3.1. General
All reactions were carried out under inert atmosphere with dry solvents under anhydrous conditions, unless otherwise stated. CH2Cl2 was distilled from calcium hydride, DMF was distilled under vacuum, CH3CN was distilled from CaH2, and THF distilled from potassium. TLC was run on Silica Gel 60 F254 (Merck) and the spots were detected in UV-light and stained with H2SO4 in ethanol and heat. Silica gel (Matrex, 60 Å, 35–70 mm, Grace Amicon) and solvents of analytical grade were used for flash column chromatography except where otherwise is stated. In these cases, Biotage Isolera OneTM with SNAP KP-SIL columns (10–50 g) and n-heptane-EtOAc gradients were used for flash column chromatography, typically with a flow rate of 12–40 mL min-1, depending on the size of the column. 1H- and 13C-NMR spectra were recorded at 298 K on a Bruker DRX-400 instrument at 400 and 100 MHz, respectively, with CDCl3 or D2O as solvents and residual CHCl3 (δH 7.27 ppm) or D2O (δH 4.79 ppm) as internal standard for 1H and CDCl3 (δC 77.23 ppm) as internal standard for 13C. Peaks that could not be assigned are not reported. J values are given in Hz. Proton decoupled 19F-NMR spectra were recorded at 298 K on the Bruker DRX-400 at 376 MHz in CDCl3 with CFCl3 (δF0.00 ppm) as internal standard. Positive and negative electrospray mass analyses were carried out on a Waters Micromass ZG 2000. Preparative HPLC separations were performed on a Beckman System Gold HPLC, using a Supelco Discovery Biowide Pore C18 column (250 × 212 mm, 5 μm) eluted with a linear gradient of CH3CN in water, both of which contained trifluoroacetic acid (0.1%). The flow rate was 11 mL min-1 and detection at 214 nm. Analytical HPLC were performed on a Beckman System Gold HPLC, using a Supelco Discovery Biowide Pore C18 column (250 × 46 mm, 5 μm) with a flow rate of 1.5 mL min-1 and detection at 214 nm.
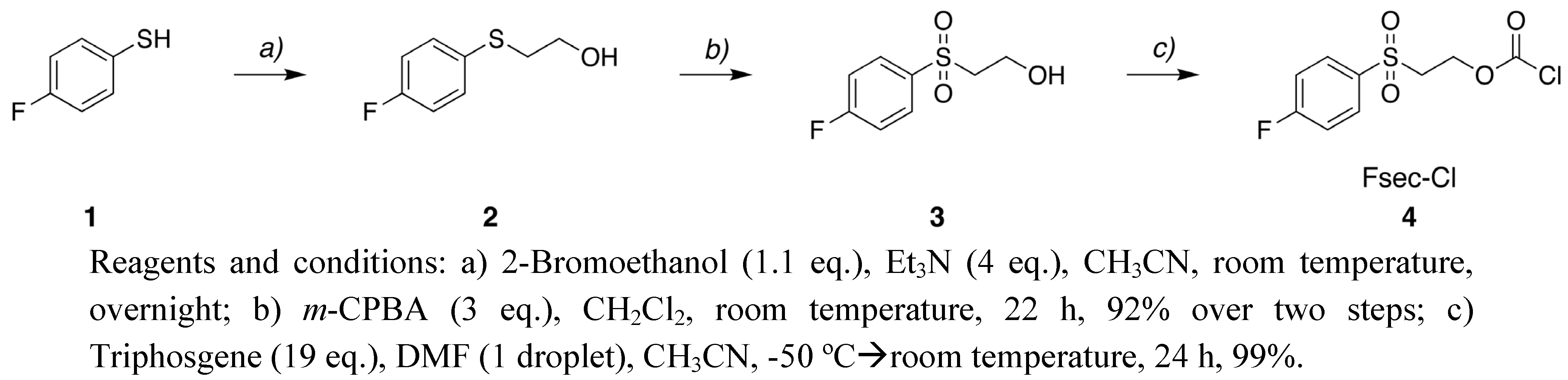
2-[(4-Fluorophenyl)thio]ethanol (2). 2-Bromoethanol (2.72 g, 21.8 mmol) was slowly added to a mixture of 4-fluorothiophenol (1, 2.56 g, 20.0 mmol) and triethylamine (11.1 mL, 80 mmol) in CH3CN (50 mL) at room temperature. The reaction mixture was stirred over night and the solvent was removed under reduced pressure. To the residue diethyl ether was added to precipitate the bromide salt. The salt was filtrated by suction and washed twice with diethyl ether. The filtration was concentrated to give 2 as a yellow oil (3.82 g) that was used without further purification. 1H-NMR (CDCl3) δ 7.43–7.35 (m, 2H, Ar-H), 7.04–6.95 (m, 2H, Ar-H), 3.70 (t, 2H, J = 6.0 Hz, -SCH2CH2OH), 3.04 (t, 2H, J = 6.0 Hz, -SCH2CH2OH); 19F-NMR (CDCl3) δ -115.1; MS(ES+) calculated for C8H10FOS (M+H+) 173.04, found 173.08.
