3.1. General
All the solvents were purchased from Aldrich Chemical Company in Sure/Seal™ bottles and were used as received. 1H-NMR spectra were recorded on a 300 MHz Bruker whereas 13C-NMR spectra were recorded on a 75 MHz Bruker NMR and TMS was used as an internal standard. Specific rotations were determined for solutions by irradiating with the sodium D line (λ= 589 nm) using a Perkin Elmer 341 polarimeter: specific rotation, [α]D values are given in units 10-1deg•cm2g-1 where the concentration c is given in g/100 mL. The chemical purity (Method A) was determined by standard HPLC (containing a PDA detector) using a Phenomenex Synergi 4 μ Fusion-RP 80 Å (150 mm × 4.6 mm) column at wavelength of 280 nm, using a gradient of 5 to 90% of acetonitrile (containing 0.01% TFA) with water (containing 0.01% TFA) up to 20 min, the column temperature was 25 °C, and the flow rate was 1 mL/min. The enantiomeric purity (Method B) was determined by chiral HPLC equipped with a PDA detector using a Chiralpak AD-RH 5μ (150 mm × 4.6 mm) column. The solvent for isocratic programs was acetonitrile/water (65/35, v/v) containing 0.01% TFA, run time 40 min, detection wavelength was 210 nm, column temperature was 60 °C, and the flow rate was 1 mL/min.
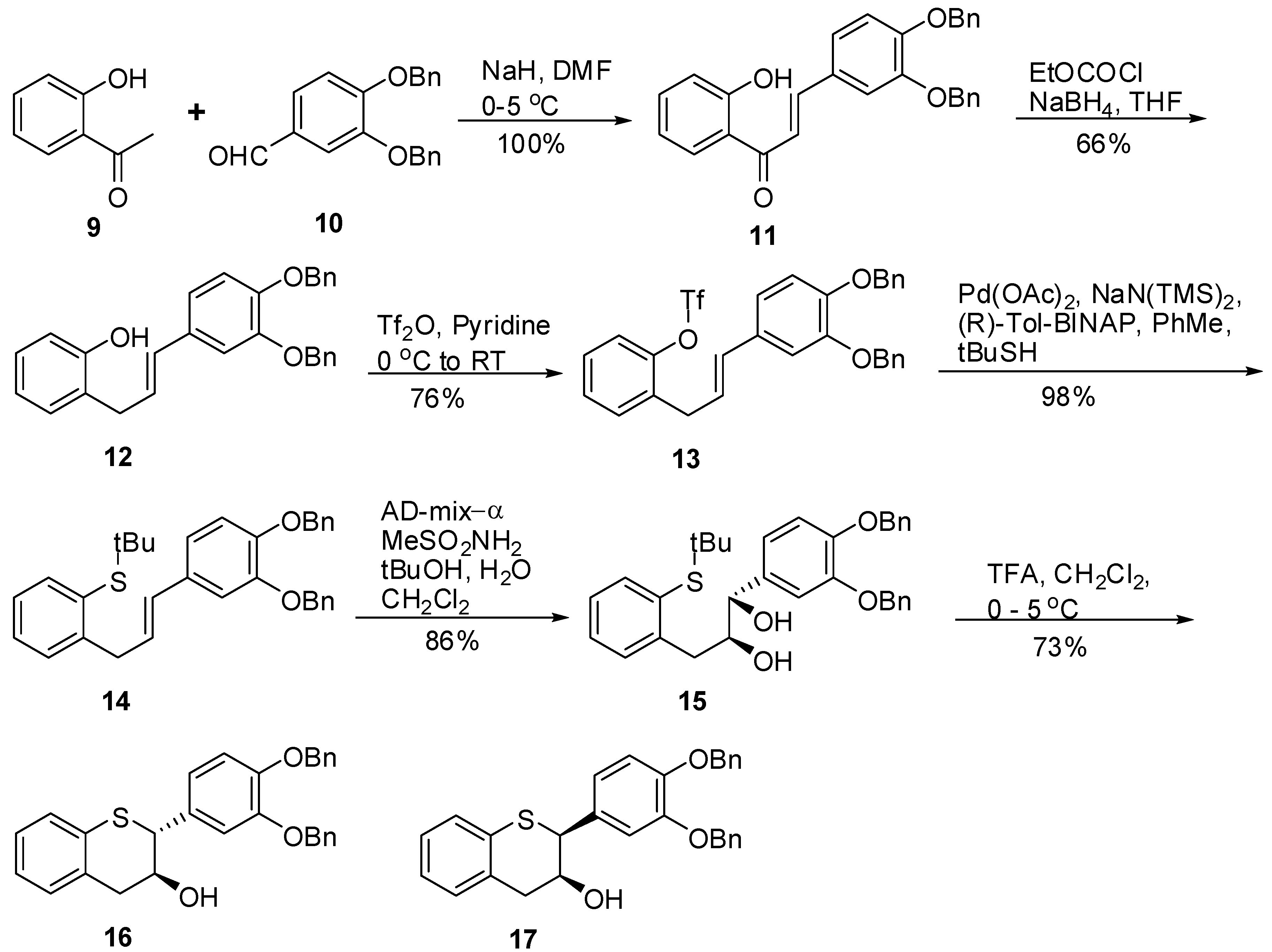
(E)-3-(3’,4’-Bis(benzyloxy)phenyl)-1-(2-hydroxyphenyl)prop-2-en-1-one (11). To a suspension of NaH (60% dispersion in oil, 3.82 g, 95.48 mmol, 1.3 eq.) in dry DMF (100 mL) was slowly added 9 (10 g, 73.45 mmol, 1 eq.) keeping the internal temperature ≤ 2 °C throughout the addition. The resulting mixture was stirred at this temperature for 10 minutes. A solution of 10 (23.38 g, 73.45 mmol, 1 eq.) in DMF (100 mL) was added over a period of 20 minutes to the reaction mixture via an addition funnel keeping the internal temperature ≤ 2 °C. The resulting red brown solution was stirred at this temperature for an additional 30 minutes before stirring at RT for 4 hours. The reaction mixture was quenched with H2O (50 mL) and diluted with EtOAc (500 mL). The organic layer was separated, washed with H2O (50 mL), sat. NaHCO3 (2 × 50 mL), brine (100 mL), dried (Na2SO4), filtered and the solvent removed in vacuo to afford a yellow solid. The solid was triturated with heptane (100 mL) at RT for 1h and filtered. The solids were dried under high vacuum at RT for 18 h to produce 11 (33.3 g, 100%) as a yellow solid with 100% AUC purity. 1H-NMR (CDCl3) δ = 5.32 (s, 4H), 6.2 (dd, 2H, J = 2.2 and 19Hz), 6.9–7.1 (m, 4H, Ar), 7.2–7.6 (m, 9H, Ar), 7.8–8.0 (m, 4H, Ar); 13C-NMR (CDCl3) δ = 31.1, 37.2, 47.3, 51.5, 117.4, 118.2, 118.4, 126.3, 127.3, 127.4, 127.5, 127.8, 128.4, 128.7, 129.1, 129.3, 130.2, 131.7, 132.1, 137.4, 138.9, 139.9, 148.6, 149.9, 150.3, 181.6; MS (m/z): 437 (M++1); HRMS calcd for C29H24O4 [M+H] 437.1753, Found: 437.1748.
