:
![Molecules 15 04815 i167]() in such a way that Kapp will only be equal to K when [Rf] = 0, that is for high enough concentrations of C for producing a negligible concentration of free reactant.
in such a way that Kapp will only be equal to K when [Rf] = 0, that is for high enough concentrations of C for producing a negligible concentration of free reactant.


Considering now the effects of cyclodextrins (and related systems) on the reactivity of bound reactants, one can find examples in which this reactivity increases or decreases in relation to free ligands. In fact, sometimes small modifications of the receptor can change the effect of binding on the reactivity. Thus, β-CD decreases the rate of oxidation of [Fe(CN)54phepy]3- by [Co(NH3)4(H2O)2]3+ whereas the rate of this reaction increases in the presence of 2-hydroxypropyl- β-CD. On the other hand, for a given cyclodextrin, changes in reactivity can also be tuned through the addition to the reaction medium of substances, such as co-solvents or salts, which change the binding constants of the reactants [93,94,95,96,97,98].
Cyclodextrins are by far the most studied saturable receptors. Thus, their effects on a variety of processes have been studied. The effects of cyclodextrins on electron transfer processes have been considered above. The effects of these kinds of receptors on biomimetic reactions have been reviewed [99] as well as studies of cyclodextrin effects on organic reactions [100,101,102,103,104,105,106], redox reactions [107,108,109], ligand substitution reactions [110], etc., in such a way that the effects of this kind of catalyst on practically any type of reaction have been studied.
Other saturable receptors have been less studied than cyclodextrins. Among these receptors calixarenes might be the most employed. These are macrocyclic compounds obtained through condensation of phenols with formaldehyde. They can act as hosts for ions and small neutral molecules. The selectivity and efficiency of the binding process depend on the ring size, on the nature of the attached groups (to the phenol component), and on the stereochemical conformation of these versatile macrocyclic molecules, that can generate a large number of conformations. This conformational freedom is a key factor in the stabilization of the particular host-guest interactions between calixarenes and their guests [111]. This conformational flexibility increases for the larger rings.
Calixarenes have been employed for alkali-metal cations transport through hydrophobic organic phases and the binding constant of the complexes have been measured [112], as well as the influence of additives on the complexation by calixarenes [104,111].
Supramolecular structures formed by the condensation of two or more calixarene units were prepared and their properties studied. Casnati et al. [113] have reviewed the synthesis and properties of these supramolecular structures. They studied specifically the structures formed by calixarenes endowed with α-amino acids. Curiously, N-linked peptidocalix[4]arenes show properties quite different from those of the corresponding C-linked derivatives. The latter show a tendency to form self-assembled nanotubes in the solid state. An interesting property of these macrostructures is that hydrogen bonding, donor and acceptor groups of the aminoacid residues and the cavity of one calyx [4]arenes act cooperatively in guest binding in nonpolar solvents but not in water, where hydrophobic interactions dominate.
The number of studies on the influence of calixarenes on the reactivity of the guest is relatively small compared to the corresponding studies on cyclodextrins. Among these studies Lopez-Cornejo et al. [114] studied the influence of p-sulfonatocalix[n]arenes (n = 4, 6, 8) on the kinetics of the oxidation of [Ru(NH3)5Pz]2+ by [Co(C2O4)3]3-. According to the authors, only the ruthenium complex interacts with the receptor. The kinetic results can be rationalized using as a starting point of the Pseudophase Model. The effect of salts on the complexation process and on reactivity was also studied. The authors concluded that the effect of the receptors on the kinetics depends on the flexibility of the host.
Lopez-Cornejo et al. [115] have studied the effects of 4-sulfocalixarene on the kinetic of the oxidation of [Ru(NH3)5Pz]2+ by [Co(C2O4)3]3- and ![Molecules 15 04815 i258]() . In this case the complex [Ru(NH3)5Pz]2+ /calixarene has a 1:2 stoichiometry. When salt (NaCl) is added, a mixture of complexes with 1:1 and 1:2 stoichiometries is formed. Again, the Pseudophase Model can be used for rationalization of the results.
. In this case the complex [Ru(NH3)5Pz]2+ /calixarene has a 1:2 stoichiometry. When salt (NaCl) is added, a mixture of complexes with 1:1 and 1:2 stoichiometries is formed. Again, the Pseudophase Model can be used for rationalization of the results.
. In this case the complex [Ru(NH3)5Pz]2+ /calixarene has a 1:2 stoichiometry. When salt (NaCl) is added, a mixture of complexes with 1:1 and 1:2 stoichiometries is formed. Again, the Pseudophase Model can be used for rationalization of the results.To conclude this section, one can say that saturable receptors do not present special differences in relation to the nonsaturable in the sense that the Pseudophase Model can be employed in the rationalization of the effects of these receptors on reactivity.
3.5. Complex Systems
In this section the effects of catalyst mixtures will be considered. Obviously, these mixtures produce more complex catalytic effects than those of simple catalysts considered previously. However, they offer the possibility of fine tuning of the catalytic effects. Moreover, they are also interesting as model for complex polytopic single catalysts.
In a mixture of catalysts two possibilities arise. The first one is that the two (or more) catalysts do not interact. This case implies that the reactant will interact with the two catalysts independently. A situation of this type arises, for example, for a mixture of neutral cyclodextrins. This system has been investigated by some of the present authors who studied the effects of mixtures of methyl-β-cyclodextrin (MβCD) and 2-hydroxypropyl-β-cyclodextrin (HβCD) on the reaction of oxidation of [Fe(CN)5(4-phepy)]3- by [Co(NH3)4(H2O)2]3+[76]. In this case, only the iron complex interacts with the cyclodextrins (by encapsulation of the 4-phepy ligand). For this reaction, when only one catalyst is present, an increase in the rate of reaction is observed, more marked for MβCD. As expected, the effect of the catalysts follows the simple law represented by Equation 3. In the case of mixtures of cyclodextrins, the experiments were performed for a fixed concentration of MβCD and a variable concentration of HβCD. For [MβCD] = 2.1 × 10−3 mol·dm-3, the effect of increasing [HβCD] is the expected one: an increase in the reaction rate. However, for [MβCD] = 3.8 × 10−3 mol·dm−3 no effect of [HβCD] was observed. Finally, if [MβCD] = 2.5 × 10-2 mol·dm−3, an increase of [HβCD] produces a diminution of the reaction rate. In all cases, the results can be described by the equation:
![Molecules 15 04815 i168]()

According to this equation, the observed behavior is the result of a competition of the two catalysts for the iron complex.
The second possibility for a mixture of catalysts corresponds to the case in which the catalysts interact among themselves. This happens, for example, in mixtures of surfactants (neutral + charged or anionic+cationic surfactants). These mixtures produce micelles in which the surface charge density can be controlled by changing the proportion of the surfactants in the mixture. This topic has been studied by Moya et al. [65,116,117,118,119,120] Thus, in reference [116] the authors studied the effects on the base hydrolysis of 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane and 2-(p-nitrophenyl)ethyl bromide of mixed micelles of tetradecyltrimethylammonium bromide and penthanol. The presence of the alcohol produces a change in the ionization degree of the micelles as well as a change in the cmc. In all cases the results can be interpreted using the Pseudophase Ion Exchange Model [55].
The spontaneous hydrolysis of phenyl chloroformate in the mixed micelles has been studied by Moya et al. [117]. An interesting result is that the effect of the charge (of micelles) is not the only factor responsible for the variation in the reaction rate: depletion of water in the interface region, as well as the structural changes in this region also influences reactivity. Among these factors, in complex systems constituted by surfactants and alcohols, are: an increase in the volume of the micellar interfacial region upon increasing alcohol concentration and a decrease of the polarity in this region caused by the presence of alcohol [118]. The applicability of the Pseudophase Model in solutions containing mixtures of surfactants (CTACl and TX-100) has been shown by Moya et al. [119] using as a probe the reaction between methyl-4-nitrobenzenesulfonate and Cl-. In this mixture, according to the authors, the neutral surfactant has little influence on the characteristics of the interfacial region where the reaction takes place.
In a kinetic and structural study in solutions containing SDS/Tween 20 and SDS/SB3-12 mixtures of surfactants, Moya et al. [120] showed that in these mixtures two types of micellar aggregates are formed upon increasing surfactant concentration. However, kinetic data do not show discontinuity, which was interpreted by the authors as a consequence of the fact that the interfacial region is similar in the two kinds of aggregates. Morphological transitions in aggregates formed in solutions containing mixed surfactants have also been observed in mixtures of single chain and gemini surfactants [70]. These transitions were reflected in kinetic data.
The systems constituted by mixed surfactants have interesting properties outside the catalysis field. Thus, micelles constituted by mixtures of anionic and cationic surfactants (catanionic micelles) have been used as a model to mimic biological membranes in the presence of anaesthetic alcohols [121]. A remarkable analogy was found between the influence of the alcohols on the solubility patterns of the catanionic micelles and on the anaesthesia of tadpoles. According to the authors, the alcohols are incorporated into the biological (lipid) membranes in the anaesthetic process. On the other hand, the changes in solubility of catanionic micelles are also due to the incorporation of alcohol into the micelles. This incorporation, in both cases, requires the presence of a relatively long chain in the alcohols, in such a way that they can enter into the hydrophobic parts of membranes and catanionic micelles.
An interesting complex system constituted by surfactants (SDS and TTABr) and β-cyclodextrin (native β-CD and capitol) has been studied by Garcia-Rio et al. [122]. The kinetic effects of such systems were explored using as a probe the hydrolysis of 4-methoxybenzenesulfonyl chloride (MBSC). Micelles and cyclodextrins (separately) influence the hydrolysis in the expected way: kinetic data follow Equation 3 when the micellar or cyclodextrin concentrations are changed. However, in mixed surfactant/CD systems the behavior is more complicated. (In this case, the receptors interact forming inclusion complexes, pseudorotaxanes). The study was performed at three fixed cyclodextrin concentrations and variable surfactants concentrations. The observed rate constant when the surfactant concentration is changed goes through a maximum, in spite of the fact that both, cyclodextrins and surfactants, separately, decrease the rate of hydrolysis. Changes in the rate of the hydrolysis follow equation [122]:
![Molecules 15 04815 i169]()

In this equation KCD represents the binding constant of the reactant to cyclodextrin and Km the binding constant to the micelles. [CDf] represents the concentration of free cyclodextrin, that is, the concentration of this receptor that does not form a pseudorotaxane with the surfactant. On the other hand, Km is the binding constant of MBSC to micelles. The concentration of free cyclodextrins is calculated by solving the equation:
![Molecules 15 04815 i170]()

[CD] being the total cyclodextrin concentration and:
![Molecules 15 04815 i171]()
![Molecules 15 04815 i172]()
![Molecules 15 04815 i173]()



In the previous equations [T] is the total surfactant concentration and KCD/T is the binding constant of the surfactant to cyclodextrin to form the pseudorotaxane:
![Molecules 15 04815 i174]()