2-[(4-Fluorophenyl)sulfonyl]ethanol (
3) [
19].
m-Chloroperbenzoic acid (8.72 g, 38.7 mmol) was slowly added to
2 (2.32 g, 13.4 mmol) in CH
2Cl
2 (100 mL) at 0 °C. The reaction mixture was stirred for 22 h at room temperature. The solvent was concentrated to precipitate
m-chlorobenzoic acid, which was removed by filtration. The crude product was purified by column chromatography (30–100% EtOAc in petroleum ether) to give
3 (2.45g, 92% yield)as a white solid in.
1H-NMR (CDCl
3) δ8.01–7.92 (m, 2H, Ar-H), 7.33–7.21 (m, 2H, Ar-H), 4.07–3.97(m, 2H, -SO
2CH
2C
H2OH), 3.35 (t, 2H,
J = 5.6 Hz, -SO
2C
H2CH
2OH), 2.64 (br. 1H, -CH
2O
H);
19F NMR (CDCl
3): δ -103.2; MS(ES
+) calculated for C
8H
10FO
3S (M+H
+) 205.03, found 205.29; m.p. 60–61 °C.
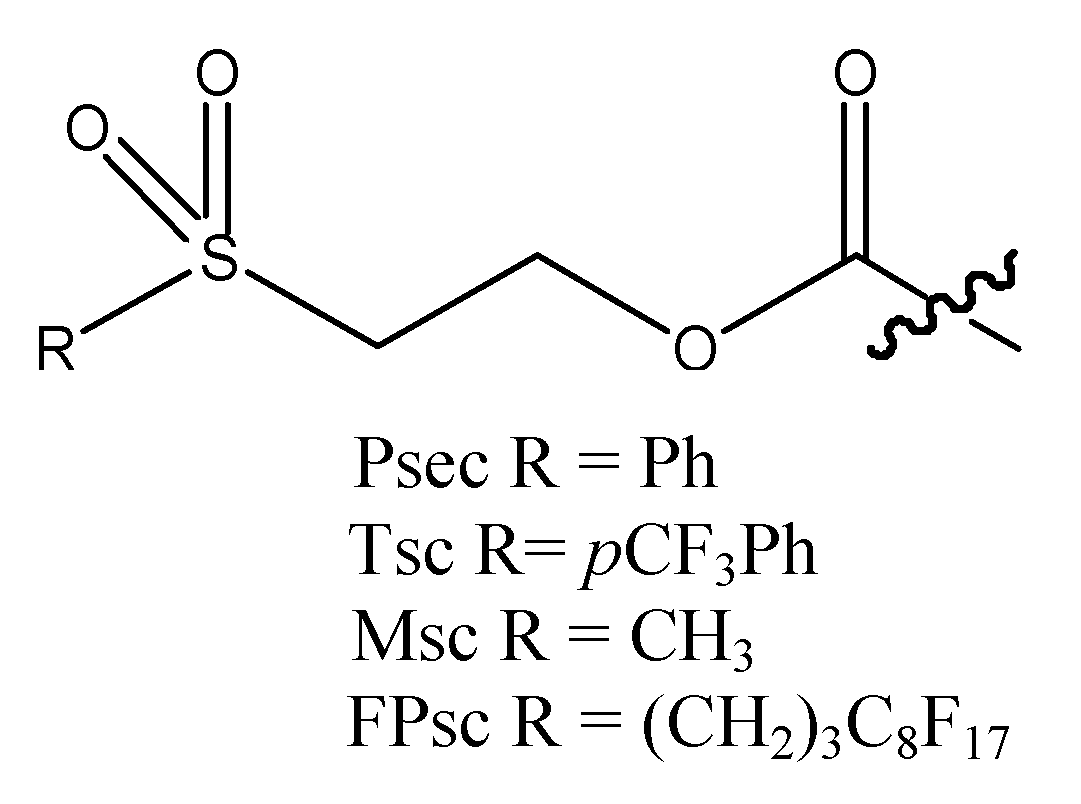
2-[(4-Fluorophenyl)sulfonyl]ethoxy carbonyl chloride (4). One drop of DMF was added to triphosgene (14.9 g, 0.15 mol) in CH3CN (40 mL) and then the solution was cooled to -50 °C. Compound 3 (2.05 g, 10.0 mmol) in CH3CN (50 mL) was slowly added to the reaction mixture and then stirred for 24 h at room temperature. Residual phosgene and hydrogen chloride were removed by purging with nitrogen gas for 40 min after which time the solvent was evaporated in vacuo to give 4 (2.65 g) as a white solid in quantitative yield. 1H-NMR (CDCl3) δ 8.00–7.91 (m, 2H, Ar-H), 7.33–7.23 (m, 2H, Ar-H), 4.65 (t, 2H, J = 5.6 Hz, -SO2CH2CH2O-), 3.53 (t, 2H, J = 5.6 Hz, -SO2CH2CH2O-);13C-NMR (CDCl3) δ 166.1 (d, J = 256.0 Hz), 116.9 (d, J = 22.5 Hz), 150.7, 135.0 (d, J = 3.1 Hz), 131.1 (d, J = 9.7 Hz), 54.3, 54.5; 19F-NMR (CDCl3): δ -102.7; m.p. 72–74 °C.