(E)-2-(3-(3’,4’-Bis(benzyloxy)phenyl)allylphenol (12). To a solution of 11 (12 g, 27.6 mmol, 1 eq.) in THF (100 mL) was added Et3N (5 mL, 35.8 mmol, 1.3 eq.) and the mixture cooled to 0 °C. Ethyl chloroformate (3.2 mL, 33 mmol, 1.2 eq.) was slowly added keeping the internal temperature at 0 °C. The resulting mixture was allowed to stir at 0 °C for 1.5 h and the progress of the reaction was monitored by TLC. The reaction mixture was suction filtered to remove the salts and the salts washed with THF (2 × 50 mL). The combined filtrate was added slowly to a cold solution of NaBH4 in water at 0 °C over 45 minutes. The resulting mixture was slowly warmed to room temperature and allowed to stir for 18h. The reaction mixture was acidified with 1N HCl (pH ~2 with a pH paper) and extracted with EtOAc (2 × 300 mL). The combined organic layers were dried (MgSO4), filtered and the solvent was removed in vacuo to afford an oil. The crude product was purified by silica gel chromatography (5–10% ethyl acetate in heptane) to afford 12 (7.7 g, 66%) as a colorless oil with 100% AUC purity. 1H-NMR (CDCl3) δ = 3.58 (d, 2H, J = 6.0 Hz), 5.2 (s, 4H), 6.1–6.25 (m, 1H), 6.33 (d, 1H, J = 8.4 Hz), 6.8–7.0 (m, 4H), 7.2–7.6 (m, 13H); 13C-NMR (CDCl3) δ = 34.0, 71.5, 71.9, 71.7, 77.5, 112.9, 115.3, 115.8, 115.9, 119.9, 120.8, 120.9, 125.8, 126.3, 127.3, 127.4, 127.8, 127.8, 127.8, 128.2, 128.5, 130.4, 131.4, 131.1, 131.4, 137.2, 148.6, 148.2, 154.1; MS (m/z) = 423.1 (M++1); HRMS calcd for C29H26O3 [M+H] 423.1962, Found: 423.1959
(E)-2-(3-(3’,4’-Bis(benzyloxy)phenyl)allyl)phenyl trifluoromethane sulfonate (13). To an ice cold solution (<5 °C) of 12 (4 g, 9.46 mmol, 1 eq.) in dry pyridine (40 mL) was slowly added Tf2O (3.22 g, 11.4 mmol, 1.2 eq.). The resulting mixture was allowed to warm to RT and allowed to stir for 18h. The reaction mixture was diluted with EtOAc (200 mL), washed with 1N HCl (4 × 100 mL), brine (100 mL), dried (Na2SO4), filtered and the solvent was removed in vacuo to give an oil. The oil was purified by silica gel chromatography (5-10% ethyl acetate in heptane) to give 13 (4 g, 76%) as a colorless oil with 100% AUC purity. 1H-NMR (CDCl3) δ = 3.6 (d, 2H, J = 6.3 Hz), 5.2 (s, 4H), 6.0–6.15 (m, 1H), 6.33 (d, 1H, J = 8.4 Hz), 6.8 (s, 2H), 7.0 (s, 1H), 7.2–7.6 (m, 14H); HRMS calcd for C30H25F3O5S [M+H] 555.1455, Found: 555.1451
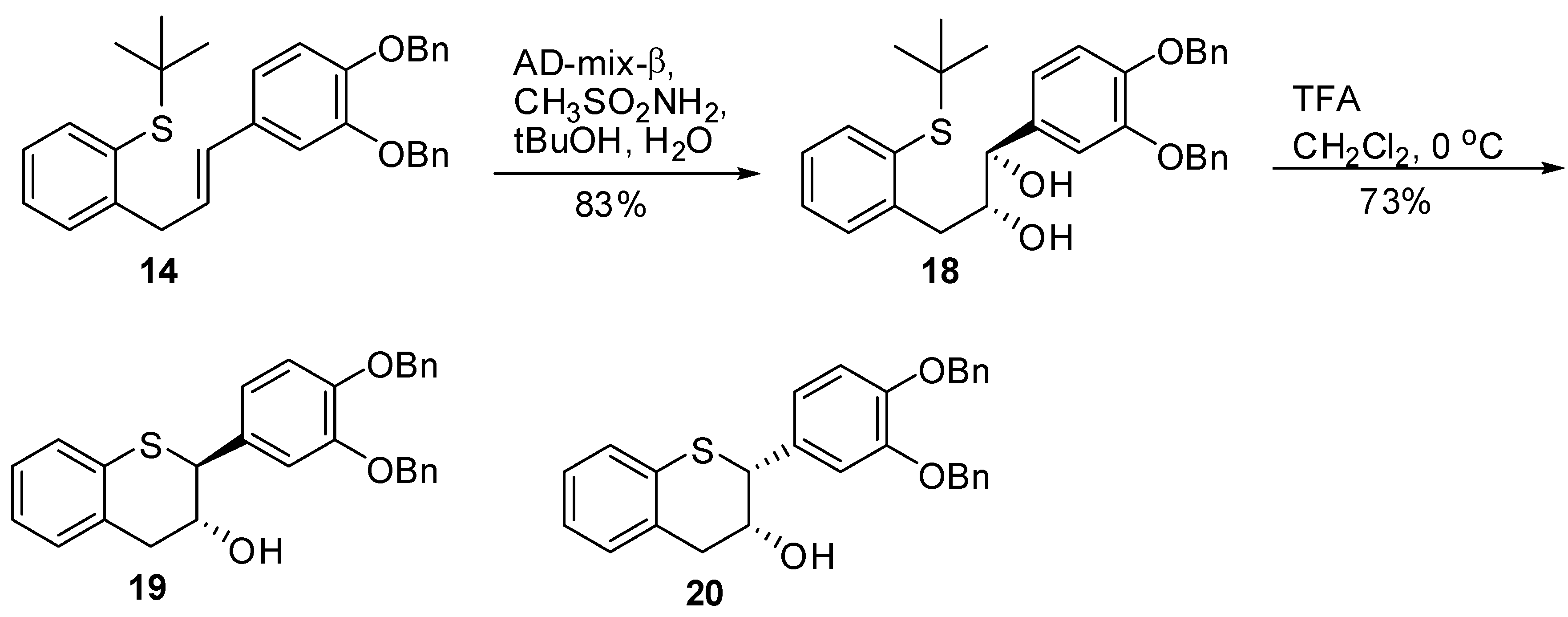
(E)-2-(3-(3’,4’-Bis(benzyloxy)phenyl)allyl)phenyl-(tert-butyl)sulfane (14). To a degassed solution of 13 (8 g, 14.43 mmol, 1 eq.) in dry toluene (80 mL) was added Pd(OAc)2 (0.194 g, 0.866 mmol, 0.06 eq.) and (R)-Tol-BINAP (0.685 g, 1.01 mmol, 0.07 eq.) at RT. The resulting reaction mixture was degassed again for an additional 15 minutes at RT. In a separate flask, a solution of NaN(TMS)2 (0.6M solution in toluene, 33.7 mL, 202.2 mmol, 1.4 eq.) was added slowly to t-BuSH (2.3 mL, 2.2 mmol, 1.4 eq.) in dry toluene (10 mL) at RT and the mixture stirred at RT under N2 for 15 minutes. This solution was transferred under N2 to the above solution. The red colored solution was heated at 100 °C under N2 for 18h. The reaction mixture was diluted with H2O (100 mL) and EtOAc (100 mL). The organic layer was separated, washed with brine (50 mL), dried (Na2SO4), filtered and the solvent was removed in vacuo to give a red viscous oil. The oil was purified by silica gel chromatography (5% EtOAc in heptane) to afford 14 (6.98g, 98%) as a yellow viscous oil with 99.8% AUC purity. 1H-NMR (CDCl3) δ = 1.22 (s, 9H), 3.88 (d, 2H, J = 6.7 Hz), 5.12 (s, 4H), 6–6.2 (m, 1H), 6.3 (d, 1H, J = 6.7 Hz), 6.77 (s, 2H), 7.0 (s, 1H), 7.1–7.6 (m, 14H); 13C-NMR (CDCl3) δ = 31.2, 37.9, 47.2, 71.5, 112.1, 115.4, 119.9, 126.2, 127.3, 127.4, 127.7, 127.8, 127.9, 128.5, 128.6, 129.2, 129.9, 130.6, 131.7, 132.1, 137.4, 138.9, 143.8, 145.1, 146.4; MS (m/z) = 495 (M++1), 439.3 (M+-t-Bu); HRMS calcd for C33H34O2S [M+H] 495.2358, Found: 495.2353
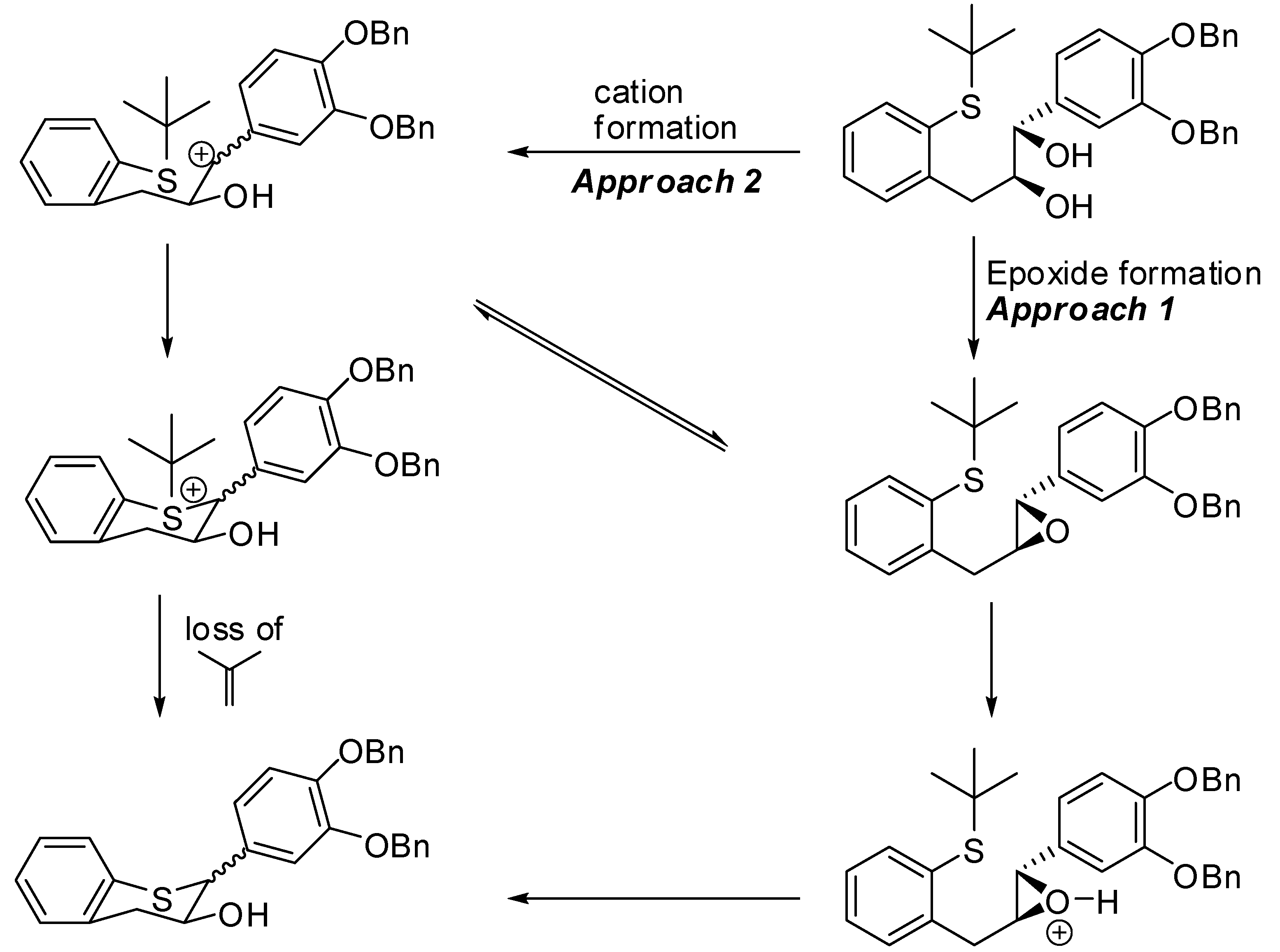
(1S,2S)-1-(3’,4’-Bis(benzyloxy)phenyl)-3-(2-(tert-butylthio)phenyl)propane-1,2-diol (15). A suspension of AD-mix-α (18g) in tBuOH/H2O (60 mL, 1/1, v/v) was stirred at RT for 15 minutes until a clear yellow solution was obtained followed by the addition of a solution of 14 (3.6 g, 7.29 mol, 1 eq.) in CH2Cl2 (15 mL). The resulting reaction mixture was cooled to 0 ºC with stirring. Then, MeSO2NH2 (0.833 g, 8.76 mmol, 1.2 eq.) was added and the mixture stirred at this temperature for 24h. The reaction was quenched by the addition of 10% Na2S2O3 (50 mL) and then extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were washed with H2O (2 × 50 mL), brine (1 × 75 mL), dried (Na2SO4), filtered, and the solvent removed in vacuo to afford the crude product. The crude product was purified by silica gel chromatography using 20% to 30% EtOAc/heptane to produce 15 (3.3g, 86%) as an oil with 100% AUC purity. [α]20 D = +34. 9 (c 1, acetone); 1H-NMR (CDCl3) δ = 1.15 (s, 9H), 2.21 (d, 1H, J = 4.45 Hz), 2.82 (dd, 1H, J = 9, 9.5 Hz), 2.88 (d, 1H, J = 4.5 Hz), 3.07 (dd, 1H, J = 4 Hz), 3.81-3.9 (m, 1H), 4.4 (dd, 1H, J = 3.4 Hz), 5.13 (s, 2H), 5.18 (s, 2H), 6.8 (s, 2H), 7.03 (s, 1H), 7.1–7.52 (m, 14H); 13C-NMR (CDCl3) δ = 30.9, 38.5, 47.6, 71.3, 71.5, 113.9, 115.1, 120.4, 126.6, 127.3, 127.5, 127.7, 127.9, 128.5, 129.1, 130.9, 132.4, 134.8, 137.3, 137.4, 139.1, 143.8, 148.8, 149.2; Optical purity = 100% ee. HRMS calcd for C33H36O4S [M+H] 529.2412, Found: 529.2338.