Interpretation of the results is quite simple: both, cyclodextrins and micelles produce a decrease in the rate of MBSC hydrolysis. So, in the presence of a given amount of cyclodextrin, the rate of reaction is lower than it is in water. When the surfactant is added to the solution, it competes with MBSC for the cyclodextrin. Thus, some MBSC is liberated and reacts more rapidly. When all the cyclodextrin is occupied by the surfactant, the maximum reaction rate is reached. After this point, a new addition of surfactant implies the formation of micelles that decrease the rate of the reaction. It is interesting to note that similar behavior is observed in mixtures of surfactants and DNA [123].
Among the aggregates formed from more than one component, the ones presented by Lee et al. [124] are interesting. These aggregates are water soluble toroids with a hydrophobic cavity formed by co-assembly of laterally grafted rod amphiphiles. It is interesting that the toroids are tubularly stacked when a guest (C60) is encapsulated in the toroids. For the moment, there are no studies on the influence of these aggregates on reaction rates.
To conclude this section: the possibility of preparing aggregates with more than one component is practically unlimited. These aggregates are of interest because they can be used in many fields, including solar energy conversions (see Section 4.5). In the field of microcatalysis, as we have seen in the examples considered in this section, the door to fine tuning of the reaction rates is opened.
4. Microheterogeneous Catalysis with Participation of Excited States of Reactants
4.1. General
The study of photochemical reactions in the presence of receptors that can catalyze them is a long standing matter. In this case, however, given that photochemical reactions are, generally speaking, rapid processes, the interest does not come from the catalytic effect itself. The main interest originated from the objective of finding artificial photosynthetic systems that mimic the natural ones [125]. So, it has been established that “any practical system for solar energy conversion and storage will involve heterogeneous reactions at some stage of the process” [126]. This follows from the need to isolate the primary products of the light-induced charge separation to avoid the rapid recombination of these rich in energy products (back reaction). Obviously, if these products are produced in different phases, the problem of the back reaction can be solved. Thus, consider a photoinduced electron transfer between two neutral reactants, which is produced in a solution containing, for example, negatively charged micelles:
![Molecules 15 04815 i175]()
![Molecules 15 04815 i176]()


The reaction products bear different charge signs in such a way that the oxidized donor, D+, will have a tendency to go to the negatively charged micelles whereas the negatively charged product, A-, would be partially or totally eliminated from the region close to the micelles. This separation of the products would eliminate (or reduce) the back electron transfer:
![Molecules 15 04815 i177]()

On the other hand, the presence of receptors in a medium where a photochemical reaction happens, can promote a true catalytic effect giving rise to product selectivity [127], which is of interest in relation to synthetic chemistry.
Photochemical reactions can be influenced by all the effects considered in Section 1 of this review in relation to thermal processes. Moreover, these changes can be detected in many cases because the incorporation of a species into a receptor produces changes in its photochemical properties, such as lifetime of the excited state, emission spectrum, etc. These changes for polytopic receptors depend on the kind of site where the excited species is located. This circumstance implies the possibility of using the changes in photophysical properties of bound species (with respect to free species) as probes for investigations of environment properties and characteristics of the receptors. Thus, the dynamics of solvation of bound species has been investigated using fluorescent probes [128]. Fluorescence depolarization studies provide information concerning the mobility of the probe and its orientation (relative to receptor) as well as the mechanical characteristics of the probe environment. In this way, the microviscosity of the region where the probe is solubilized can be estimated [129]. In the field of micellar solutions, the use of fluorescent probes is well documented. Thus, critical micellar concentration, aggregation or the distribution of solutes among the different pseudophases have been studied using fluorescence techniques [130].
An important question in relation to microheteogeneous catalysis, when one of the reactants is in an excited state, is related to the applicability of the models developed in previous sections. These models are based on the assumption of an equilibrium distribution of the reactant between bound and free states (Equation 1). This, of course, implies that the rate of the reaction in which R participates is slow in relation to the forward and reverse processes in Equation 1. This introduces the question: are previous models applicable given that the photochemical reactions are generally rapid reactions? Moreover, in the case these equations hold, in the sense that experimental data can be fitted to them, do the equilibrium constants appearing in these equations correspond to ground or to excited states? In relation to these questions, two limiting cases will be considered:
(1) In the first case (fast exchange limit) it will be assumed that the kinetics of the interaction of the excited species with the receptors is rapid enough, in the forward and reverse directions, to assume that this process:
![Molecules 15 04815 i178]() is at equilibrium.
is at equilibrium.

(a) It will be considered, firstly, that the reactant, R*(free or bound) can react with a second species (the quencher) Q. This reaction is followed by measuring the fluorescence decay of R* after (for example) a laser pulse that produces it:
![Molecules 15 04815 i179]()

It can be shown [131] that under these circumstances the observed rate constant for the decay of the emission is given by:
![Molecules 15 04815 i180]()

In Equation 143,
![Molecules 15 04815 i181]()
![Molecules 15 04815 i182]()


koi being the spontaneous decay rate constant and kqi the rate constant corresponding to the quenching reactions (i = f, b).
Thus, one can write:
![Molecules 15 04815 i183]() where:
where:
![Molecules 15 04815 i184]()
![Molecules 15 04815 i185]()



Consequently, the spontaneous (ko,obs) as well as the quenching (kq,obs) rate constants will follow the equations of the Pseudophase Model, but with K*, the equilibrium constant corresponding to the partition of the excited species, instead of K (see Equation 1).
(b) Now suppose that one is performing steady-state measurements, in such a way that the kinetics is monitored making use of the Stern-Volmer equation (and the lifetimes in the absence of the quencher). Under the same circumstance that in the previous case, the observed Stern-Volmer constant is given by [131]:
![Molecules 15 04815 i186]()

In this case, the observed Stern-Volmer constant is also consistent with the equation of the Pseudophase Model. However, only if kof = kob = ko one can write:
![Molecules 15 04815 i187]() in such a way that one recovers Equation 146b.
in such a way that one recovers Equation 146b.

(2) In the second case, the slow exchange limit of R*, will be considered.
(a) In this limit, the decay of emission intensity (after a laser pulse) will be biexponential [132]:
![Molecules 15 04815 i188]() as corresponding to the fact that, once the fluorophore is excited at t = 0, the concentrations of Rf* and Rb* correspond to the equilibrium distribution of the ground state and there is no exchange between the excited state populations.
as corresponding to the fact that, once the fluorophore is excited at t = 0, the concentrations of Rf* and Rb* correspond to the equilibrium distribution of the ground state and there is no exchange between the excited state populations.

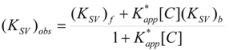
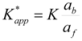
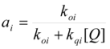
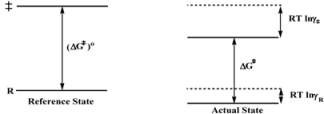{kind=link}
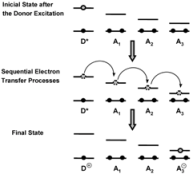{kind=link}
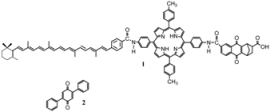{kind=link}
Equation 150 is formally identical to the one predicted by the Pseudophase Model. However, ![Molecules 15 04815 i260]() depends on [Q] (through af and ab). Thus, unless koi>>kqi (i = f, b), curved Stern-Volmer plots may result in this case.
depends on [Q] (through af and ab). Thus, unless koi>>kqi (i = f, b), curved Stern-Volmer plots may result in this case.
 depends on [Q] (through af and ab). Thus, unless koi>>kqi (i = f, b), curved Stern-Volmer plots may result in this case.
depends on [Q] (through af and ab). Thus, unless koi>>kqi (i = f, b), curved Stern-Volmer plots may result in this case.Previous examples, in which the equation of the Pseudophase Model holds, at least formally, are, in fact, extreme and simplified cases. Apart from the assumptions specifically indicated, in the deduction of previous equations it was assumed that the quencher does not interact with the receptor, in such a way that only one concentration of this species is used.
In the general case, when the kinetics of the union/separation of the reactant (and or the quencher) to the catalyst are coupled to the kinetics of the spontaneous decay and/or quenching process the situation is much more complex. Thus, Yoshida et al. [133] obtained the equation of Io/I for the case in which only R interacts with the catalyst:
![Molecules 15 04815 i192]()

Previous paragraphs give an idea of the complexity of microheteogeneous catalysis when the reactant is in an excited state. This complexity implies that in order to obtain information on this kind of system one must take care and make a judicious choice of reactant (or the reactant/quencher couple). However, if this is done, as will be seen in the following sections, one can obtain rich information on the process and on the catalyst and its environment.
4.2. Micellar Solutions
As mentioned previously, the kinetics of microheterogeneous catalysis when one of the reactants is in an excited state is complicated by the coupling of the reactive and diffusive steps. This complication, however, gives information on the characteristics of the diffusive steps. In order to explore this field, several treatments have been developed.
Thus, Infelta et al. [134,135] considered the case in which the reactant, in its excited state, is present only in the micellar pseudophase and the quencher is distributed between the two pseudophases present in the system:
![Molecules 15 04815 i193]()

Under these circumstances, the decay of R* follows the equation:
![Molecules 15 04815 i194]() where F(t) represents the rate of decay of R* in the absence of the quencher, given by:
where F(t) represents the rate of decay of R* in the absence of the quencher, given by:
![Molecules 15 04815 i195]() and G(t) is the contribution of the reaction (quenching) to the decay of Rb*:
and G(t) is the contribution of the reaction (quenching) to the decay of Rb*:
![Molecules 15 04815 i196]() where [Q]f represents the quencher concentration in the aqueous pseudophase. From the previous equations it follows that:
where [Q]f represents the quencher concentration in the aqueous pseudophase. From the previous equations it follows that:
![Molecules 15 04815 i197]()
![Molecules 15 04815 i198]()
![Molecules 15 04815 i199]()






The previous equation gives an idea of the complexity resulting from the coupling of the reactive and diffusive events. However, these equations simplify under some circumstances. Thus, for long times, when ![Molecules 15 04815 i259]() , it follows that:
, it follows that:
![Molecules 15 04815 i200]()
 , it follows that:
, it follows that:

This equation, working at different [Q]f , permit us to have C, B and kob and, once one has determined these parameters, it is possible to calculated kq+ and kq-:
![Molecules 15 04815 i201]()
![Molecules 15 04815 i202]() (notice that KQ can be determined in an independent experiment). So, all the rate constants determining the kinetics of the decay of Rb* can be obtained.
(notice that KQ can be determined in an independent experiment). So, all the rate constants determining the kinetics of the decay of Rb* can be obtained.


Infelta et al. considered other scenarios, but as in the example considered previously, it is always possible, by selecting the conditions of the experiments, to determine all the kinetic parameters in the system. In fact, the model of Infelta et al. is an approximate model, as pointed out by Barzykin and Tachiya, due to the fact that a Poisson occupation of the micelles is assumed, and many-body effects, which can be important at high micelle concentration, are neglected. However, Barzykin and Tachiya [136] concluded that the approximate Infelta model works fairly well in most of the cases.
Almgren et al. [137] considered the situation in which the reactant can exchange its location whereas the quencher is only soluble in the aqueous pseudophase:
![Molecules 15 04815 i203]()
![Molecules 15 04815 i204]()
![Molecules 15 04815 i205]() and, when the quencher is added to the system:
and, when the quencher is added to the system:
![Molecules 15 04815 i206]()




According to this reaction scheme,
![Molecules 15 04815 i207]() and the decay of [Rb*(t)] is given by:
and the decay of [Rb*(t)] is given by:
![Molecules 15 04815 i208]()
![Molecules 15 04815 i209]()



This equation permits us to obtain the different rate constants working at different concentrations of Q and/or C. Extensions of the Almgren model to other scenarios can be found in references [137] and [138].
Another reaction scheme was considered by Quina and Toscano:
![Molecules 15 04815 i210]()

In this case there is no exchange between R* in the micellar and aqueous pseudophases, that is, the situation corresponds to the slow exchange limit. If, as assumed by Quina and Toscano, the quencher has no access to the bound reactant, it is obvious that the decay of Rb* is independent of the quencher concentration. Thus, for the aqueous pseudophase the Stern-Volmer equation holds:
![Molecules 15 04815 i211]()

If the excitation wavelength corresponds to the isosbestic point of the absorption spectrum (in the presence of micelles), the total quantum yield, in the absence of the quencher, is:
![Molecules 15 04815 i212]() and when the quencher is present:
and when the quencher is present:
![Molecules 15 04815 i213]()


From the previous equations it follows that:
![Molecules 15 04815 i214]()

Quina and Toscano also considered the case in which exchange in the excited state is possible, but this exchange is not rapid enough to be at equilibrium:
![Molecules 15 04815 i215]()

If, as the authors assumed, [Qf] = [Qb] = [Q], the apparent rate constant for the fluorescence decay is given by:
![Molecules 15 04815 i216]()

In this equation kof and kqf can be obtained directly from experiments in the absence of receptors, C. The other rate constants in Equation 170 can be obtained directly from data corresponding to experiments at different [C] and/or [Q].
The equations of Quina and Toscano have been checked in micellar solutions [139] as well as in bile salt aggregates [140,141] and cyclodextrins [142]. All previous models for processes in micellar solutions have been checked many times with experimental results. According to this, it seems reasonable to conclude that these models work well provided that their basic assumptions are accomplished in the system under study. However, micellar solutions with more than two components can have complex behavior, with complicated phase diagrams. It is clear that, if one moves to different regions of the phase diagram, the behavior of the reaction can change, this reflecting the structural complexity of the system. This circumstance is clearly seen in the case of the anthracene quenching by N,N-diethylaniline in the SDS/benzyl alcohol/water system [143].
In the opposite extreme of simplicity, one can mention the quenching of the excited state of [Ru (phen)2bps] by 4,4’-diheptylviologen in the presence of SDS, studied by Hackett and Turro [144]. This system was selected to obtain “the electron transfer rate constants at the micelles surface without the use of complex equations”. In this case, the neutral ruthenium complex is expected to reside in the aqueous phase, whereas the quenchers are expected to be anchored to the hydrophobic interior of the micelles through their heptil chains, with their redox active groups at the surface of the micelles. So the quenching is only possible at this surface.
In relation to electron transfer processes, other authors studied this kind of reaction in micellar systems, using the Marcus treatment [2].. Thus, Dressik et al. [145] have studied the oxidation of Ru(II) photoexcited complexes in micellar solutions. According to them, there is an increase of the reorganization free energy as a consequence of the interaction complex/micelle. This behavior has been observed by other authors [11]. On the other hand, Weidemaier et al. [146,147,148] have developed a model for electron transfer at micelles. This model can explain the influence of the length of the surfactant on the reorganization free energy of the electron transfer process.
Barzikyn and Tachiya [149] have also studied the problem of electron transfer reactions in micellar systems. They presented a theory of diffusion assisted reaction on the micelles’ surfaces and applied this treatment to photoinduced electron transfer. The results of the treatment can be obtained in a closed matrix form that, according to the authors, can describe virtually any reaction in any geometry. A relatively similar treatment has been applied by Ranagathan et al. [150].
4.3. Polymer Solutions
Polymers can act as catalyst showing behavior similar to micelles, as established in section 3.3. However, the interest in photochemistry and photophysics of ligands bound to polymers comes not so much in relation to catalysis as in relation to some applications such as the fabrication of sensors [151]. Moreover, in many cases photochemical behavior has been studied under conditions in which the ligand is covalently bound to polyelectrolytes. Thus, Morrison et al. [152] studied the photochemistry of arenes covalently bound to polyelectrolytes (so, only Rb* is present in the system), when the quencher is distributed between the polymer and the continuous phase. In this case, the arenes can be placed in different kinds of sites, in the sense that they are not equally accessible to the quencher. These differences in accessibility, in this case, are the consequence of conformational changes of the polymers, in such a way that the photochemistry of the ligand gives structural information on the polymer. It is interesting to note that, in this case, for low quencher concentration, Stern-Volmer behavior is observed. However, when the quencher concentration increases, deviation of this behavior is observed, in such a way that:
![Molecules 15 04815 i217]()

Correspondingly, the fluorescence decay is monoexponential at low quencher concentrations and multiexponential at higher concentrations.
According to the authors, there are two possible causes for this behavior: (1) conformational changes in the receptor, caused by the quencher, or (2) a non-homogeneous quencher distribution close to the polymer. Calling A and B the two sites and using a two state model in which the probe is protected from the quencher at site B, the authors derived the following equation:
![Molecules 15 04815 i218]()
![Molecules 15 04815 i219]() which is in agreement with the experimental facts.
which is in agreement with the experimental facts.


On the other hand, Chu and Thomas [153] also studied the quenching of chromophore covalently bound to the polyelectrolyte. The chromophore (pyrene) was located in poly(methacrylic acid) in three different ways: (a) as a guest molecule by simple solubilization in its closed compact form; (b) covalently bound to the polymers ends and (c) randomly bound to the polymer chain (with no more than one pyrene unit per polymer chain in this and b cases). In this case, the charge of the polymer and its conformation can be changed through pH variations. As quenchers, Tl+ and I− were used. These quenchers behave similarly when the polymer is in compact conformation (until pH = 4). However, once the polymer opens, there are differences between Tl+ and I−. In the first case, the negative charge of the polymer increases the local concentration of the quencher, which leads to a dramatic increase of the quenching. On the contrary, iodide ions are repelled by the open (negatively charged) polymer, thus decreasing the quenching process.
The same system, pyrene/poly(methacrylic acid) was also studied by Turro et al. [154,155] and at higher pyrene density (on the polymer), in such a way that pyrene/pyrene interactions were possible. As quenchers, nitromethane, Tl+, Cu+2, I− and 4-dimethylaminopyridine were employed. Additionally, the quenching of pyrene bound to the monomer was studied. In the latter case, plots of Io/I vs. [Q] were linear for all the quenchers, but the Stern-Volmer constants only agreed with those from τo/τ vs. [Q] when Q = nitromethane, Tl+, and I− but not in the other cases.
The behavior of these two quenchers was attributed by the authors to static quenching in spite of the linear character of their corresponding Stern-Volmer plots. In fact, a case of static quenching would imply:
![Molecules 15 04815 i220]() where KQ/R is the binding constant fluorophore/quencher (in the ground state) and KSV the true Stern-Volmer constant (that would be obtained from τo/τ vs. [Q]).
where KQ/R is the binding constant fluorophore/quencher (in the ground state) and KSV the true Stern-Volmer constant (that would be obtained from τo/τ vs. [Q]).

Now consider the case of an efficient quencher present at a low concentration. In this case the term containing [Q]2 can be small, in such a way that a linear Stern-Volmer plot would be obtained. When pyrene is bound to polymer (instead of monomer) the quenching is less efficient, which was considered to be a consequence of the slow diffusion of the quenchers towards Rb* in the case of the polymer (this implies a total or partial control by diffusion). In this case, differences between the two types of quenchers also appear, the Stern-Volmer plots being linear for nitromethane, Tl+ and I− and curved for Cu+2 and 4-dimethylaminopyridine. Excimer fluorescence is also observed due to the intrachain interaction of pyrene groups [154,155]. In fact, polymers can produce an enhancement of formation of excimers and exciplexes if they produce an increase of the local chromophore concentration. This question has been considered by Turro et al. [156] who studied the influence of poly(styrene sulfonate) on the excimer formation of (α-naphtylmethyl)ammonium chloride. The kinetic analysis of excimer formation, in pure water and in solutions containing the polymer, was made from the following reaction scheme:
![Molecules 15 04815 i221]()

According to the authors the decay of fluorescence of monomer, IM, and excimer, ID, are given by:
![Molecules 15 04815 i222]()
![Molecules 15 04815 i223]()


CM and CD being the concentrations of monomer and excimer, respectively and A, λ1 and λ2 are the function of the rate constants shown in the scheme. In fact, λ1 and λ2 can be obtained from the decay curves. From them, taking into account that:
![Molecules 15 04815 i224]()

k1 can be obtained from the (linear) plot of λ1 + λ2 vs. [M]. Results in this paper reveal that the polymer increases k1 by a factor of 103, although its effect on k-1 is insignificant.An interesting case in which R* is long-lived enough for maintaining the equilibrium distribution between Rb* and Rf* has been studied by Wensel et al. [157] who considered the effect of DNA on the rates of energy transfer between ions. According to the authors, in the circumstance of rapid exchange between Rb* and Rf* the decay is monoexponential, as given by:
![Molecules 15 04815 i225]() in such a way that, in the presence of DNA the following equation holds:
in such a way that, in the presence of DNA the following equation holds:
![Molecules 15 04815 i226]()


k being the decay rate constant for a given quencher concentration and ko = 1/τo. C+ is the total concentration of cations (R, Q and counterions). On the other hand, Arkin et al. [158] report the quenching of photoexcited [Ru(phen)2ddpz]+2, which binds by intercalation by [Ru(phi)2bpy]+3 and [Ru(phe)2phe]+3. The Stern Volmer equation holds for these processes in micellar (SDS) solutions, but the plot of Io/Ivs. [Q] are curved in the presence of DNA, as corresponds to a static quenching process. In the case of micelles, the observed rate constant is given by:
![Molecules 15 04815 i227]() where V is the partial molar volume of the surfactant in the micelle and Ka and Kb are the binding constants of the fluorophore and the quencher to the micelle. The differences observed for DNA and micellar solutions are indicative of the fact that the structure of DNA plays a central role in mediating electron transfer reactions: the π-stack of DNA permits an intimate electron coupling between the donor and the acceptor which is absent in the micelles.
where V is the partial molar volume of the surfactant in the micelle and Ka and Kb are the binding constants of the fluorophore and the quencher to the micelle. The differences observed for DNA and micellar solutions are indicative of the fact that the structure of DNA plays a central role in mediating electron transfer reactions: the π-stack of DNA permits an intimate electron coupling between the donor and the acceptor which is absent in the micelles.

In relation to photoinduced electron transfer between intercalated reactants in DNA, Fukuzumi et al. [159] investigated the driving force dependence of the quenching (by electron transfer) of ethidium bromide to and a series of intercalators. They observed that the (electron transfer) quenching rate constants showed behavior in agreement with the Marcus treatment of nonadiabatic intramolecular electron transfer reactions. By using intercalators with different redox potentials they showed that some of these reactions were in the so-called inverted region, that is, kq decreases as the driving force increases.
Additional complications that the use of polymers as receptors introduces into the system (cooperativity and conformational changes being the most stressed), are reflected in the lower number of papers concerning this topic compared to the number concerning micelles. Besides, the bulk of research is based on the development of polyelectrolyte theories or systems to probe biological important polymers, such as DNA.
4.4. Solutions Containing Saturable Receptors
As mentioned above when dealing with the effects of this kind of receptor in processes in which the reactant(s) are in the ground state, for a given guest molecule, cyclodextrins and related receptors can influence reactivity in different ways. One of the factors influencing the action of this kind of catalyst is the cavity size of the host. This is clearly seen in the work of Yorozu et al. [160] who studied the effect of α, β and γ cyclodextrins on the photochemistry of β-naphthol. In this case, given the lifetime of the excited reactant, the excited naphthol decay in the same situation (bound or free) where it appears. Under these circumstances, parameters ab and af, defined in Equation 151b, are the same (ab = af = 1) in such a way that the distribution of free and bound naphthol in the excited state is the same as in the ground state. This is supported by the fact that the distribution of the β-naphthol determined from absorbance measurements is the same as fluorescence techniques.
The authors performed their studies in two limiting situations: (i) [Rf] = [R], [Rb] = 0 and (ii) [Rb] = [R], [Rf] = 0 (at a high enough cyclodextrin concentration for encapsulating all the naphthol). The acid dissociation constant of β-naphthol (Ka*) decreases in the presence of cyclodextrins and depends on the type of receptors (α-, β- or γ-CD). However, the diminution of Ka* is different for the three receptors ((Ka*)α<(Ka*)β<(Ka*)γ), which is due to the differences in the inclusion geometry: for α-CD the OH group of naphthol is placed in the cavity of the receptor, whereas in the case of β- and γ-CD the OH group is partially exposed to water and more so for γ-CD. The authors also studied the quenching of β-naphthol by iodide ion. In this case, the naphthalene group is more exposed in α-CD and the quenching is more effective for this receptor.
The presence of substituents also has influence on the action of cyclodextrins. Thus, Andrade-Dias et al. [161] have shown the differences between the interactions of sodium decanoate with native and methylated cyclodextrins. According to the authors, the differences arise from the distorted conformations associated with the strong tilt of the axes of one or two glucopyranose units which, in turn, reduces the surfactant access to the cavities and the available space for inclusion.
A complication of the interpretation of photochemical (as well as thermal) data in cyclodextrins solutions arises when there is the possibility of formation of several complexes. Thus, naphthalene and β-CD can form two kinds of inclusion complexes, 1:1 and 2:2. Under these circumstances, the quenching of the excited state of naphthalene by anionic quenchers (I- and IO3-) was analyzed by Hamai [162] who obtained the quenching rate constants for the different kind of complexes.However, Nelson and Warner [163] considered the quenching of naphthalene in solutions containing β-CD and 1% (v/v) of benzyl alcohol. The data can be interpreted considering that only the 1:1 complex is formed. According to the authors:
![Molecules 15 04815 i228]() where rmax is given by:
where rmax is given by:
![Molecules 15 04815 i229]()