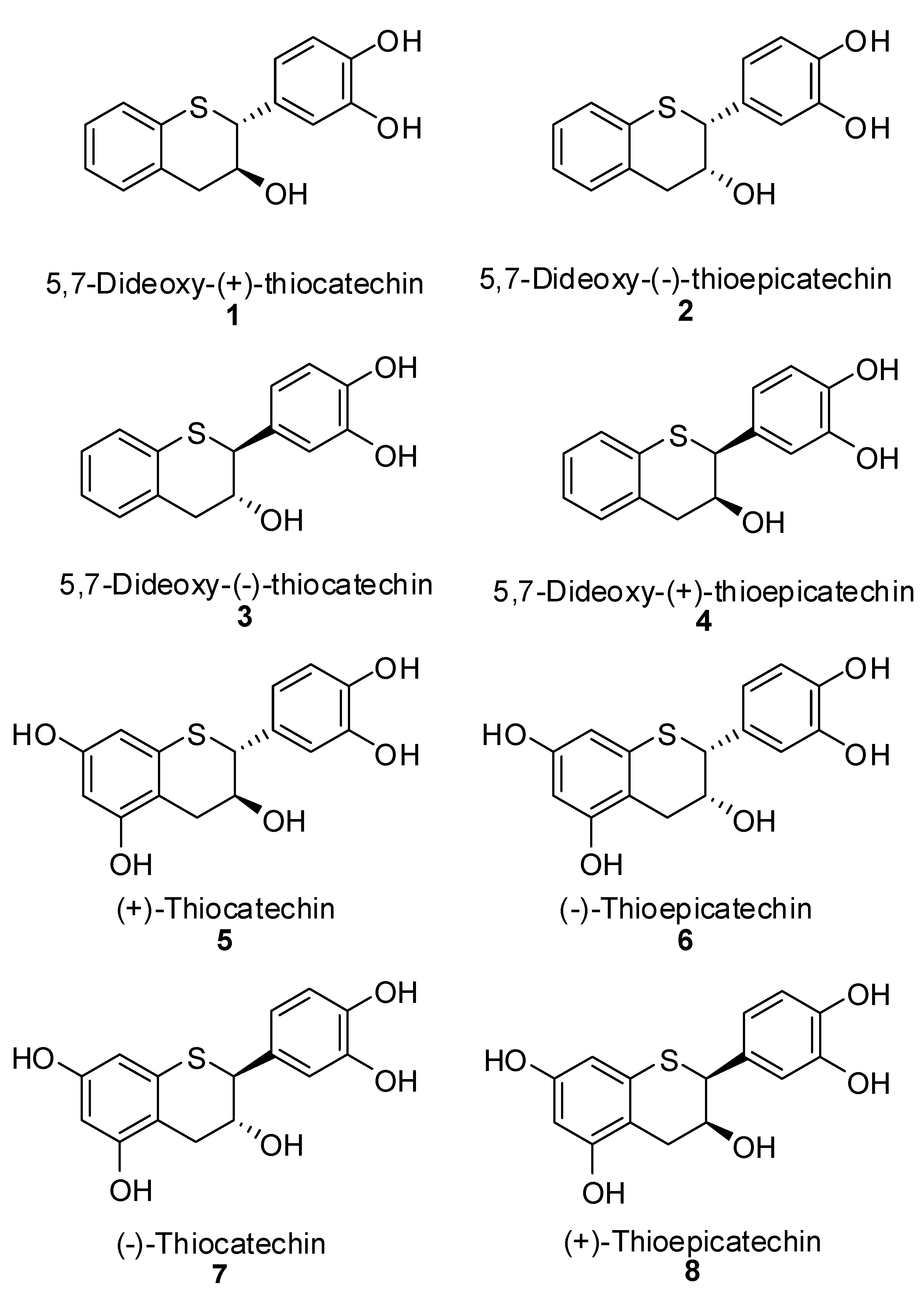
3’,4’-Bis(benzyloxy)-5,7-dideoxy-(+)-thiocatechin (16) and 3’,4’-Bis(benzyloxy)-5,7-dideoxy-(+)-thioepicatechin (17). To a cold solution of 15 (1.42 g, 2.7 mmol, 1 eq.) in CH2Cl2 (50 mL) was slowly added TFA (494 μL, 2.4 eq.). The resulting mixture was kept at 0 °C. The reaction was quenched by addition of Et3N (890 μL, 2.4 eq.) followed by cold H2O (15 mL). The organic layer was separated and washed with brine solution (15 mL), dried (Na2SO4), filtered, and the solvent removed in vacuo. The crude product was then purified by silica gel chromatography (10–30% EtOAc in heptane) to produce the desired major diastereomer 16 (830 mg, 68%) and the minor diastereomer 17 (60 mg, 4.9%).
3’,4’-Bis(benzyloxy)-5,7-dideoxy-(+)-thiocatechin (16). [α]20 D = +108.9 (c 1, acetone); 1H-NMR (CDCl3) δ = 1.98 (d, 2H, J = 4.8 Hz), 2.52 (dd, 1H, H4a, J = 8.4, 7.3 Hz), 3.08 (dd, 1H, H4b, J = 4, 3.9 Hz), 4.16–4.3 (m, 1H), 5.1 (s, 2H), 5.16 (s, 2H), 6.85–7.2 (m, 6H), 7.22–7.5 (m, 11H); 13C-NMR (CDCl3) δ = 36.6, 51.7, 56.4, 70.1, 71.3, 77.5, 115.2, 121.8, 124.6, 124.8, 127.3, 127.5, 127.9, 127.9, 128.5, 128.5, 130.5, 130.9, 131.2, 131.9, 132.7, 136.9, 137.5, 148.4, 149.2; Optical purity = 96% ee; Chemical purity = 100% (AUC); MS (m/z) = 455.1 (M++1), 437.2 (M+-OH), 345.3 (M+-OH-Bn); HRMS calcd for C29H26O3S [M+H] 455.1681, Found: 455.1677.
3’,4’-Bis(benzyloxy)-5,7-dideoxy-(+)-thioepicatechin (17). [α]20 D = +38.6 (c 1, Acetone); 1H NMR (300 MHz, CDCl3) δ = 2.2 (d, 1H, J = 9.4 Hz), 2.96 (dd, 1H, H4a, J = 5, 17 Hz), 3.1 (dd, 1H, H4b, J = 3.5, 17 Hz), 4.3–4.42 (m, 1H), 4.45 (d, 1H, J = 1.6 Hz), 5.12 (s, 2H), 5.14 (s, 2H), 6.85–7.2 (m, 6H), 7.28–7.5 (m, 11H); 13C NMR (75 MHz, CDCl3) δ = 37.9, 50.1, 66.4, 71.3, 76.6, 114.9, 115.6, 121.7, 124.8, 125.9, 126.8, 127.3, 127.5, 127.8, 128.5, 128.5, 130.1, 131.2, 131.4, 132.4, 137.1, 137.3, 148.9, 148.9; Optical purity = 100% ee; HPLC purity = 100% (AUC); MS (m/z) = 455.1 (M++1), 437.2 (M+-OH), 345.3 (M+-OH-Bn); HRMS calcd for C29H26O3S [M+H] 455.1681, Found: 455.1679.
(1R,2R)-1-(3’,4’-Bis(benzyloxy)phenyl)-3-(2-(tert-butylthio)phenyl)propane-1,2-diol (18). A suspension of AD-mix-β (18 g) in tBuOH/H2O (60 mL, 1/1, v/v) was stirred at RT for 15 minutes until a clear yellow solution was obtained. This was followed by the addition of a solution of 14 (3.6g, 7.29 mol, 1 eq.) in CH2Cl2 (15 mL). The resulting reaction mixture cooled to 0 °C with stirring. Then, MeSO2NH2 (0.833 g, 8.76 mmol, 1.2 eq.) was added and the mixture stirred at this temperature for 24 h. The reaction was quenched by the addition of 10% Na2S2O3 (50 mL) and then extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were washed with H2O (2 × 50 mL), brine (1 × 75 mL), dried (Na2SO4), filtered, and the solvent removed in vacuo to afford the crude product. The crude product was purified by silica gel chromatography using 20% to 30% EtOAc/heptane to produce 18 (3.2g, 83%) as an oil with 99.8% AUC purity. [α]20 D = -34.3 (c 1, acetone); 1H-NMR (CDCl3) δ = 1.15 (s, 9H), 2.21 (d, 1H, J = 4.5 Hz), 2.82 (dd, 1H, J = 9, 9.5 Hz), 2.88 (d, 1H, J = 4.5 Hz), 3.07 (dd, 1H, J = 3.4, 4 Hz), 3.81-3.9 (m, 1H), 4.4 (dd, 1H, J = 3.4, 4 Hz), 5.13 (s, 2H), 5.18 (s, 2H), 6.8 (s, 2H), 7.03 (s, 1H), 7.1–7.52 (m, 14H); 13C-NMR (CDCl3) δ = 30.9, 38.6, 47.6, 71.4, 71.5, 113.9, 115.1, 120.3, 126.7, 127.3, 127.5, 127.8, 127.8, 128.5, 129.2, 130.9, 132.4, 134.8, 137.3, 137.4, 139.1, 143.8, 148.8, 149.2; Optical purity = 100% ee; HRMS calcd for C33H36O4S [M+H] 529.2412, Found: 529.2337.
3’,4’-Bis(benzyloxy)-5,7-dideoxy-(-)-thiocatechin (19) and 3’,4’-bis(benzyloxy)-5,7-dideoxy-(-)-thio-epicatechin (20). To a cold solution of 18 (1.6 g, 3.04 mmol, 1 eq.) in CH2Cl2 (60 mL) was slowly added TFA (617 μL, 3.0 eq.). The resulting mixture was kept at 0 °C. The reaction was quenched by addition of Et3N (1.1 mL, 2.4 eq.) followed by cold H2O (25 mL). The organic layer was separated and washed with brine solution (25 mL), dried (Na2SO4), filtered, and the solvent removed in vacuo. The crude product was then purified by silica gel chromatography (10–30% EtOAc in heptane) to produce the desired major diastereomer 19 (925 mg, 68%), and minor diastereomer 20 (72 mg, 4.8%).
3’,4’-Bis(benzyloxy)-5,7-dideoxy-(-)-thiocatechin (19). [α]20 D = -110.4 (c 1, acetone); 1H-NMR (CDCl3) δ = 1.98 (d, 1H, J = 4.3 Hz), 2.9 (dd, 1H, H4a, J = 8.3, 14.2, 17 Hz), 3.08 (dd, 1H, H4b, J = 4, 16.5 Hz), 4.15–4.32 (m, 2H), 5.12 (s, 2H), 5.16 (s, 2H), 6.8–7.15 (m, 6H), 7.2–7.5 (m, 11H); 13C-NMR (CDCl3) δ = 31.8, 51.6, 70.1, 71.3, 71.4, 77.2, 115.1, 115.3, 121.8, 124.5, 125.3, 126.8, 127.3, 127.5, 127.9, 127.9, 128.1, 128.5, 128.5, 130.5, 130.9, 131.9, 132.7, 136.9, 137.1, 148.1, 148.1; Optical purity = 99% ee; Chemical purity = 100% (AUC); MS (m/z) = 455.1 (M++1), 345.3 (M+-OH-Bn); HRMS calcd for C29H26O3S [M+H] 455.1681, Found: 455.1678.
3’,4’-Bis(benzyloxy)-5,7-dideoxy-(-)-thioepicatechin (20). [α]20 D = -74.2 (c 1, acetone); 1H-NMR (CDCl3) δ = 2.2 (d, 1H, J = 9.4Hz), 2.95 (dd, 1H, H4a, J = 5, 17 Hz), 3.1 (dd, 1H, H4b, J = 3.5, 17 Hz), 4.3-4.41 (m, 1H), 4.42 (d, 1H, J = 1.4 Hz), 5.1 (s, 2H), 5.12 (s, 2H), 6.8–7.2 (m, 6H), 7.28–7.5 (m, 11H); 13C-NMR (CDCl3) δ = 37.9, 50.1, 66.4, 71.3, 76.6, 114.9, 115.6, 121.7, 124.8, 125.9, 126.7, 127.2, 127.9, 127.8, 128.0, 128.5, 128.5, 130.1, 131.2, 131.4, 132.4, 137.1, 137.3, 148.9, 148.9; Optical purity = 99.5% ee; Chemical purity = 100% (AUC); MS (m/z) = 455.1 (M++1), 345.3 (M+-OH-Bn); HRMS calcd for C29H26O3S [M+H] 455.1681, Found: 455.1679.
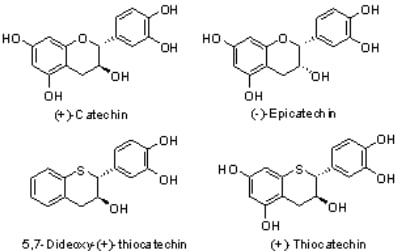
5,7-Dideoxy-(+)-thiocatechin (1). To an ice cold solution of compound 16 (614 mg, 1.35 mmol, 1 eq.) in dry CH2Cl2 (50 mL) was added N,N-dimethylaniline (1.37 mL, 10.82 mol, 8 eq.) under N2. The solution was stirred at this temperature for ~10 minutes before AlCl3 (1.8 g, 13.52 mmol, 10 eq.) was added in one portion. The flask was covered with aluminum foil to protect it from light and stirred at ice bath temperature for 3h. EtOAc (100 mL) and silica gel (8.5 g) were added to the reaction mixture and stirred for 15 minutes. The reaction mixture was filtered through a pad of silica gel. The silica gel pad was washed with EtOAc (5 × 50 mL). The filtrates were combined and the solvent was removed in vacuo to ~5 mL of volume keeping the bath temperature < 25 °C. The crude mixture was diluted with purified water (50 mL), frozen and lyophilized to give the crude product. The crude product was further purified by preparative HPLC to give the desired compound 1 (180mg, 54%, 100% AUC) as an off-white solid. [α]20 D = + 22.2 (c 1, acetone); 1H-NMR (acetone-d6) δ = 2.9 (dd, 1H, J = 9, 16.1 Hz), 3.1 (dd, 1H, J = 4, 16.2 Hz), 4.1 – 4.35 (m, 2H), 6.7 (s, 2H, Ar), 6.9 (s, 1H, Ar), 6.92–7.2 (m, Ar, 4H), 7.4–8.3 (br s, 2H); 13C-NMR (acetone-d6) δ = 39.2, 52.7, 70.9, 116.0, 116.5, 121.3, 124.9, 125.7, 127.3, 130.9, 131.7, 134.3, 134.8, 145.6, 145.9; MS= 255.2 [M+-H2O-H, 100%]; HRMS calcd for C15H14O3S [M+H] 275.0742, Found: 275.0737.
5,7-Dideoxy-(-)-thioepicatechin (2). To an ice cold solution of compound 17 (790 mg, 1.74 mmol, 1 eq.) in dry CH2Cl2 (80 mL) was added N,N-dimethylaniline (1.76 mL, 13.92 mol, 8 eq.) under N2. The solution was stirred at this temperature for ~10 minutes before AlCl3 (2.32 g, 17.4 mmol, 10 eq.) was added in one portion. The flask was covered with aluminum foil to protect it from light and stirred at the ice bath temperature for 3h. EtOAc (50 mL) and silica gel (10 g) were added to the reaction mixture and stirred for 15 minutes. The reaction mixture was filtered through a pad of silica gel. The silica gel pad was washed with EtOAc (4 × 50 mL). The filtrates were combined and the solvent removed in vacuo to ~20 mL of volume keeping the bath temperature < 25 °C. The crude mixture was diluted with purified water (100 mL), cooled and lyophilized to give the crude product. The crude product was further purified by preparative HPLC to give the desired compound 2 (270 mg, 64%, 98.5% AUC) as an off-white solid. [α]20 D = -28.6 (c 1, acetone); 1H-NMR (acetone-d6) δ = 2.88 (dd, 1H, J = 5.9 and 16.7 Hz), 3.04 (d, 1H, J = 14.5 Hz), 4.25–4.4 (m, 2H), 6.5–7.2 (m, 7H, Ar), 7.4–8.3 (br s, 2H); 13C-NMR (acetone-d6) δ = 37.9, 49.3, 66.4, 114.5, 116.1, 120.5, 123.8, 124.9, 126.1, 130.7, 130.9, 131.1, 133.3, 144.3, 144.4; MS= 255.2 [M+-H2O-H, 100%]; HRMS calcd for C15H14O3S [M+H] 275.0742, Found: 275.0739.
5,7-Dideoxy-(-)-thiocatechin (3). To an ice cold solution of compound 19 (506 mg, 1.11 mmol, 1 eq.) in dry CH2Cl2 (40 mL) was added N, N-dimethylaniline (1.13 mL, 8.92 mol, 8 eq.) under N2. The solution was stirred at this temperature for ~10 minutes before AlCl3 (1.5 g, 11.14 mmol, 10 eq.) was added in one portion. The flask was covered with aluminum foil to protect it from light and stirred at the ice bath temperature for 2.5h. EtOAc (100 mL) and silica gel (8 g) were added to the reaction mixture and stirred for 15 minutes. The reaction mixture was filtered through a pad of silica gel. The silica gel pad was washed with EtOAc (3 × 50 mL). The filtrates were combined and the solvent was removed in vacuo to ~20 mL of volume keeping the bath temperature < 25 °C. The crude mixture was diluted with purified water (50 mL), cooled and lyophilized to give the crude product. The crude product was further purified by preparative HPLC to give the desired compound 3 (180 mg, 59%, 99% AUC) as an off-white solid. [α]20 D = -21.3 (c 1, acetone); 1H-NMR (acetone-d6) δ = 2.9 (dd, 1H, J = 9, 16.1 Hz), 3.1 (dd, 1H, J = 4, 16.2 Hz), 4.1–4.35 (m, 2H), 6.7 (s, 2H, Ar), 6.9 (s, 1H, Ar), 6.92-7.2 (m, Ar, 4H), 7.4–8.3 (br s, 2H); 13C-NMR (acetone-d6) δ = 39.2, 52.7, 70.9, 116.0, 116.5, 121.3, 124.9, 125.7, 127.3, 130.9, 131.7, 134.3, 134.8, 145.6, 145.9; MS = 255.2 [M+-H2O-H, 100%]; HRMS calcd for C15H14O3S [M+H] 275.0742, Found: 275.0735.