α1 and α2 being the proportionality constants defined as:
![Molecules 15 04815 i230]()
![Molecules 15 04815 i231]()


These equations hold in the presence of the alcohol, but not when it is component is absent.
An interesting possibility arises when both, the quencher and the fluorophore can be included in the same cyclodextrin, this leading to the acceleration of the (bimolecular) quenching. This possibility was first reported by Kano et al. [164] and later by other authors. [165,166,167]. The formation of a ternary complex R/CD/Q is clearly shown by the strong component of the static quencher, not observed in the absence of CD [168]. These kinds of ternary complexes are of interest in relation to analytical applications. Thus, 1-bromonaphthalene has been studied as a probe for detection of menthol [169].
Cyclodextrins, contrary to polymers, are receptors with a well-defined structure in a great range of concentration and environments. Their cavities are suitable to accommodate a great variety of luminescent guests with a well-defined stoichiometry. For these reasons, cyclodextrins are quite adequate receptors to check kinetic models. Besides, the possibility of having substituted cyclodextrins (becoming them in charged receptors, for example) offers a wide framework for research. For these reasons, cyclodextrins are quite adequate receptors to check kinetic models.
4.5. Complex Systems
As established in Section 4a the main interest in the field of microheterogeneous catalysis with participation of excited states has been to developed artificial photosynthetic systems that could reproduce behavior of the natural ones. In photosynthetic process one of the key steps is a photoinduced charge separation:
![Molecules 15 04815 i232]()
![Molecules 15 04815 i233]()


Processes a and b are rapid reactions. Thus, the main problem is not to produce a charge separated state. However, in most of the cases, reaction (183b) is followed by the (also rapid) charge recombination (the back reaction):
![Molecules 15 04815 i234]()

This reaction implies that the energy accumulated in the D+ + A− pair is lost. In this regard, a good part of the efforts for producing artificial photosynthetic systems has been related to the production of donors (D) or acceptors (A) that produce long-lived charge separated states. This part of the process has reached important advances. Thus, Fukuzumi et al. [170] have prepared donors and acceptors that participate in photoinduced electron transfer processes leading to products with lifetimes longer than the lifetime of natural systems, and accumulating a higher energy. These two characteristics are important but something more is necessary: although the D+ + A− pair was long-lived, it is necessary to extract the energy accumulated by this pair before the back reaction happens. A way to obtain this stabilization is to maintain a series of acceptors (or donors) close to the primary pair able to experience a charge transfer process. This situation is depicted in the Scheme 2.
Scheme 2.
Simplified scheme of sequential electron transfer process in a photosynthetic system.

It is clear from this scheme that in the successive electron transfer there is a loss of energy. But, at the same time, the distance between the constituent, D+ and A− pair, increases. Given that the rate of an electron transfer decreases exponentially with this distance [171] the energetic loss is compensated by a much longer lifetime, long enough to permit the energy transfer in the correct direction and, thus, opening the possibility of been stored. An important question is that, in order to reach these objectives, the donor and the acceptors must be fixed to avoid their diffusional movements that could produce the contacts D+/A-, with the subsequent back electron transfer. For this reason, in the natural systems, D and A are fixed in the cellular membranes. In artificial photosynthetic systems, consequently the D/A system must be fixed to some kind of membrane. As an example the system developed by Gust et al. [125] will be considered. This system is constituted by a triade having a quinone, a porphyrin and a carothene (1). It is placed in an artificial membrane (a vesicle) together with a second quinone (2).
Scheme 3.
Formulas of the triade and the quinone employed in reference 125.