5,7-Dideoxy-(+)-thioepicatechin (4). To an ice cold solution of compound 20 (790 mg, 1.74 mmol, 1 eq.) in dry CH2Cl2 (80 mL) was added N, N-dimethylaniline (1.76 mL, 13.92 mol, 8 eq.) under N2. The solution was stirred at this temperature for ~10 minutes before AlCl3 (2.32 g, 17.4 mmol, 10 eq.) was added in one portion. The flask was covered with aluminum foil to protect it from light and stirred at the ice bath temperature for 3 h. EtOAc (50 mL) and silica gel (10 g) were added to the reaction mixture and stirred for 15 minutes. The reaction mixture was filtered through a pad of silica gel. The silica gel pad was washed with EtOAc (4 × 50 mL). The filtrates were combined and the solvent was removed in vacuo to ~20 mL of volume keeping the bath temperature < 25 °C. The crude mixture was diluted with purified water (100 mL), cooled and lyophilized to give the crude product. The crude product was further purified by preparative HPLC to give the desired compound 4 (270mg, 64%, 98.5% AUC) as an off-white solid. [α]20 D = +28.4 (c 1, acetone); 1H-NMR (acetone-d6) δ = 2.88 (dd, 1H, J = 5.9 and 16.7 Hz), 3.04 (d, 1H, J = 14.5 Hz), 4.25–4.4 (m, 2H), 6.5–7.2 (m, 7H, Ar), 7.4–8.3 (br s, 2H); 13C-NMR (acetone-d6) δ = 37.9, 49.3, 66.4, 114.5, 116.1, 120.5, 123.7, 124.9, 126.1, 130.7, 130.9, 131.1, 133.3, 144.3, 144.4; MS = 255.2 [M+-H2O-H, 100%]; HRMS calcd for C15H14O3S [M+H] 275.0742, Found: 275.0736.
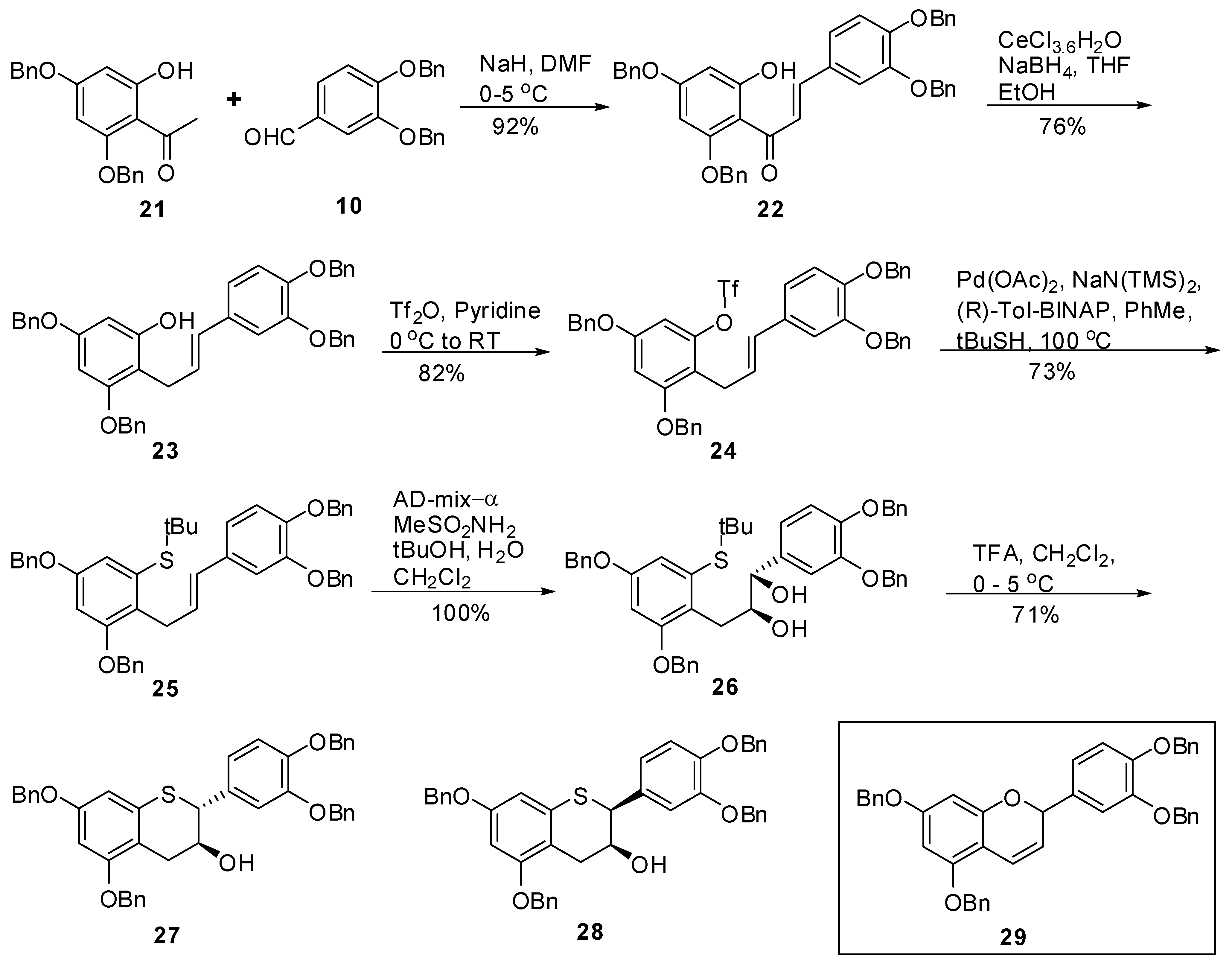
(E)-1-(2,4-Bis(benzyloxy)-6-hydroxyphenyl)-3-(3’,4’-bis(benzyloxy)phenyl)prop-2-en-1-one (22). To a cold solution (<5 °C) of NaH (60% dispersion in oil, 0.69 g, 17.23 mmol, 1.2 eq.) in DMF (50 mL) was added 21 (5 g, 14.36 mmol, 1 eq.) keeping the internal temperature < 5 °C throughout the addition. The resulting mixture was allowed to stir at this temperature for 20 minutes before addition of a solution of 10 (4.57g, 14.36 mmol, 1 eq.) in DMF (75 mL) over 15 minutes. The resulting mixture was allowed to warm to RT and stirred for 16 h. A gel was obtained. Water (75 mL) and EtOAc (75 mL) were added with stirring whereupon a solid started to appear. Heptane (200 mL) was then added, the solids were suction filtered and washed with heptane (2 × 50 mL). The solids were dried under high vacuum at 40–45 °C for 24 h to produce 22 (8.6g, 92%) as a yellow solid with 100% AUC purity. 1H- NMR (CDCl3) δ = 4.93 (s, 2H), 5.06 (s, 2H), 5.09 (s, 2H), 5.2 (s, 2H), 6.2 (dd, 2H, J = 1.6 and 4.6 Hz), 6.6–6.8 (m, 2H), 6.9 (s, 1H), 7.2–7.42 (m, 25H), 7.71 (q, 2H, J = 15.5 Hz); 13C-NMR (CDCl3) δ = 70.3, 70.8, 70.9, 71.4, 96.2, 97.8, 105.8, 114.1, 114.9, 123.1, 125.3, 126.9 (2C), 127.3 (2C), 127.2 (2C), 127.6 (2C), 127.8, 127.9, 128.0, 128.3 (2C), 128.4, 128.5 (4C), 128.6 (2C), 136.2, 136.3, 136.8, 136.9, 142.8, 149.5, 149.6, 161.1, 164.7, 166.3, 191.5. Anal. calcd for C43H36O6, C 79.61, H 5.59 Found C 79.58, H 5.36.
(E)-3,5-Bis(benzyloxy)-2-(3’,4’-bis(benzyloxy)phenyl)allyl)phenol (23). To a solution of ethanol (236 mL) and THF (800 ml) was added CeCl3.7H2O (74 g, 198.0 mmol, 2.5 eq.) at room temperature and the mixture was stirred at this temperature until a clear solution was obtained. To this was added chalcone 22 (51.4 g, 79.23 mmol, 1 eq.) followed by THF (500 mL). The solution was stirred at room temperature for ~10 minutes and then cooled to −1.5 to −0.2 °C (internal temperature) with agitation. Solid NaBH4 (7.5 g, 197.37 mmol, 2.5 eq.) was added in portions over 0.5 h keeping the internal temperature ≤ 0.3 °C throughout the addition. The mixture was stirred at this temperature (−0.8 to −0.3 °C) for ~2.5 h. The reaction mixture was quenched with 5% aqueous citric acid (167 mL) followed by EtOAc (1.5 L). The mixture was stirred as the internal temperature rose to ~12 °C. The organic layer was separated and washed with H2O (2 × 1L, 1 × 800 mL), brine (1 × 500 mL), dried (Na2SO4), filtered and the solvent was removed in vacuo to give a semi solid. HPLC analysis of the crude product indicated 86% product and 14% by-product (AUC). The crude product was purified by silica gel chromatography using heptane/CH2Cl2/EtOAc (25/25/0.5, v/v/v) to give 23 (38g, 76%) as an-off white solid with 99.5% AUC purity. 1H-NMR (CDCl3) δ = 3.55 (d, J = 5.4 Hz, 2H), 4.94–5.08 (m, 5H), 5.12 (d, J = 4.4 Hz, 4H), 6.04–6.2 (m, 2H), 6.22–6.4 (m, 2H), 6.82 (s, 2H), 6.97 (d, J = 1.2 Hz, 1H), 7.18–7.5 (m, 20H); 13C-NMR (CDCl3) δ = 26.4, 70.2, 70.4, 71.5, 93.6, 95.3, 107.0, 112.9, 115.3, 119.9, 126.7, 127.3, 127.3, 127.4, 127.5, 127.8, 127.8, 127.9, 128.0, 128.5, 128.6, 128.5, 128.6, 130.9, 136.5, 137.3, 137.4, 146.6, 148.2, 155.8, 157.9, 158.8. Anal. calcd for C43H38O5, C 81.36, H 6.03, Found C 81.22, H 5.86.
(E)-3,5-Bis(benzyloxy)-2-(3’,4’-bis(benzyloxy)phenyl)allylphenyl trifluoromethane sulfonate (24). To an ice cold solution of 23 (6 g, 9.45 mmol, 1 eq.) in pyridine (35 mL) was slowly added Tf2O (1.75 mL, 10.4 mmol, 1.1 eq). The resulting mixture was allowed to warm to RT and stirred for 18h. The reaction mixture was concentrated in vacuo and the residue was dissolved in EtOAc (250 mL) and washed with 1N HCl (2 × 100 mL). The aqueous layer was back washed with EtOAc (100 mL). The organic layers were combined and washed with brine (2 × 75 mL), dried (Na2SO4), filtered and the solvent removed in vacuo to produce the crude product. The crude product was purified by silica gel chromatography (10–15% EtOAc in heptane) to produce the 24 (4.1 g, 82%) as an off-white solid with 100% AUC purity. 1H-NMR (CDCl3) δ = 3.55 (d, 2H, J = 6.4 Hz), 5.0 (s, 2H), 5.05 (s, 2H), 5.08 (s, 2H), 5.1 (s, 2H), 5.95–6.1 (m, 1H), 6.28 (d, 1H, J = 16 Hz), 6.5 (d, 1H, J = 2.3 Hz), 6.6 (d, 1H, J = 2.3 Hz), 6.7–6.86 (m, 2H), 6.9 (d, 1H, J = 1.8 Hz), 7.3–7.5 (m, 20H); 13C-NMR (CDCl3) δ = 70.7, 71.5, 77.4, 100.4, 115.3, 125.1, 127.3, 127.4, 127.5, 127.6, 128.19, 128.5, 125.7, 128.8. Anal. calcd for C44H37F3O7S, C 68.92, H 4.86, F 7.43 Found C 68.89, H 4.77, F 7.22.
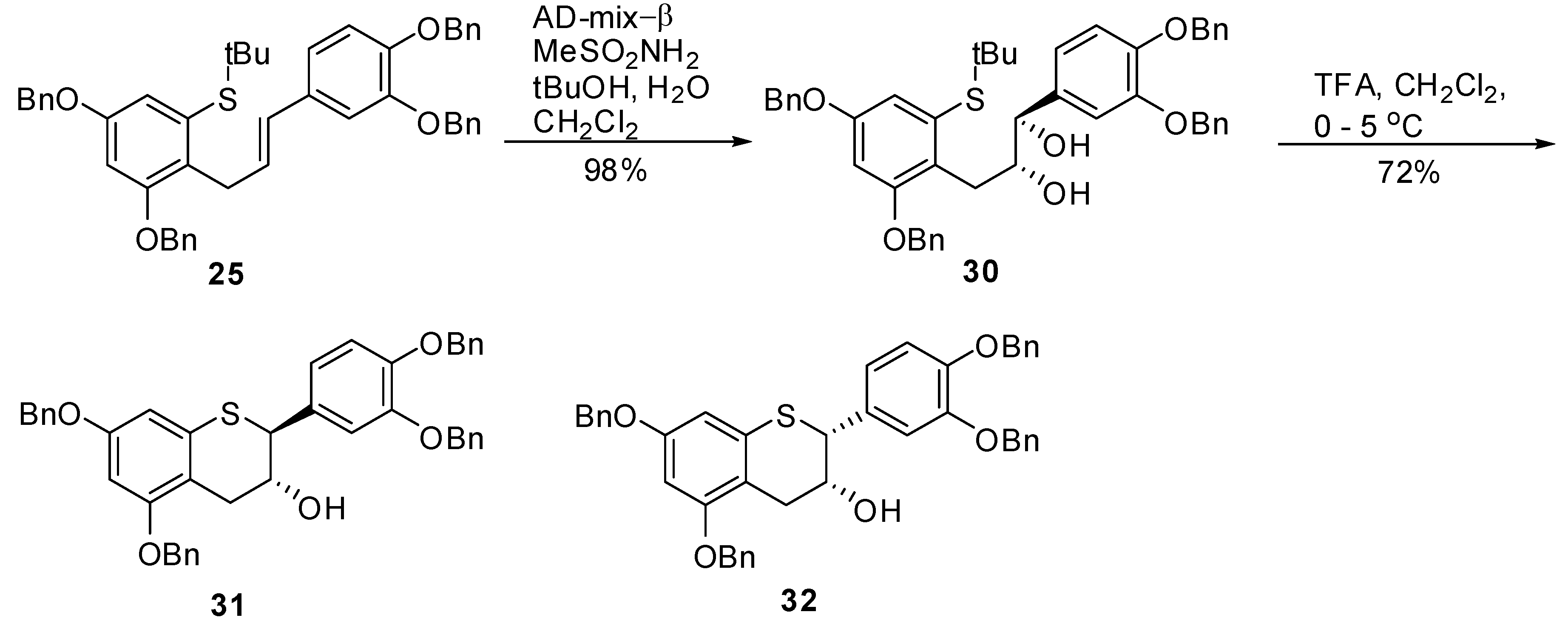
(E)-(3,5-Bis(benzyloxy)-2-(3-(3’,4’-bis(benzyloxy)phenyl)allyl)phenyl) (tert-butyl)sulfane (25). To a degassed solution of triflate 24 (1.86 g, 2.4 mmol, 1 eq.) in toluene (50 mL) was added Pd(OAc)2 (33 mg, 0.14 mmol, 0.06 eq.) and (R)-Tol-BINAP (115 mg, 0.17 mmol, 0.07 eq.) at room temperature. The mixture was again degassed for an additional 15 minutes at room temperature. In a separate flask, KN(TMS)2 (0.5M solution in toluene, 7 mL, 3.4 mmol, 1.4 eq.) was added to t-BuSH (0.39 mL) followed by stirring the mixture at RT for 15 minutes before adding this solution to the mixture of triflate containing catalyst. The resulting reaction mixture was kept at 100–105 ºC (bath temperature) for 36 h. The reaction was quenched by adding H2O (50 mL) and extracted with EtOAc (2 × 100 mL). The combined organic layers were washed with H2O (50 mL), brine (50 mL), dried, (Na2SO4), filtered and the solvent removed in vacuo to give the crude product. The crude product was then purified by silica gel chromatography (20–40% CH2Cl2 in heptane) to afford 25 (1.25 g, 73%) as an off-white solid with 98.3% AUC purity; 1H-NMR (CDCl3) δ = 1.28 (s, 9H), 3.85 (d, 2H, J = 5.9 Hz), 4.98 (s, 2H), 5.01 (s, 2H), 5.06 (s, 2H), 5.12 (s, 2H), 6.05–6.25 (m, 2H), 6.6 (d, 1H, J = 2.4 Hz), 6.7–6.85 (m, 3H), 6.9 (d, 1H, J = 1.8 Hz), 7.2–7.5 (m, 20H); 13C-NMR (CDCl3) δ = 31.3, 31.6, 47.4, 70.3, 70.3, 71.5, 71.6, 77.5, 101.8, 112.7, 113.4, 115.5, 119.7, 127.1, 127.4, 127.5, 127.5, 127.7, 127.7, 127.9, 128.1, 128.4, 128.4, 128.5, 128.6, 129.6, 132.3, 133.8, 136.9, 137.5, 139.5, 148.1, 149.2, 156.1, 157.1; MS (m/z) = 707.1 (M++1), 651.2 (M+-tBu). Anal. calcd for C47H46O4S, C 79.85, H 6.56, Found C 79.64, H 6.49.
(1S,2S)-(3-(2,4-Bis(benzyloxy)-6-(tert-butylthio)phenyl)-1-(3’,4’-bis(benzyloxy)phenyl)propane-1,2-diol (26). To a solution of tBuOH/H2O (1/1, v/v, 100 mL) was added AD-mix-α (16.5 g) and the suspension was stirred at RT until a clear solution was obtained. The solution was then cooled to 0-5 ºC and a solution of 25 (3.3 g, 4.67 mmol) in CH2Cl2 (100 mL) was added in one portion followed by CH3SO2NH2 (0.533 g, 5.6 mol, 1.2 eq.). The resulting reaction mixture was stirred at this temperature for 24 h. After 24h and 30h, additional amounts of AD-mix-α (16.5 g) and CH3SO2NH2 (0.533 g, 5.6 mol, 1.2 eq.) were added respectively and the stirring was continued for an additional 24h at this temperature. HPLC analysis indicated the completion of the reaction. To the reaction mixture was added H2O (100 ml) and EtOAc (150 mL). The organic layer was separated and washed with 10% aqueous sodium metabisulfite (2 x 100 mL), H2O (100 mL), brine (50 mL), dried (Na2SO4), filtered and the solvent removed in vacuo to afford the crude product as an off-white solid. The solid was triturated with heptane (50 mL) at room temperature and filtered to give 26 (3.48 g, 100%) as an off-white solid with 97% AUC purity; [α]20 D = -17.4 (c 1, acetone); Optical purity = 95% ee; 1H-NMR (CDCl3) δ = 0.2 (s, 6H), 1.2 (s, 9H), 2.4 (d, 2H, J = 6 Hz), 3.1 (d, 2H, J = 6.05 Hz), 3.15 (s, 1H), 3.8–3.95 (m, 1H), 4.35 (d, 1H, J = 4.5 Hz), 4.96 (s, 2H), 5.0 (s, 2H), 5.05 (s, 2H), 6.65 (d, 1H, J = 2.4 Hz), 6.75–6.9 (m, 2H), 7.05 (d, 1H, J = 1.4 Hz), 7.25–7.5 (m, 20H); 13C-NMR (CDCl3) δ = 31.1, 41.9, 70.3, 70.8, 71.4, 71.6, 77.5, 102.1, 115.2, 116.1, 120.2, 125.2, 127.3, 127.4, 127.5, 127.5 127.7, 128.1, 128.2, 129.4, 128.4, 128.7, 128.8, 134.5, 135.3, 137.4, 149.1; MS (m/z) = 723.1 (M+-OH). Anal. calcd for C47H48O6S, C 76.19, H 6.53, Found C 75.98, H 6.44.