The triad (1) is inserted into the membrane vectorially, with the quinone pointing towards the exterior of the vesicle. After excitation of the porphyrin, there is a charge separation process, in such a way that the quinone acquires a positive charge whereas the quinone in the triad acquires a negative charge. After this, the free quinone (2) is reduced by the quinone in the triad, producing a semiquinone anion. This anion can accept a proton from the solution where the vesicles are dispersed. The protonated (and neutral) quinone can move freely in the membrane. If this quinone contacts the carothene, it is oxidized forming a protonated quinone positively charged, which is acidic, pKa = −6. This protonated quinone can release a proton into the interior of the vesicle, in such a way that a protonic gradient appears. The free energy corresponding to this gradient, that is, to the difference of proton chemical potential, is used to produce ATP from ADP and phosphate. The yield of the system is relatively low, one ATP molecule is produced for every fourteen incident photons. However, it is a promising system that opens the possibility of future improvement.
There are alternatives to the production of fuels in artificial photosynthetic systems. Some of these alternatives have been considered in a recent review article by Gust et al. [172]. The alternatives go from systems able to produce ATP employing systems like those previously described to other systems devoted to photoreforming of biofuels to hydrogen. The latter systems employ photochemical biofuel cells. These cells consume biofuels such as glucose, ethanol or methanol to generate hydrogen. The photoactive system is the anode of the cell constituted by a glass covered with a transparent conductor where a thin layer of nanoparticles of SnO2 or TiO2 are inserted. These nanoparticles, in turn, are covered with a sensitizer (a porphyrin). The resulting electrode is immersed in a solution containing NADH/NAD+ (NADH = β-nicotinamida adenine dinucleotide). Upon illumination, the excited porphyrin, P*, inject an electron into the electrode, generating P+·. These electrons flow to the cathode through an external wire: in the cathodic compartment (which is separated from the rest of the solution by an ion-selective membrane that allows hydrogen ions to diffuse through the solution) protons are reduced to hydrogen. On the other hand, P+· are reduced in the anodic compartment by NADH to produce P and NAD+. In the anodic compartment, NAD+ is reduced by a dehydrogenase enzymatic catalyst, in such a way that the oxidized form of the enzyme oxidizes the biofuel. In this way, the cell functions to reform the biofuel, and hydrogen is produced through a reduction of H+, employing electrons that, at the end, come from the biofuel.
An interesting photochemical device is based on the idea of employing water instead of biofuel. In this case, the biofuel oxidation catalyst is replaced by a water oxidation catalyst (nanoparticle IrO2·nH2O). Also the porphyrin is replaced by ruthenium polypyridyl dye, which is colled to the electrode through adequate functional groups.
The principle of operation in this cell is similar to that of the photochemical biofuel cell. In this case, the electrons injected into the anode by the excited ruthenium complex again are employed (in the cathode) to reduce protons to hydrogen. On the other hand, the oxidized ruthenium complex is reduced by the iridium oxide nanoparticles, which, in turn, oxidize water to O2 and protons. This solar water splitting system is clearly inefficient and unstable to be useful for practical purposes. Nevertheless, the system has the possibility of hydrogen production via water oxidation, using ideas drawn from natural photosynthesis and molecular photochemistry.
Other strategies for solar energy conversion have been developed. Among these is the approach based on dye sensitized solar cells that involve the immobilization of a single molecular specie to nanocristalyne TiO2. Choi et al. [173] have demonstrated that the yield of these cells can be increased if the dyes are, in turn, encapsulated by cyclodextrins. An interesting consequence of the encapsulation is an increase of the stability of the system.
As we have seen nanoparticles are crucial components of the systems described previously for solar energy conversion. Other applications of nanoparticles in the field of catalysis are also possible: thus, in the field of chemical catalysis they offer interesting possibilities since chemical selectivity can be determined controlling the shape and size of the nanoparticles. An excellent review on this topic can be found in reference [174].
A conclusion emerges from the previous paragraph. Although simple systems constituted by only one kind of receptors can be useful, and they are indeed useful to discover the basic characteristics of microcatalysis, it appears that from a practical point of view, more complex systems will be needed probably to achieve some functions, such as photochemical water splitting. This would actually open new doors to basic and applied research. In this regard, the rapid development of new experimental techniques will play an important role [174].
5. Conclusions
In this work the topic of microheterogeneous catalysis has been reviewed.The interest in microheterogeneous catalysis arises from different reasons. On the one hand, it can mimic catalysis of biological systems. On the other hand, microheterogeneous catalysis is also of interest in the fields of inorganic and organic synthesis in order to obtain a desired product, favoured in the presence of a microheterogeneous system. The main research of the microheterogeneously catalyzed reactions in the excited state has been centred on finding practical systems for solar energy conversion and storage.
It has been shown that the equations describing microheterogeneous catalysis are particular cases of the Brönsted equation and can be derived from the latter if one expresses the activity coefficient taking the correct reference state. Besides, catalysis in the homogeneous media, as well as in the heterogeneous media and the fundamental equation in the field of the electrocatalysis can also be derived from the Brönsted equation.
When in the catalyzed reaction the reactants are in their ground state, the Pseudophase Model has been used extensively because this formulation is flexible enough to take into account the peculiarities of each system. For example, in micellar solutions, the effects on the rate constants of the ion condensation at the micellar surface can be taken into account through decomposition of the binding constant in an electrostatic and a non-electrostatic component. This separation can be accomplished by using Lippard’s equation. The effects of the ion condensation have also been focused from other points of view. In this regard, Quina and Chaimovich developed the Ion Exchange Pseudophase Model, and Rodenas et al. studied this effect employing a treatment based on a cell model and the non-linearized Poisson-Boltzmann equation. The effect of ion-pair formation between the reactants can be also taken into account through a formal modification of the Pseudophase Model formulation.
At premicellar concentration, a sigmoidal dependence of the rate constant on the substrate concentration is observed. This behavior, also observed in enzyme solutions, has been rationalized by Piszkiewicz and Biresaw-Bunton.
In reverse micelles, a modified Pseudophase Model which takes into account the number of binding sites in a droplet has been employed satisfactorily.
For saturable receptors, the Pseudophase Model can be modified to take into account the existence of four possible reactions paths when both of the reactants can bind to the receptors.
Cooperative effects, present in polymer solutions, can be introduced in the Pseudophase Model expressing the binding constant as a parameter which depends on polymer concentration.
Use of the Pseudophase Model requires that the free and bound reactants are in equilibrium. However, in photochemical processes this premise is not always accomplished. It depends on the relation between the lifetime of the excited state and the rates of the forward and reverse processes in the union to the catalyst. However, within the limits of fast or slow exchange, the Pseudophase Model holds, at least formally. For other situations, the most important models treating the main possible scenarios have been reviewed. Basically, the Infelta model treats the case in which the excited species are present only in the micellar pseudophase and the quenchers are distributed between the aqueous and micellar pseudophases, following a Poisson distribution. In the Almgren model there are two cases. In both, the excited species are present in aqueous and micellar pseudophases but the quencher is only present in the aqueous pseudophase in one case and, in the other, the quencher is also present in the micellar pseudophase. The differences between these models arise from the quencher distribution in a given micelle containing one excited probe. On the other hand, the Quina model treats the cases in which the exchange of the probe is or not possible. These models were developed to study photochemical reactions in micelles as receptor, but their applicability to polymers and cyclodextrins has also been considered.
Acknowledgements
This work was financed by D.G.I.C.Y.T. (CTQ2008-00008/BQU) and the Consejeria de Educación y Ciencia de la Junta de Andalucia. M. Marchena acknowledges the Spanish Ministerio de Educacion y Ciencia for a doctoral fellowship.
References
- Bunton, C.A.; Nome, F.; Quina, F.H.; Romsted, L.S. Ion binding and reactivity at charged aqueous interfaces. Acc. Chem. Res. 1991, 24, 357–364. [Google Scholar] [CrossRef]
- Marcus, R.A. Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. [Google Scholar] [CrossRef]
- Böttcher, C.J.F. Theory of Dielectric Polarization, 2nd ed; Elsevier: Amsterdam, Netherlands, 1973; Volume 1, Chapter 7; Volume 2, p. 318 and ff. [Google Scholar]
- Lao, K.Q.; Franzen, S.; Stanley, R.J.; Lambright, D.G.; Boxer, S.G. Effects of applied electric-fields on the quantum yields of the initial electron-transfer steps in bacterial photosynthesis . 1. Quantum yield failure. J. Phys. Chem. 1993, 97, 13165–13171. [Google Scholar] [CrossRef]
- Weaver, M.J. Dynamic solvent effects on activated electron-transfer reactions: principles, pitfalls, and progress. Chem. Rev. 1992, 92, 463–480. [Google Scholar] [CrossRef]
- Hazra, P.; Sarkar, N. Intramolecular charge transfer processes and solvation dynamics of Coumarin 490 in reverse micelles. Chem. Phys. Lett. 2001, 342, 303–311. [Google Scholar] [CrossRef]
- Biswas, R.; Rohman, N.; Pradhan, T.; Buchner, R. Intramolecular charge transfer reaction, polarity, and dielectric relaxation in AOT/water/heptane reverse micelles: pool size dependence. J. Phys. Chem. B 2008, 112, 9379–9388. [Google Scholar] [CrossRef]
- Bagchi, B.; Chandra, A. Collective orientational relaxation in dense dipolar liquids. Adv. Chem. Phys. 1991, 80, 1–126. [Google Scholar]
- Furse, K.E.; Corcelli, S.A. The dynamics of water at DNA interfaces: computational studies of Hoechst 33258 Bound to DNA. J. Am. Chem. Soc. 2008, 130, 13103–13109. [Google Scholar] [CrossRef]
- Moilanen, D.E.; Fenn, E.E.; Wong, D.; Fayer, M.D. Water dynamics at the interface in AOT reverse micelles. J. Phys. Chem. B 2009, 113, 8560–8568. [Google Scholar] [CrossRef]
- Sanchez, F.; Lopez-Lopez, M.; Perez-Tejeda, P. Effect of the micellar electric field on electron-transfer processes. a study of the metal-to-metal charge transfer within the binuclear complex pentaammineruthenium(III)-(mu-cyano)pentacyanoruthenium(II) in micellar solutions of sodium dodecylsulfate (SDS) and hexadecyltrimethylammonium chloride (CTACl). Langmuir 1998, 14, 3762–3766. [Google Scholar]