(1R,2R)-(3-(2,4-Bis(benzyloxy)-6-(tert-butylthio)phenyl)-1-(3’,4’-bis(benzyloxy)phenyl)propane-1,2-diol (30). To a solution of tBuOH/H2O (1/1, v/v, 90 mL) was added AD-mix-β (6.1 g) and the suspension was stirred at RT until a clear solution was obtained. The solution was then cooled to 0–5 ºC and a solution of 25 (1.22 g, 1.73 mmol) in CH2Cl2 (60 mL) was added in one portion followed by CH3SO2NH2 (0.197 g, 2.07 mol, 1.2 eq.). The resulting reaction mixture was stirred at this temperature for 24 h and the progress of the reaction monitored by HPLC. After 24 h and 30 h, additional amounts of AD-mix-α (6.1 g) and CH3SO2NH2 (0.197 g, 2.07 mol, 1.2 eq.) were added respectively and stirring continued for an additional 24h at this temperature. To the reaction mixture was added H2O (50 mL) and EtOAc (100 mL). The organic layer was separated and washed with 10% aqueous sodium metabisulfite (2 × 100 mL), H2O (50 mL), brine (50 mL), dried (Na2SO4), filtered and the solvent removed in vacuo to afford the crude product as an off-white solid. The solid was purified by silica gel chromatography (30% EtOAc in heptane) to give 30 (1.2 g, 98%) as an off-white solid with 99.4% AUC purity. [α]20 D = +16.4 (c 1, acetone); Optical purity =95% ee; 1H-NMR (CDCl3) δ = 0.2 (s, 6H), 1.2 (s, 9H), 2.4 (d, 1H, J = 6.2 Hz), 3.05 (d, 2H, J = 6.2 Hz), 3.15 (d, 1H, J = 3.1 Hz), 3.75–3.82 (m, 1H), 4.3–4.4 (m, 1H), 4.95 (s, 2H), 5.0 (s, 2H), 5.1 (s, 4H), 6.6 (d, 1H, J = 2.4 Hz), 6.7–6.85 (m, 2H), 7.0 (s, 1H), 7.15–7.5 (m, 20H); 13C-NMR (CDCl3) δ = 31.1, 41.9, 70.3, 70.8, 71.4, 71.6, 77.5, 102.1, 115.2, 116.1, 120.2, 125.2, 127.3, 127.4, 127.5, 127.5, 127.7, 128.1, 128.2, 129.4, 128.4, 128.7, 128.8, 134.5, 135.3, 137.4, 149.1; MS (m/z) = 723.1 (M+-OH). Anal. calcd for C47H48O6S, C 76.19, H 6.53, Found C 76.11, H 6.39.
5,7,3’,4’-Tetra-O-benzyl-(+)-thiocatechin (27) and 5,7,3’,4’-Tetra-O-benzyl-(+)-thioepicatechin (28). To a cold solution (0–5 °C) of 26 (3.46 g, 4.68 mmol, 1 eq.) in CH2Cl2 (150 mL) was added TFA (1.28 g, 11.22 mmol, 2.4 eq.). The mixture was kept at 0-5 °C for 24 hours. Upon consumption of the starting material, the reaction was quenched by addition of Et3N (1.5 mL) and cold H2O (50 mL). The organic layer was separated and washed with brine (50 mL), dried (Na2SO4), filtered and the solvent removed in vacuo. The crude product was then purified by silica gel chromatography (heptane/CH2Cl2/EtOAc, 25/25/1, v/v/v) to afford 27 (1.75 g, 57%), and 28 (440 mg, 14%).
5,7,3’,4’-Tetra-O-benzyl-(+)-thiocatechin (27). [α]20 D = +55.2 (c 1, CH2Cl2); Chemical purity = 97% (AUC); Optical purity = 96% ee; 1H-NMR (CDCl3) δ = 2.0 (d, 1H, J = 5.5 Hz), 2.7 (dd, 1H, J = 8.4, 17.2 Hz), 3.1 (dd, 1H, J = 4.5, 17.2 Hz), 4.1 (d, 1H, J = 8.5 Hz), 4.15-4.3 (m, 1H), 4.95 (s, 4H), 5.1 (s, 2H), 5.12 (s, 2H), 6.4 (s, 2H), 6.85–6.95 (m, 2H), 7.01 (d, 1H, J = 1.5 Hz), 7.2–7.5 (m, 20H); 13C-NMR (CDCl3) δ = 29.8, 31.9, 51.3, 70.0, 70.2, 70.3, 71.3, 71.4, 77.5, 97.4, 103.3, 113.8, 115.2, 115.4, 121.9, 127.3, 127.5, 127.6, 127,9, 127.9, 127.9, 128.1, 128.5, 128.5, 128.6, 128.6, 130.62, 133.9, 136.8, 136.8, 137.0, 137.2, 148.1, 149.2, 157.1, 158.1; MS (m/z) = 667.5 (M++1). Anal. calcd for C43H38O5S, C 77.45, H 5.74, S 4.81 Found C 77.36, H 5.59, S 4.62.
5,7,3’,4’-Tetra-O-benzyl-(+)-thioepicatechin (28). [α]20 D = +61.3 (c 1, CH2Cl2); Optical purity = 96.6% ee; Chemical purity = 100% (AUC); 1H-NMR (CDCl3) δ = 2.2 (d, 1H, J = 9.6 Hz), 2.9 (q, ABq, JA = 3.9, 4.1, 17.4 Hz, JB = 4.9, 5.3 Hz), 4.3–4.42 (m, 2H), 4.95 (s, 2H), 5.12 (s, 2H), 5.14 (s, 2H), 6.3 (s, 2H), 6.82 (d, 1H, J = 8.3 Hz), 6.98 (dd, 1H, J = 2, 8.3 Hz), 7.12 (d, 1H, J = 2 Hz), 7.2–7.5 (m, 20H); 13C-NMR (CDCl3) δ = 31.4, 31.9, 49.8, 66.4, 70.0, 70.3, 71.3, 71.4, 77.5, 97.7, 102.3, 112.3, 114.9, 115.65, 121.7, 127.2,. 127.3, 127.5, 127.6, 127.8, 127.9, 128.1, 128.5, 128.5, 128.6, 128.6, 131.4, 133.7, 136.8, 136.8, 137.2, 137.3, 148.4, 148.9, 158.1, 158.5; MS (m/z) = 667.5 (M++1). Anal. calcd for C43H38O5S, C 77.45, H 5.74, S 4.81 Found C 77.28, H 5.63, S 4.66.
5,7,3’,4’-Tetra-O-benzyl-(-)-thiocatechin (31) and 5,7,3’,4’-tetra-O-benzyl-(-)-thioepicatechin (32). To a cold solution (0–5 °C) of 30 (1.26 g, 1.62 mmol, 1 eq.) in CH2Cl2 (60 mL) was added TFA (300 μL, 3.89 mmol, 2.4 eq.). The mixture was kept at 0–5 °C. Upon consumption of the starting material, the reaction was quenched by addition of Et3N (515 μL) and cold H2O (25 mL). The organic layer was separated and washed with brine (25 mL), dried (Na2SO4), filtered and the solvent removed in vacuo. The crude product was then purified by silica gel chromatography (heptane/CH2Cl2/EtOAc, 1/1/0 to 25/25/1, v/v/v) to afford the desired compounds 31 (0.82 g, 76%), and 32 (170 mg, 16%).
5,7,3’,4’-Tetra-O-benzyl-(-)-thiocatechin (31). [α]20 D= -62.5 (c 1, CH2Cl2); Chemical purity = 98.6% (AUC); Optical purity = 96% ee; 1H-NMR (CDCl3) δ = 1.95 (d, 1H, J = 4.4 Hz), 2.7 (dd, 1H, J = 8.4, 17.2 Hz), 3.12 (dd, 1H, J = 4.5, 17.2 Hz), 4.15 (d, 1H, J = 8.5 Hz), 4.18–4.32 (m, 1H), 4.98 (s, 4H), 5.1 (s, 2H), 5.12 (s, 2H), 6.3 (s, 2H), 6.8–6.92 (m, 2H), 7.02 (d, 1H, J = 1.5 Hz), 7.2–7.5 (m, 20H); 13C-NMR (CDCl3) δ = 29.8, 51.3, 70.0, 70.2, 70.3, 71.3, 71.4, 77.5, 97.4, 102.3, 113.8, 115.4, 121.9, 127.3, 127.5, 127.6, 127.9, 127.9, 127.9, 128.1, 128.2, 128.5, 128.5, 128.6, 128.6, 129.1, 130.6, 133.9, 136.8, 136.9, 137., 137.2, 149.2, 149.2, 157.8, 159.0; MS (m/z) = 667.5 (M++1). Anal. calcd for C43H38O5S, C 77.45, H 5.74, S 4.81 Found C 77.41, H 5.66, S 4.78.
5,7,3’,4’-Tetra-O-benzyl-(-)-thioepicatechin (32). [α]20 D= -58.4 (c 1, CH2Cl2); Chemical purity = 100% (AUC); Optical purity = 97% ee; 1H-NMR (CDCl3) δ = 2.2 (d, 1H, J = 10.5 Hz), 2.9 (ABq, JA = 4, 17.7 Hz, JB = 5, 17.9 Hz), 4.3–4.42 (m, 2H), 4.92 (s, 4H), 5.06 (s, 2H), 5.09 (s, 2H), 6.36 (s, 2H), 6.86 (d, 1H, J = 8.3 Hz), 6.95 (dd, 1H, J = 2.1, 8.3 Hz), 7.12 (d, 1H, J = 2 Hz), 7.2–7.5 (m, 20H); 13C-NMR (CDCl3) δ= 29.8, 51.3, 70.0, 70.2, 70.3, 71.3, 71.4, 76.6, 97.4, 102.3, 113.8, 115.2, 115.4, 121.9, 127.3, 127.5, 127.6, 127.9, 127.9, 127.9, 128.1, 128.2, 128.5, 128.5, 128.6, 128.6, 129.1, 130.6, 133.9, 136.8, 136.8, 137.0, 137.2, 140.2, 140.2, 157.3, 158.0; MS (m/z) = 667.5 (M++1). Anal. Calcd for C43H38O5S, C 77.45, H 5.74, S 4.81 Found C 77.37, H 5.65, S 4.58.