- Choudhury, S.D.; Kumbhakar, M.; Nath, S.; Sarkar, S.K.; Mukherjee, T.; Pal, H. Compartmentalization of reactants in different regions of sodium 1,4-bis(2-ethylhexyl)sulfosuccinate/heptane/water reverse micelles and its influence on bimolecular electron-transfer kinetics. J. Phys. Chem. B 2007, 111, 8842–8853. [Google Scholar]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Pure and Applied Chem. 1997, 69, 13–29. [Google Scholar] [CrossRef]
- Muriel-Delgado, F.; Jimenez, R.; Gomez-Herrera, C.; Sanchez, F. Use of the Bronsted equation in the interpretation of micellar effects in kinetics (II). Study of the reaction [Fe(CN)5 4-CNpy] 3- + CN-reversible arrow Fe(CN)64- + 4-CNpy in CTACl micellar solutions. Langmuir 1999, 15, 4344–4350. [Google Scholar] [CrossRef]
- Grueso, E.; Prado-Gotor, R.; Lopez, M.; Gomez-Herrera, C.; Sanchez, F. DNA effects upon the reaction between acetonitrile pentacyanoferrate(II) and ruthenium pentammine pyrazine: Kinetic and thermodynamic evidence of the interaction of DNA with anionic species. Chem. Phys. 2005, 314, 101–107. [Google Scholar] [CrossRef]
- Bonan, C.; Germani, R.; Ponti, P.P.; Savelli, G.; Cerichelli, G.; Bacaloglu, R.; Bunton, C.A. Micellar headgroup size and anion nucleophilicity in SN2 reactions. J. Phys. Chem. 1990, 94, 5331–5336. [Google Scholar] [CrossRef]
- Mitra, R.K.; Sinha, S.S.; Verma, P.K.; Pal, S.K. Modulation of dynamics and reactivity of water in reverse micelles of mixed surfactants. J. Phys. Chem. B 2008, 112, 12946–12953. [Google Scholar] [CrossRef]
- Sinha, S.S.; Mitra, R.K.; Pal, S.K. Temperature-dependent simultaneous ligand binding in human serum albumin. J. Phys. Chem. B 2008, 112, 4884–4891. [Google Scholar] [CrossRef]
- Ramamurthy, V.; Eaton, D.F. Photochemistry and photophysics within cyclodextrin cavities. Acc. Chem. Res. 1988, 21, 300–306. [Google Scholar] [CrossRef]
- Zepik, H.H.; Walde, P.; Ishikawa, T. Vesicle formation from reactive surfactants. Angew. Chem. Int. Ed. 2008, 47, 1323–1325. [Google Scholar] [CrossRef]
- Hanczyc, M.M.; Fujikawa, S.M.; Szostak, J.W. Experimental models of primitive cellular compartments: encapsulation, growth, and division. Science 2003, 302, 618–622. [Google Scholar] [CrossRef]
- Carrasco, M.; Coca, R.; Cruz, I.; Daza, S.; Espina, M.; Garcia-Fernandez, E.; Guerra, F.J.; Leon, R.; Marchena, M.J.; Perez, I.; Puente, M.; Suarez, E.; Valencia, I.; Villalba, I.; Jimenez, R. Determination of the electrostatic potential difference between DNA and the solution containing it: a kinetic approach. Chem. Phys. Lett. 2007, 441, 148–151. [Google Scholar] [CrossRef]
- Lopez, P.; Sanchez, F.; Moya, M.L.; Jimenez, R. Common basis for salt, micelle and microemulsion effects upon the ionic reaction of hexachloroiridate(IV) with thiosulfate. J. Chem. Soc.Faraday Trans. 1996, 92, 3381–3384. [Google Scholar]
- Menger, F.M.; Portnoy, C.E. On chemistry of reactions proceeding inside molecular aggregates. J. Am. Chem. Soc. 1967, 89, 4698–4703. [Google Scholar] [CrossRef]
- Olson, A.R.; Simonson, T.R. Rates of ionic reactions in aqueous solutions. J. Chem. Phys. 1949, 17, 1167–1173. [Google Scholar] [CrossRef]
- Frost, A.A.; Pearson, R.G. Kinetics and Mechanism: A Study of Homogeneous Chemical Reactions; John Wiley & Sons: New York, NY, USA, 1996; p. 181, and ff. [Google Scholar]
- Scatchard, G. Equilibria and reaction rates in dilute electrolyte solutions. Natl. Bur. Standar U.S. Circ. 1953, 524, 185–192. [Google Scholar]
- Bronsted, J.N. On the theory of the chemical reaction rate. Z. Phys. Chem. 1922, 102, 169–207. [Google Scholar]
- Marchena, M.; Sanchez, F. The Bronsted equation: The universal equation? Prog. React. Kinet. Mech. 2006, 31, 221–248. [Google Scholar]
- Langmuir, I. The constitution and fundamental properties of solids and liquids. Part I. solids. J. Am. Chem. Soc. 1916, 38, 2221–2295. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Bockris, J.O’M.; Reddy, A.K.N. Modern Electrochemistry; Plenum Press: New York, NY, USA, 1970; Volume 2, p. 1141, and ff. [Google Scholar]
- Butler, J.A.V. Studies in heterogeneous equilibria. Part II: the kinetic interpretation of the Nernst theory of electromotive force. Trans. Faraday Soc. 1924, 19, 729–733. [Google Scholar] [CrossRef]
- Erdey-Gruz, T.; Volmer, M. The theory of hydrogen high tension. Z. Physik. Chem. 1930, 150, 203–213. [Google Scholar]
- Lopez-Lopez, M.; Lopez-Cornejo, P.; Garcia, A.; Sanchez, F. Determination of substrate/ligand binding constants from electromotrive force measurements. J. Sol. Chem. 2008, 37, 519–526. [Google Scholar] [CrossRef]
- Almgren, M.; Grieser, F.; Thomas, J.K. One-electron redox potentials and rate of electron-transfer in aqueous micellar solution - partially solubilized quinones. J. Phys. Chem. 1979, 83, 3232–3236. [Google Scholar] [CrossRef]
- Davies, K.; Hussam, A. Electrochemical studies of metal-complexes in sodium dodecyl-sulfate micellar solution. Langmuir 1993, 9, 3270–3276. [Google Scholar] [CrossRef]
- Carbone, A.I.; Cavasino, F.P.; Sbriziolo, C.; Pelizzetti, E. Equilibrium and kinetic-studies of the electron-transfer reactions involving ferrocene and cobalt(III) complexes in micellar solutions. J. Phys. Chem. 1985, 89, 3578–3582. [Google Scholar] [CrossRef]
- Herrera, C.G.; Jimenez, R.; Perez-Tejeda, P.; Lopez-Cornejo, P.; Prado-Gotor, R.; Sanchez, F. On the equivalence of the pseudophase related models and the Bronsted approach in the interpretation of reactivity under restricted geometry conditions. Prog. React. Kinet. Mech. 2004, 29, 289–310. [Google Scholar]
- Miller, D.D.; Evans, D.F. Fluorescence quenching in double-chained surfactants theory of quenching in micelles and vesicles. J. Phys. Chem. 1989, 93, 323–333. [Google Scholar] [CrossRef]
- Miller, D.D.; Magid, L.J.; Evans, D.F. Fluorescence quenching in double-chained surfactants 2. Experimental results. J. Phys. Chem. 1990, 94, 5921–5930. [Google Scholar]
- de la Vega, R.; Lopez-Cornejo, P.; Perez-Tejeda, P.; Sanchez, A.; Prado, R.; Lopez, M.; Sanchez, F. Influence of the micellar electric field on electron-transfer processes (ii): a study of the Ru(NH3)5Pz2++Co(C2O4)33- reaction in SDS micellar solution containing NaCl. Langmuir 2000, 16, 7986–7990. [Google Scholar] [CrossRef]
- Lopes-Costa, T.; Sanchez, F.; Lopez-Cornejo, P. Cooperative and noncooperative binding of *[Ru(Bpy)3]2+ to DNA and SB4.5G dendrimers. J. Phys. Chem. B 2009, 113, 9373–9378. [Google Scholar]
- Lopez-Comejo, P.; Perez, P.; Garcia, F.; de la Vega, R.; Sanchez, F. Use of the pseudophase model in the interpretation of reactivity under restricted geometry conditions. an application to the study of the [Ru(NH3)Pz]2++ S2O82- electron-transfer reaction in different microheterogeneous systems. J. Am. Chem. Soc. 2002, 124, 5154–5164. [Google Scholar] [CrossRef]
- Armitage, B.A. DNA Binders and Related Subjects; Waring, M.J., Chaires, J.B., Eds.; Springer Verlas: New York, Berlin, 2005; (Top. Curr. Chem. 2005, 263, 55). [Google Scholar]
- Lavery, R. Multiple Aspects of DNA and RNA: from Biophysics to Bioinformatics; Chatenay, D., Cocco, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Garcia-Rio, L.; Mejuto, J.C.; Perez-Lorenzo, M. Simultaneous effect of microemulsions and phase-transfer agents on aminolysis reactions. J. Phys. Chem. B 2007, 111, 11149–11156. [Google Scholar] [CrossRef]
- Graciani, M.D.; Rodriguez, M.A.; Moya, M.L. Study of the ligand substitution reaction Fe(CN)5H2O3- plus pyrazine in micellar solutions. Int. J. Chem. Kinet. 1997, 29, 377–384. [Google Scholar] [CrossRef]
- Prado-Gotor, R.; Jimenez, R.; Perez-Tejeda, P.; Lopez-Cornejo, P.; Lopez-Lopez, M.; Sanchez, A.; Muriel-Delgado, F.; Sanchez, F. Electron transfer reactions in micellar systems. Prog. React. Kinet. Mech. 2000, 25, 371–407. [Google Scholar]
- Barzykin, A.V.; Tachiya, M. Unified treatment of luminescence quenching kinetics in micelles with quencher migration on the basis of a generalized Smoluchowski approach. J. Phys. Chem. B 1998, 102, 1296–1300. [Google Scholar] [CrossRef]
- Alargova, R.G.; Petkov, J.T.; Petsev, D.N. Micellization and interfacial properties of alkyloxyethylene sulfate surfactants in the presence of multivalent counterions. J. Colloid Interface Sci. 2003, 261, 1–11. [Google Scholar] [CrossRef]
- Benjamin, I. Chemical reactions and solvation at liquid interfaces: A microscopic perspective. Chem. Rev. 1996, 96, 1449–1475. [Google Scholar] [CrossRef]
- Abuin, E.; Lissi, E.; Ceron, A.; Rubio, M.A. Spectroscopic probing of the effect of alkanols on the properties of the head group region in reverse micelles of AOT-heptane-water. J. Colloid Interface Sci. 2003, 258, 363–366. [Google Scholar] [CrossRef]
- Hartley, G.S.; Roe, J.W. Ionic concentrations at interfaces. Trans. Faraday Soc. 1940, 35, 101–109. [Google Scholar] [CrossRef]
- Quina, F.H.; Chaimovich, H. Ion-exchange in micellar solutions .1. Conceptual-framework for ion-exchange in micellar solutions. J. Phys. Chem. 1979, 83, 1844–1850. [Google Scholar] [CrossRef]
- Lopez-Cornejo, P.; Sanchez, F. Micellar effects on the kinetics of the oxidation of the excited state of the [Ru(Bpy)3]2+ complex by S2O82-. A comparison of different approaches for the interpretation of micellar effects on kinetics. J. Phys. Chem. B 2001, 105, 10523–10527. [Google Scholar] [CrossRef]
- Mayer, J.E. The theory of ionic solutions. J. Chem. Phys. 1950, 18, 1426–1436. [Google Scholar] [CrossRef]
- de la Vega, R.; Perez-Tejeda, P.; Lopez-Cornejo, P.; Sanchez, F. Kinetic study of the oxidation of [Ru(NH3)5Pz]2+ by [Co(C2O4)3]3- in AOT-oil-water microemulsions and in CTAC1 micellar solutions. Langmuir 2004, 20, 1558–1563. [Google Scholar]
- Howegrant, M.; Lippard, S.J. Binding of platinum(II) intercalation reagents to deoxyribonucleic-acid - dependence on base-pair composition, nature of the intercalator, and ionic-strength. Biochemistry 1979, 18, 5762–5769, (N. Y.). [Google Scholar] [CrossRef]
- Grueso, E.; Alcantara, D.; Martinez, J.; Mancera, M.; Penades, S.; Sanchez, F.; Pradogotor, R. Kinetic approach for the study of noncovalent interaction between [Ru(Nh3)5pz]2+ and gold nanoparticles. J. Phys. Chem. A 2007, 111, 9769–9774. [Google Scholar]
- Lopez-Cornejo, P.; Prado-Gotor, R.; Garcia-Santana, A.; Perez, F.; Sanchez, F. Comparative study of micellar and DNA effects on the reaction [Ru(NH3)5py]2++S2O82-. Langmuir 2003, 19, 3185–3189. [Google Scholar]
- Rodenas, E.; Ortega, F.; Vera, S.; Otero, C.; Maestro, S. Surfactants in solution; Mittal, K.L., Ed.; Plenum Pub. Co: New York, NY, USA, 1989; volume. 9, p. 211, and ff. [Google Scholar]
- Ortega, F.; Rodenas, E. An electrostatic approach for explaining the kinetic results in the reactive counterion surfactants CTAOH and CTACN. J. Phys. Chem. 1987, 91, 837–840. [Google Scholar] [CrossRef]
- Rodriguez, A.; del Mar Graciani, M.; Bitterman, K.; Carmona, A.T.; Moya, M.L. Micellar kinetic effects in gemini micellar solutions: influence of sphere-to-rod transitions on kinetics. J. Colloid Interface Sci. 2007, 313, 542–550. [Google Scholar] [CrossRef]
- Graciani, M.D.; Rodriguez, A.; Moya, M.L. Study of the reaction between methyl 4-nitrobenzenesulfonate and bromide ions in mixed single-chain-gemini micellar solutions: kinetic evidence for morphological transitions. J. Colloid Interface Sci. 2008, 328, 324–330. [Google Scholar] [CrossRef]
- Rodriguez, A.; Graciani, M.D.; Munoz, M.; Robina, I.; Moya, M.L. Effects of ethylene glycol addition on the aggregation and micellar growth of gemini surfactants. Langmuir 2006, 22, 9519–9525. [Google Scholar] [CrossRef]
- Lopez-Cornejo, P.; Prado-Gotor, R.; Gomez-Herrera, C.; Jimenez, R.; Sanchez, F. Influence of the charge and concentration of coreactants on the apparent binding constant of the reactant to micelles. Langmuir 2003, 19, 5991–5995. [Google Scholar] [CrossRef]