(+)-Thiocatechin (5). To an ice cold solution of 27 (1.43 g, 2.15 mmol, 1 eq.) in dry CH2Cl2 (60 mL) was added N,N-dimethylaniline (1.63 mL, 12.88 mol, 8 eq.) under N2. The solution was stirred at this temperature for ~10 minutes before AlCl3 (2.29 g, 17.18 mmol, 10 eq.) was added in one portion. The flask was covered with aluminum foil to protect it from light and stirred at ice bath temperature for 2 h. EtOAc (50 mL) and silica gel (5 g) were added to the reaction mixture and stirred for 5 minutes. The reaction mixture was filtered through a pad of silica gel. The silica gel pad was washed with EtOAc (5 × 50 mL). The filtrates were combined and the solvent was removed in vacuo to ~10 mL of volume, keeping the bath temperature < 25 °C. The crude mixture was diluted with purified water (50 mL), cooled and lyophilized to give the crude product. The crude product was further purified by preparative HPLC to give 5 (380 mg, 57%) as an off-white solid in 99% AUC purity. [α]20 D = +32.2 (c 1, Acetone); 1H-NMR (acetone-d6) δ = 2.58 (dd, 1H, J = 8.3, 15.5 Hz), 3.18 (d, 1H, J = 16.3 Hz), 4.17 (d, 1H, J = 8.3 Hz), 4.32 (s, 1H), 6.08 (s, 1H), 6.2 (s, 1H), 6.8 (s, 2H), 6.92 (s, 1H); 13C-NMR (acetone-d6) δ = 32.3, 52.3, 71.1, 99.9, 103.6, 112.3, 116.0, 116.5, 121.4, 131.3, 135.7, 145.6, 145.8, 157.0; MS = 307.1 [M++H]; HRMS Calcd for C15H15O5S [M+H] 307.0642 Found 307.0644.
(-)-Thioepicatechin (6). To an ice cold solution of 32 (0.67 g, 1.01 mmol, 1 eq.) in dry CH2Cl2 (35 mL) was added N,N-dimethylaniline (1.02 mL, 8.05 mol, 8 eq.) under N2. The solution was stirred at this temperature for ~10 minutes before AlCl3 (1.37 g, 10.1 mmol, 10 eq.) was added in one portion. The flask was covered with aluminum foil to protect it from light and stirred at the ice bath temperature for 2 h. EtOAc (50 mL) and silica gel (5 g) were added to the reaction mixture and stirred for 5 minutes. The reaction mixture was filtered through a pad of silica gel. The silica gel pad was washed with EtOAc (3 × 50 mL). The filtrates were combined and the solvent was removed in vacuo to ~10 mL of volume keeping the bath temperature < 25 °C. The crude mixture was diluted with purified water (50 mL), cooled and lyophilized to give the crude product. The crude product was further purified by preparative HPLC to give 6 (100 mg, 57%) as an off-white solid in 99.8% AUC purity. [α]20 D= -28.9 (c 1, acetone); 1H-NMR (acetone-d6) δ = 2.7 (dd, 1H, J = 6.7, 17 Hz), 2.82 (dd, 1H, J = 4.1, 17 Hz), 4.32 (d, 1H, J = 2.3 Hz), 4.33–4.5 (br s, 1H), 6.16 (dd, 2H, J = 2.3, 11.8 Hz), 6.72 (d, 1H, J = 8 Hz), 6.82 (dd, 1H, J = 2.3, 11.8 Hz), 7.08 (d, 1H, J = 2 Hz), 7.7–8.3 (br s, 4H); 13C-NMR (acetone-d6) δ = 50.0, 68.0, 99.9, 104.1, 110.6, 115.5, 117.1, 121.7, 133.0, 135.2, 145.3, 145.4, 156.9, 157.5; MS = 307.1 [M++H]; HRMS Calcd for C15H15O5S [M+H] 307.0642 Found 307.0634.
(-)-Thiocatechin (7). To an ice cold solution of 31 (0.92 g, 1.38 mmol, 1 eq.) in dry CH2Cl2 (40 mL) was added N, N-dimethylaniline (1.08 mL, 8.5 mol, 6.2 eq.) under N2. The solution was stirred at this temperature for ~10 minutes before AlCl3 (1.51 g, 11 mmol, 8.2 eq.) was added in one portion. The flask was covered with aluminum foil to protect it from light and stirred at the ice bath temperature for 2 h. EtOAc (50 mL) and silica gel (5 g) were added to the reaction mixture and stirred for 5 minutes. The reaction mixture was filtered through a pad of silica gel. The silica gel pad was washed with EtOAc (3 × 50 mL). The filtrates were combined and the solvent was removed in vacuo to ~10 mL of volume keeping the bath temperature < 25 °C. The crude mixture was diluted with purified water (50 mL), cooled and lyophilized to give the crude product. The crude product was further purified by preparative HPLC to give 7 (205 mg, 56%) as an off-white solid in 99% AUC purity. [α]20 D= -28.6 (c 1, acetone); 1H-NMR (acetone-d6) δ = 2.58 (dd, 1H, J = 5.9, 16.7 Hz), 3.18 (dd, 1H, J = 4.7, 16.7 Hz), 3.8–3.96 (br s, 1H), 4.1 (d, 1H, J = 9 Hz), 4.16–4.3 (m, 1H), 6.12 (d, 1H, J = 2.2 Hz), 6.2 (d, 1H, J = 2.2 Hz), 6.72 (s, 2H, J = 0.8 Hz), 6.9 (s, 1H), 7.6–8.3 (br s, 4H); 13C-NMR (acetone-d6) δ = 32.4, 52.3, 71.1, 98.5, 103.5, 112.0, 115.9, 116.5, 121.4, 131.3, 135.8, 145.5, 145.8, 157.0; MS = 307.1 [M++H]; HRMS Calcd for C15H15O5S [M+H] 307.0642 Found 307.0633.
(+)-Thioepicatechin (8). To an ice cold solution of 28 (0.4 g, 0.6 mmol, 1 eq.) in dry CH2Cl2 (20 mL) was added N,N-dimethylaniline (0.6 mL, 4.81 mol, 8 eq.) under N2. The solution was stirred at this temperature for ~10 minutes before AlCl3 (0.8 g, 6.01 mmol, 10 eq.) was added in one portion. The flask was covered with aluminum foil to protect it from light and stirred at the ice bath temperature for 2 h. EtOAc (50 mL) and silica gel (5 g) were added to the reaction mixture and stirred for 5 minutes. The reaction mixture was filtered through a pad of silica gel. The silica gel pad was washed with EtOAc (3 × 50 mL). The filtrates were combined and the solvent was removed in vacuo to ~10 mL of volume keeping the bath temperature < 25 °C. The crude mixture was diluted with purified water (50 mL), cooled and lyophilized to give the crude product. The crude product was further purified by preparative HPLC to give 8 (124 mg, 52%) as an off-white solid in 100% AUC purity. [α]20 D= +29.7 (c 1, acetone); 1H-NMR (acetone-d6) δ = 2.7 (dd, 1H, J = 6.7, 17 Hz), 2.82 (dd, 1H, J = 4.1, 17 Hz), 4.32 (d, 1H, J = 2.3 Hz), 4.33-4.5 (br s, 1H), 6.16 (dd, 2H, J = 2.3, 11.8 Hz), 6.72 (d, 1H, J = 8 Hz), 6.82 (dd, 2H, J = 2.3, 11.8 Hz), 7.08 (d, 1H, J = 2 Hz), 7.7–8.3 (br s, 4H); 13C-NMR (acetone-d6) δ = 50.0, 68.0, 99.9, 104.1, 110.6, 115.5, 117.1, 121.7, 133.0, 135.2, 145.3, 145.4, 156.9, 157.5; MS = 307.1 [M++H]; HRMS Calcd for C15H15O5S [M+H] 307.0642 Found 307.0631.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
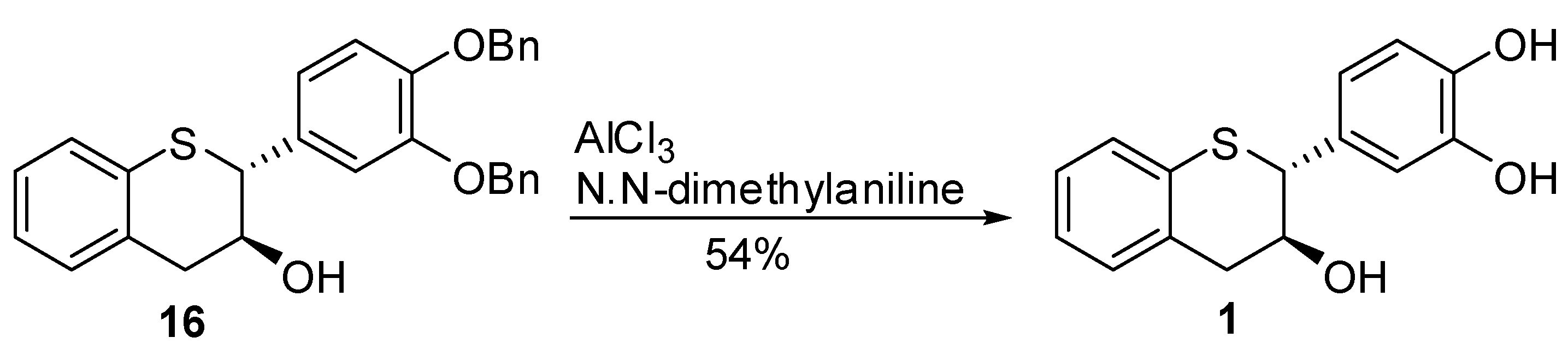{kind=link}
{kind=link}
{kind=link}