- Piszkiewicz, D. Micelle catalyzed reactions are models of enzyme catalyzed reactions which show positive homotropic interactions. J. Am. Chem. Soc. 1976, 98, 3053–3055. [Google Scholar] [CrossRef]
- Piszkiewicz, D. Positive cooperativity in micelle-catalyzed reactions. J. Am. Chem. Soc. 1977, 99, 1550–1557. [Google Scholar] [CrossRef]
- Piszkiewicz, D. Cooperativity in bimolecular micelle-catalyzed reactions: inhibition of catalysis by high-concentrations of detergent. J. Am. Chem. Soc. 1977, 99, 7695–7697. [Google Scholar] [CrossRef]
- Hill, A.V. The possible effects of the aggregation of molecules of hemoglobin on its dissociation curve. J. Physiol. (Lond) 1910, 40, 4. [Google Scholar]
- Lopez, P.; Sanchez, F.; Moya, M.L.; Jimenez, R. Common basis for salt, micelle and microemulsion effects upon the ionic reaction of hexachloroiridate(iv) with thiosulfate. J. Chem. Soc. Faraday Trans. 1996, 92, 3381–3384. [Google Scholar] [CrossRef]
- Lopez-Cornejo, P.; Mozo, J.D.; Roldan, E.; Dominguez, M.; Sanchez, F. Kinetic study of the reaction *[Ru(Bpy)3]2++S2O82- in solutions of brij-35 at premicellar and micellar concentrations. Chem. Phys. Lett. 2002, 352, 33–38. [Google Scholar] [CrossRef]
- Biresaw, G.; Bunton, C.A. Size vs reactivity in organized assemblies-deacylation and dephosphorylation in functionalized assemblies. J. Phys. Chem. 1986, 90, 5849–5853. [Google Scholar] [CrossRef]
- Valero, M.; Sanchez, F.; Gomez-Herrera, C.; Lopez-Cornejo, P. Study of water solubilized in AOT/n-decane/water microemulsions. Chem. Phys. 2008, 345, 65–72. [Google Scholar] [CrossRef]
- Marchena, M.; Sanchez, F. Kinetic effects of methyl-beta-cyclodextrin and 2-hydroxypropyl-beta-cyclodextrin and their mixtures on the reaction [Fe(CN)5(4-Phepy)]3- + [Co(NH3)4(H2O)2]3+. Chem. Phys. Lett. 2009, 471, 234–238. [Google Scholar] [CrossRef]
- Fernandez, E.; Garcia-Rio, L.; Knorl, A.; Leis, J.R. Metal-ligand complexation in water-in-oil microemulsions. i. thermodynamic approach. Langmuir 2003, 19, 6611–6619. [Google Scholar]
- Fernandez, E.; Garcia-Rio, L.; Knorl, A.; Leis, J.R. Kinetic study of ni2+ and co2+ complexation by pada in AOT-based water-in-oil microemulsions. Eur. J. Org. Chem. 2005, 4, 740–748. [Google Scholar]
- Secco, F.; Venturini, M.; Lopez, M.; Perez, P.; Prado, R.; Sanchez, F. Effect of DNA on the rate of electron transfer reactions between non-intercalated reactants: kinetic study of the reactions Reactions [Ru(NH3)5Pz]2++[Co(C2O4)3]3- and [Ru(NH3)5Py]2++[Co(NH3)4pzCO2]2+ in aqueous solutions in the presence of DNA. Phys. Chem. Chem. Phys. 2001, 3, 4412–4417. [Google Scholar]
- De la Vega, R.; Perez, P.; Prado-Gotor, R.; Sanchez, F. DNA interactions with small solutes: change in the character of the binding of [Ru(NH3)5Pz]2+ to DNA as a consequence of changes in the solvent. Chem. Phys. 2004, 297, 163–169. [Google Scholar] [CrossRef]
- Sun, Y.S.; Landry, J.P.; Fei, Y.Y.; Zhu, X.D. Effect of fluorescently labeling protein probes on kinetics of protein-ligand reactions. Langmuir 2008, 24, 13399–13405. [Google Scholar] [CrossRef]
- Grueso, E.; Sanchez, F.; Martin, V. I.; Garcia-Fernandez, E.; Prado-Gotor, R. Quantification of salts and cosolvents-DNA interactions in terms of free energies: a study using the pyren-1-carboxyaldehyde as fluorescent probe. Chem. Phys. 2008, 352, 306–310. [Google Scholar] [CrossRef]
- Tan, Z.J.; Chen, S.J. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys. J. 2006, 90, 1175–1190. [Google Scholar] [CrossRef]
- Grueso, E.; Sanchez, F. DNA-surfactant interactions: a procedure for determination group contributions. J. Phys. Chem. B 2008, 112, 698–702. [Google Scholar] [CrossRef]
- Hays, M.E.; Jewell, C.M.; Lynn, D.M.; Abbott, N.L. Reversible condensation of DNA using a redox-active surfactant. Langmuir 2007, 23, 5609–5614. [Google Scholar] [CrossRef]
- Haynil, D.T. Biological thermodynamic; Cambridge University Press, 2004; p. 223, and ff. [Google Scholar]
- Ortiz, J.; Guichou, J.F.; Chavanieu, A.; Sanchez, F.; Prado-Gotor, R. Polymerization-induced enhancement of binding and binding-induced polymerization. Chem. Phys. Lett. 2004, 384, 266–270. [Google Scholar] [CrossRef]
- Chavanieu, A.; Guichou, J.F.; Prado-Gotor, R.; Perez-Tejeda, P.; Jimenez, R.; Lopez-Cornejo, P.; Sanchez, F. Strength and character of peptide/anion interactions. J. Phys. Chem. B 2005, 109, 19676–19680. [Google Scholar]
- de la Vega, R.; Perez-Tejeda, P.; Prado-Gotor, R.; Lopez-Cornejo, P.; Jimenez, R.; Perez, F.; Sanchez, F. Effects of SB1.5G and SB4.5G dendrimers on the rate of the electron transfer reaction between [Ru(NH3)5Pz]2+ and [Co(C2O4)3]3-. Chem. Phys. Lett. 2004, 398, 82–86. [Google Scholar] [CrossRef]
- Jimenez, R.; Garcia-Fernandez, E.; Sanchez, F. Dendrimer effects upon the reaction between (acetonitrile) pentacyano-ferrate(II) and pentaamminepyrazine-ruthenium(II). Chem. Phys. Lett. 2006, 420, 372–376. [Google Scholar] [CrossRef]
- Sanchez, R.; Villar, M.; Guiraum, A.; Prado-Gotor, R. Restricted geometry conditions promoted by alooh nanoparticles: variable strength and character of alooh-cluster/charged ligand interactions as a consequence of changes in the solvent. J. Phys. Chem. C 2008, 112, 9240–9246. [Google Scholar]
- Miyoshi, H.; Matsuo, Y.; Liu, Y.Y.; Sakata, T.; Mori, H. Behavior of fluorescent molecules bound to the interior of silica nanocapsules in various solvents. J. Colloid Interface Sci. 2009, 331, 507–513. [Google Scholar] [CrossRef]
- Martin, C.; Sanchez, F.; Jimenez, R.; Prado, R.; Perez-Tejeda, P.; Lopez-Cornejo, P. Salt and solvent effects on the kinetics and thermodynamics of the inclusion of the ruthenium complex [Ru(NH3)5(4,4 '-Bpy)]2+ in beta-cyclodextrin. J. Phys. Chem. B 2006, 110, 12959–12963. [Google Scholar]
- Connors, K.A. The stability of cyclodextrin complexes in solution. Chem. Rev. 1997, 97, 1325–1357. [Google Scholar] [CrossRef]
- de Namor, A.F.D.; Chahine, S.; Castellano, E.E.; Piro, O.E. Thermodynamics of host-guest interactions in lower rim functionalized calix[4]arenes and metal cations: the medium effect. J. Phys. Chem. B 2004, 108, 11384–11392. [Google Scholar]
- Dey, J.; Roberts, E.L.; Warner, I.M. Effect of sodium perchlorate on the binding of 2-(4'-aminophenyl)- and 2-(4'-(n,n'-dimethylamino)phenyl)benzothiazole with beta-cyclodextrin in aqueous solution. J. Phys. Chem. A 1998, 102, 301–305. [Google Scholar]
- Lima, S.; Goodfellow, B.J.; Teixeira-Dias, J.J.C. How inorganic anions affect the inclusion of hexanoic and decanoic acid in beta-cyclodextrin. J. Phys. Chem. A 2004, 108, 10044–10049. [Google Scholar] [CrossRef]
- Rekharsky, M.V.; Inoue, Y. Solvent and guest isotope effects on complexation thermodynamics of alpha-, beta-, and 6-amino-6-deoxy-beta-cyclodextrins. J. Am. Chem. Soc. 2002, 124, 12361–12371. [Google Scholar] [CrossRef]
- Breslow, R.; Dong, S.D. Biomimetic reactions catalyzed by cyclodextrins and their derivatives. Chem. Rev. 1998, 98, 1997–2011. [Google Scholar] [CrossRef]
- Takahashi, K. Organic reactions mediated by cyclodextrins. Chem. Rev. 1998, 98, 2013–2033. [Google Scholar] [CrossRef]
- Iglesias, E. Cyclodextrins as enzyme models in nitrosation and in acid-base-catalyzed reactions of alkyl nitrites. J. Am. Chem. Soc. 1998, 120, 13057–13069. [Google Scholar] [CrossRef]
- Ramachandran, M.S. Lathakannan Beta-cyclodextrin-catalyzed decomposition of caro's acid. Int. J. Chem. Kinet. 2002, 34, 508–513. [Google Scholar] [CrossRef]
- Sueishi, Y.; Hishikawa, H. Complexation of 4-dimethylaminoazobenzene with various kinds of cyclodextrins: effects of cyclodextrins on the thermal cis-to-trans isomerization. Int. J. Chem. Kinet. 2002, 34, 481–487. [Google Scholar] [CrossRef]
- Tee, O.S.; Gadosy, T.A. Acyl transfer mediated by complexation: The effect of cyclodextrins on the reaction of nucleophiles with p-nitrophenyl acetate and hexanoate. J.e Chem. Soc. Perkin Transactions 2 1994, 2307–2311. [Google Scholar]
- Tee, O.S. The stabilization of transition states by cyclodextrins and other catalysts. Adv.n Phys. Org. Chem. 1994, 29, 1–85. [Google Scholar] [CrossRef]
- Tee, O.S. The binding of transition-states by cyclomalto-oligosaccharides. Carbohydr. Res. 1989, 192, 181–195. [Google Scholar] [CrossRef]
- Imonigie, J.A.; Macartney, D.H. Effects of cyclodextrin inclusion on the kinetics of the outer-sphere oxidation of 4-tert-butylcatechol by transition-metal complexes in acidic aqueous-media. Inorg. Chem. 1993, 32, 1007–1012. [Google Scholar] [CrossRef]
- Kaifer, A.E. Interplay between molecular recognition and redox chemistry. Acc. Chem. Res. 1999, 32, 62–71. [Google Scholar] [CrossRef]
- Nielson, R.M.; Hupp, J.T.; Yoon, D.I. Modulation of outer-sphere electron-transfer reactivity via primitive molecular recognition effects:Ru(NH3)5(4-Methyl-Pyridine)3+/2+ self-exchange kinetics in the presence of macrocyclic polyether species. J. Am. Chem. Soc. 1995, 117, 9085–9086. [Google Scholar] [CrossRef]
- Shortreed, M.E.; Wylie, R.S.; Macartney, D.H. Inclusion of (N-adamantan-1'-ylpyrazinium)pentacyanoferrate(II) ion in alpha-cyclodextrins and beta-cyclodextrins. Effects of inclusion on the spectroscopic properties and ligand substitution kinetics. Inorg. Chem. 1993, 32, 1824–1829. [Google Scholar] [CrossRef]
- Lonetti, B.; Lo Nostro, P.; Ninham, B.W.; Baglioni, P. Anion effects on calixarene monolayers: a hofmeister series study. Langmuir 2005, 21, 2242–2249. [Google Scholar] [CrossRef]
- Izatt, R.M.; Pawlak, K.; Bradshaw, J.S.; Bruening, R.L. Thermodynamic and kinetic data for macrocycle interaction with cations and anions. Chem. Rev. 1991, 91, 1721–2085. [Google Scholar]
- Casnati, A.; Sansone, F.; Ungaro, R. Peptido- and glycocalixarenes: Playing with hydrogen bonds around hydrophobic cavities. Acc. Chem. Res. 2003, 36, 246–254. [Google Scholar] [CrossRef]
- Sanchez, A.; Jimenez, R.; Ternero, F.; Mesa, R.; Pinero, C. A.; Muriel, F.; Lopez-Cornejo, P. Rigidity and/or flexibility of calixarenes. effect of the p-sulfonatocalix[n]arenes (n = 4, 6, and 8) on the electron transfer process [Ru(NH3)5Pz]2++[Co(C2O4)3]3-. J. Phys. Chem. B 2007, 111, 10697–10702. [Google Scholar]
- Lopez-Cornejo, P.; Bote, B.; Felix, R.; Infantes, I.; Lopez, P.; Martin, A.; Mateos, E.; Perez, M.; Rojas, A.; Suarez, R. Binding of [Ru(NH3)5Pz]2+to 4-sulfocalix[4]arene sodium salt. effects of the host-guest interaction on electron transfer processes. J. Phys. Chem. B 2009, 113, 12721–12726. [Google Scholar]
- Rodriguez, A.; Munoz, M.; Graciani, M.D.M.; Moya, M.L. Kinetic micellar effects in tetradecyltrimethylammonium bromide-pentanol micellar solutions. J. Colloid Interface Sci. 2002, 248, 455–461. [Google Scholar] [CrossRef]
- Munoz, M.; Rodriguez, A.; Graciani, M.D.; Moya, M.L. Micellar medium effects on the hydrolysis of phenyl chloroformate in ionic, zwitterionic, nonionic, and mixed micellar solutions. Int. J. Chem. Kinet. 2002, 34, 445–451. [Google Scholar] [CrossRef]
- Munoz, M.; Graciani, M.D.; Rodriguez, A.; Moya, M.L. Influence of the addition of alcohol on the reaction methyl-4-nitrobenzenesulfonate plus Br- in tetradecyltrimethylammonium bromide aqueous micellar solutions. J. Colloid Interface Sci. 2003, 266, 208–214. [Google Scholar] [CrossRef]
- Fernandez, G.; Rodriguez, A.; Graciani, M.D.; Munoz, M.; Moya, M.L. Study of the reaction methyl 4-nitrobenzene-sulfonate plus cl- in mixed hexadecyltrimethyl-ammonium chloride-triton x-100 micellar solutions. Int. J. Chem. Kinet. 2003, 35, 45–51. [Google Scholar] [CrossRef]
- Munoz, M.; Rodriguez, A.; Graciani, M.D.; Moya, M.L. Conductometric, surface tension, and kinetic studies in mixed SDS-tween 20 and SDS-SB3-12 micellar solutions. Langmuir 2004, 20, 10858–10867. [Google Scholar] [CrossRef]
- Mahiuddin, S.; Zech, O.; Raith, S.; Touraud, D.; Kunz, W. Catanionic micelles as a model to mimic biological membranes in the presence of anesthetic alcohols. Langmuir 2009, 25, 12516–12521. [Google Scholar] [CrossRef]
- Garcia-Rio, L.; Mendez, M.; Paleo, A.R.; Sardina, F.J. New insights in cyclodextrin: surfactant mixed systems from the use of neutral and anionic cyclodextrin derivatives. J. Phys. Chem. B 2007, 111, 12756–12764. [Google Scholar] [CrossRef]
- Lopez-Cornejo, P. Personal communication. Universidad de Sevilla: Sevilla, Spain, 2010. [Google Scholar]
- Lee, E.; Kim, J.K. ; Lee, M. Tubular stacking of water-soluble toroids triggered by guest encapsulation. J. Am. Chem. Soc. 2009, 131, 18242–18243. [Google Scholar]
- Gust, D.; Moore, T.A.; Moore, A.L. Mimicking photosynthetic solar energy transduction. Acc. Chem. Res. 2001, 34, 40–48. [Google Scholar] [CrossRef]
- Clark, C.D.; Hoffman, M.Z. Effect of solution medium on the rate constants of excited-state electron-transfer quenching reactions of ruthenium(II)-diimine photosensitizers. Coord. Chem. Rev. 1997, 159, 359–373. [Google Scholar]
- Pattabiraman, M.; Kaanumalle, L.S.; Natarajan, A.; Ramamurthy, V. Regioselective photodimerization of cinnamic acids in water: templation with cucurbiturils. Langmuir 2006, 22, 7605–7609. [Google Scholar] [CrossRef]
- Bhattacharyya, K. Solvation dynamics and proton transfer in supramolecular assemblies. Acc. Chem. Res. 2003, 36, 95–101. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Photophysics of molecules in micelle-forming surfactant solutions. Chem. Soc. Rev. 1978, 7, 453–472. [Google Scholar]
- Ananthapadmanabhan, K.P.; Goddard, E.D.; Turro, N.J.; Kuo, P.L. Fluorescence probes for critical micelle concentration. Langmuir 1985, 1, 352–355. [Google Scholar] [CrossRef]
- Marchena, M.; Sanchez, F. Kinetic of photochemical reaction under restricted geometry conditions. Prog. Reac. Kinet. Mech. 2010, 35, 27–80. [Google Scholar] [CrossRef]
- Maiti, N.C.; Krishna, M.M.G.; Britto, P.J.; Periasamy, N. Fluorescence dynamics of dye probes in micelles. J. Phys. Chem. B 1997, 101, 11051–11060. [Google Scholar]
- Yoshida, N.; Takechi, M.; Asano, T.; Moroi, Y.; Humphry-Baker, R.; Grätzel, M. Thermodynamics and kinetics of solubilization of naphthalene into anionic micelles. Chem. Phys. Lett. 2000, 332, 265–270. [Google Scholar] [CrossRef]
- Infelta, P.P.; Gratzel, M. Statistics of solubilizate distribution and its application to pyrene fluorescence in micellar systems : Concise kinetic-model. J. Chem. Phys. 1979, 70, 179–186. [Google Scholar] [CrossRef]
- Maestri, M.; Infelta, P.P.; Gratzel, M. Kinetics of fast light-induced redox processes in micellar systems: Intra-micellar electron-transfer. J. Chem. Phys. 1978, 69, 1522–1526. [Google Scholar] [CrossRef]
- Barzykin, A.V.; Tachiya, M. Unified treatment of luminescence quenching kinetics in micelles with quencher migration on the basis of a generalized Smoluchowski approach. J. Phys. Chem. B 1998, 102, 1296–1300. [Google Scholar] [CrossRef]
- Almgren, M.; Grieser, F.; Thomas, J.K. Dynamic and static aspects of solubilization of neutral arenes in ionic micellar solutions. J. Am. Chem. Soc. 1979, 101, 279–291. [Google Scholar] [CrossRef]
- Almgren, M.; Lofroth, J.E.; Vanstam, J. Fluorescence decay kinetics in monodisperse confinements with exchange of probes and quenchers. J. Phys. Chem. 1986, 90, 4431–4437. [Google Scholar] [CrossRef]
- Quina, F.H.; Toscano, V.G. Photophenomena in surfactant media-quenching of a water-soluble fluorescence probe by iodide-ion in micellar solutions of sodium dodecyl-sulfate. J. Phys. Chem. 1977, 81, 1750–1754. [Google Scholar] [CrossRef]
- Doughert, S.J.; Berg, J.C. Distribution equilibria in micellar solutions. J. Colloid Interface Sci. 1974, 48, 110–121. [Google Scholar] [CrossRef]
- Rinco, O.; Nolet, M.C.; Ovans, R.; Bohne, C. Probing the binding dynamics to sodium cholate aggregates using naphthalene derivatives as guests. Photochem. Photobiol. Sci. 2003, 2, 1140–1151. [Google Scholar] [CrossRef]
- Turro, N.J.; Okubo, T.; Chung, C.J. Analysis of static and dynamic host-guest associations of detergents with cyclodextrins via photo-luminescence methods. J. Am. Chem. Soc. 1982, 104, 1789–1794. [Google Scholar] [CrossRef]
- Guo, X.; Xu, H.; Guo, R. Fluorescence quenching of anthracene by N,N-diethylaniline in the sodium dodecyl sulfate/benzyl alcohol/water system. J. Colloid Interface Sci. 2001, 240, 559–565. [Google Scholar] [CrossRef]
- Hackett, J.W.; Turro, C. Bimolecular electron transfer quenching of neutral *[Ru(phen)2bps] by 4,4 -diheptyl viologen in water and bound to SDS micelles. J. Phys. Chem. A 1998, 102, 5728–5733. [Google Scholar] [CrossRef]
- Dressick, W.J.; Hauenstein, B.L.; Demas, J.N.; Degraff, B.A. Electron-transfer quenching of ruthenium(II) photosensitizers by mercury(II) chlorides . 2. reactions in aqueous sodium lauryl sulfate micellar solutions. Inorg. Chem. 1984, 23, 1107–1113. [Google Scholar] [CrossRef]
- Weidemaier, K.; Fayer, M.D. Role of diffusion in photoinduced electron transfer on a micelle surface: Theoretical and monte carlo investigations. J. Phys. Chem. 1996, 100, 3767–3774. [Google Scholar] [CrossRef]
- Weidemaier, K.; Tavernier, H.L.; Fayer, M.D. Photoinduced electron transfer on the surfaces of micelles. J. Phys. Chem. B 1997, 101, 9352–9361. [Google Scholar] [CrossRef]
- Weidemaier, K.; Fayer, M.D. Photoinduced electron-transfer and geminate recombination on a micelle surface: Analytical theory and monte-carlo simulations. J. Chem. Phys. 1995, 102, 3820–3829. [Google Scholar] [CrossRef]
- Barzykin, A.V.; Seki, K.; Tachiya, M. Theory of diffusion-assisted reactions on micelle surfaces: photoinduced electron transfer followed by back transfer. J. Phys. Chem. B 1999, 103, 9156–9160. [Google Scholar] [CrossRef]
- Ranganathan, R.; Vautier-Giongo, C.; Bales, B.L. Toward a hydrodynamic description of bimolecular collisions in micelles. an experimental test of the effect of the nature of the quencher on the fluorescence quenching of pyrene in SDS micelles and in bulk liquids. J. Phys. Chem. B. 2003, 107, 10312–10318. [Google Scholar] [CrossRef]
- Ami, T.; Fujimoto, K. Fluorescence labeling of DNA based on photochemical ligation. Sci. Tech. Adv. Mat. 2006, 7, 249–254. [Google Scholar] [CrossRef]
- Morrison, M.E.; Dorfman, R.C.; Clendening, W.D.; Kiserow, D.J.; Rossky, P.J.; Webber, S.E. Quenching kinetics of anthracene covalently bound to a polyelectrolyte .1. effects of ionic-strength. J. Phys. Chem. 1994, 98, 5534–5540. [Google Scholar] [CrossRef]
- Chu, D.Y.; Thomas, J.K. Photophysical studies of a water-soluble copolymer of methacrylic-acid and 1-pyreneacrylic acid. Macromolecules 1984, 17, 2142–2147. [Google Scholar] [CrossRef]
- Turro, N.J.; Arora, K.S. Pyrene as a photophysical probe for intermolecular interactions of water-soluble polymers in dilute-solutions. Polymer 1986, 27, 783–796. [Google Scholar] [CrossRef]
- Arora, K.; Turro, N.J. Polyelectrolyte effects on the quenching of pyrene fluorescence in solutions of a pyrene substituted poly(acrylic acid). J. Pol. Sci. Part A-Polymer Chemistry 1987, 25, 259–269. [Google Scholar] [CrossRef]
- Turro, N.J.; Okubo, T.; Chung, C.J.; Emert, J.; Catena, R. Polyelectrolyte-enhanced excimer formation of bis(alpha-naphthylmethyl)ammonium chloride and (alpha-naphthylmethyl)ammonium chloride. J. Am. Chem. Soc. 1982, 104, 4799–4803. [Google Scholar] [CrossRef]
- Wensel, T.G.; Meares, C.F.; Vlachy, V.; Matthew, J.B. Distribution of ions around DNA, probed by energy-transfer. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 3267–3271. [Google Scholar] [CrossRef]
- Arkin, M.R.; Stemp, E.D.A.; Turro, C.; Turro, N.J.; Barton, J.K. Luminescence quenching in supramolecular systems: a comparison of DNA- and SDS micelle-mediated photoinduced electron transfer between metal complexes. J. Am. Chem. Soc. 1996, 118, 2267–2274. [Google Scholar]
- Fukuzumi, S.; Nishimine, M.; Ohkubo, K.; Tkachenko, N.V.; Lemmetyinen, H. Driving force dependence of photoinduced electron transfer dynamics of intercalated molecules in DNA. J. Phys. Chem. B 2003, 107, 12511–12518. [Google Scholar]
- Yorozu, T.; Hoshino, M.; Imamura, M.; Shizuka, H. Photo-excited inclusion complexes of beta-naphthol with alpha-cyclodextrin, beta-cyclodextrin, and gamma-cyclodextrin in aqueous-solutions. J. Phys. Chem. 1982, 86, 4422–4426. [Google Scholar] [CrossRef]
- Andrade-Dias, C.; Lima, S.; Teixeira-Dias, J.J.C.; Teixeira, J. Why do methylated and unsubstituted cyclodextrins interact so differently with sodium decanoate micelles in water? J. Phys. Chem B. 2008, 112, 15327–15332. [Google Scholar] [CrossRef]
- Hamai, S. Association of inclusion-compounds of beta-cyclodextrin in aqueous-solution. Bull. Chem. Soc. Jpn. 1982, 55, 2721–2729. [Google Scholar] [CrossRef]
- Nelson, G.; Warner, I.M. Fluorescence quenching studies of cyclodextrin complexes of pyrene and naphthalene in the presence of alcohols. J. Phys. Chem. 1990, 94, 576–581. [Google Scholar] [CrossRef]
- Kano, K.; Takenoshita, I.; Ogawa, T. 3 Component complexes of cycloheptaamylose- fluorescence quenching of pyrenes and naphthalenes in aqueous-media. Chem. Lett. 1980, 1035–1038. [Google Scholar]
- Rideout, D.C.; Breslow, R. Hydrophobic acceleration of diels-alder reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. [Google Scholar]
- Kobashi, H.; Takahashi, M.; Muramatsu, Y.; Morita, T. Effects of beta-cyclodextrin on fluorescence quenching of sodium 1-pyrenesulfonate by aniline in aqueous-media. Bull. Chem. Soc. Jpn. 1981, 54, 2815–2816. [Google Scholar] [CrossRef]
- Breslow, R.; Czarniecki, M.F.; Emert, J.; Hamaguchi, H. Improved acylation rates within cyclodextrin complexes from flexible capping of the cyclodextrin and from adjustment of the substrate geometry. J. Am. Chem. Soc. 1980, 102, 762–770. [Google Scholar]
- Kano, K.; Takenoshita, I.; Ogawa, T. Fluorescence quenching of pyrene and naphthalene in aqueous cyclodextrin solutions:Evidence of 3-component complex-formation. J. Phys. Chem. 1982, 86, 1833–1838. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Hu, X. S.; Ariese, F.; Gooijer, C. Enantioselective room temperature phosphorescence detection of non-phosphorescent analytes based on interaction with beta-cyclodextrin/1-bromonaphthalene complexes. Talanta 2005, 66, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Fukuzumi, S.; Kotani, H.; Ohkubo, K.; Ogo, S.; Tkachenko, N.V.; Lemmetyinen, H. Electron-transfer state of 9-mesityl-10-methylacridinium ion with a much longer lifetime and higher energy than that of the natural photosynthetic reaction center. J. Am. Chem. Soc. 2004, 126, 1600–1601. [Google Scholar]
- Creutz, C. Nonadiabatic, short-range, intramolecular electron transfer from ruthenium(II) to cobalt(III) complexes. J. Phys. Chem. B. 2007, 111, 6713–6717. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef]
- Choi, H.; Kang, S.O.; Ko, J.; Gao, G.; Kang, H.S.; Kang, M.S.; Nazeeruddin, M.K.; Gratzel, M. An efficient dye-sensitized solar cell with an organic sensitizer encapsulated in a cyclodextrin cavity. angew. Chem. Int. 2009, 48, 5938–5941. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Frei, H.; Park, J.Y. Advancing the frontiers in nanocatalysis, biointerfaces, and renewable energy conversion by innovations of surface techniques. J. Am. Chem. Soc. 2009, 131, 16589–16605. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Bernal, E.; Marchena, M.; Sánchez, F. Microheterogeneous Catalysis. Molecules 2010, 15, 4815-4874. https://doi.org/10.3390/molecules15074815
AMA Style
Bernal E, Marchena M, Sánchez F. Microheterogeneous Catalysis. Molecules. 2010; 15(7):4815-4874. https://doi.org/10.3390/molecules15074815
Chicago/Turabian StyleBernal, Eva, María Marchena, and Francisco Sánchez. 2010. "Microheterogeneous Catalysis" Molecules 15, no. 7: 4815-4874. https://doi.org/10.3390/molecules15074815