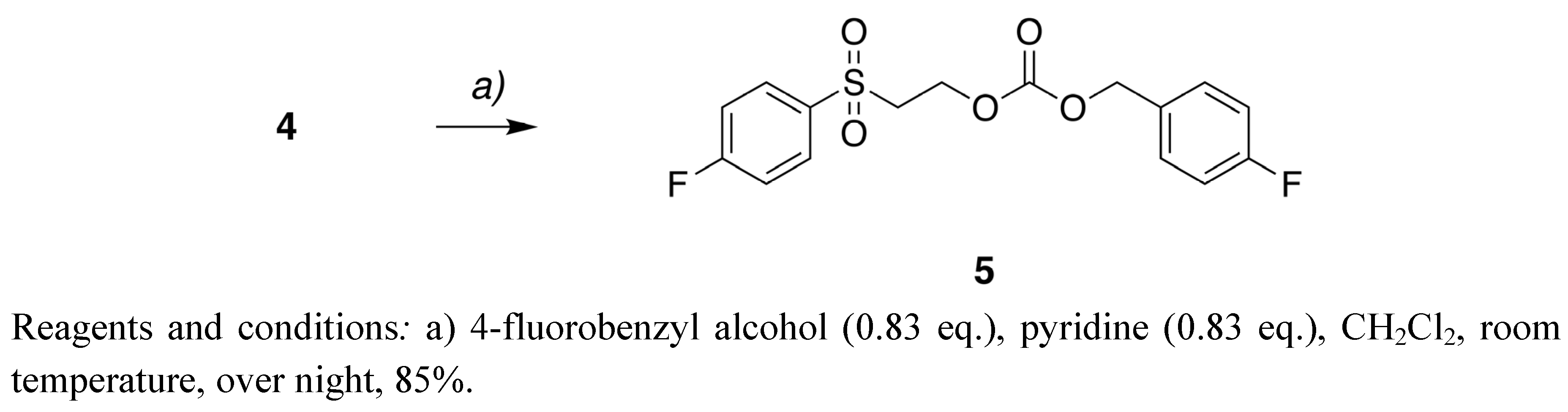
4-Fluorobenzyl 2-[(4-fluorobenzyl)sulfonyl]ethyl carbonate (5). Compound 4 (0.16 g, 0.60mmol) was added to pyridine (0.047 g, 0.60 mmol) and 4-fluorobenzyl alcohol (0.064 g, 0.50 mmol) in CH2Cl2 (2 mL). The mixture was stirred over night at room temperature. The solvent was removed under reduced pressure and the residue was purified by column chromatography (petroleum ether-EtOAc 3:1) to give 5 (0.151 g) in 85% yield.1H-NMR (CDCl3) δ 7.97–7.86 (m, 2H, Ar-H), 7.34–7.24 (m, 2H, Ar-H), 7.22–7.13 (m, 2H, Ar-H), 7.08–6.99 (m, 2H, Ar-H), 5.01 (s, 2H, -CH2O-), 4.46 (t, 2H, J = 6.0 Hz, -SO2CH2CH2O-), 3.48 (t, 2H, J = 6.0 Hz, -SO2CH2CH2O-); 13C-NMR (CDCl3) δ 166.0 (d, J = 255.6 Hz), 163.0 (d, J = 246.0 Hz), 154.2, 135.4 (d, J = 3.3 Hz), 131.2 (d, J = 9.8 Hz), 130.7 (d, J = 3.3 Hz), 130.5 (d, J = 8.4 Hz), 116.7 (d, J = 22.6 Hz), 115.7 (d, J = 21.4 Hz), 69.3, 69.3, 55.1; 19F-NMR (CDCl3): δ -113.2, -103.3; MS(ES−) calculated for C17H15F2O7S (M+HCO2−) 401.05, found 401.12.
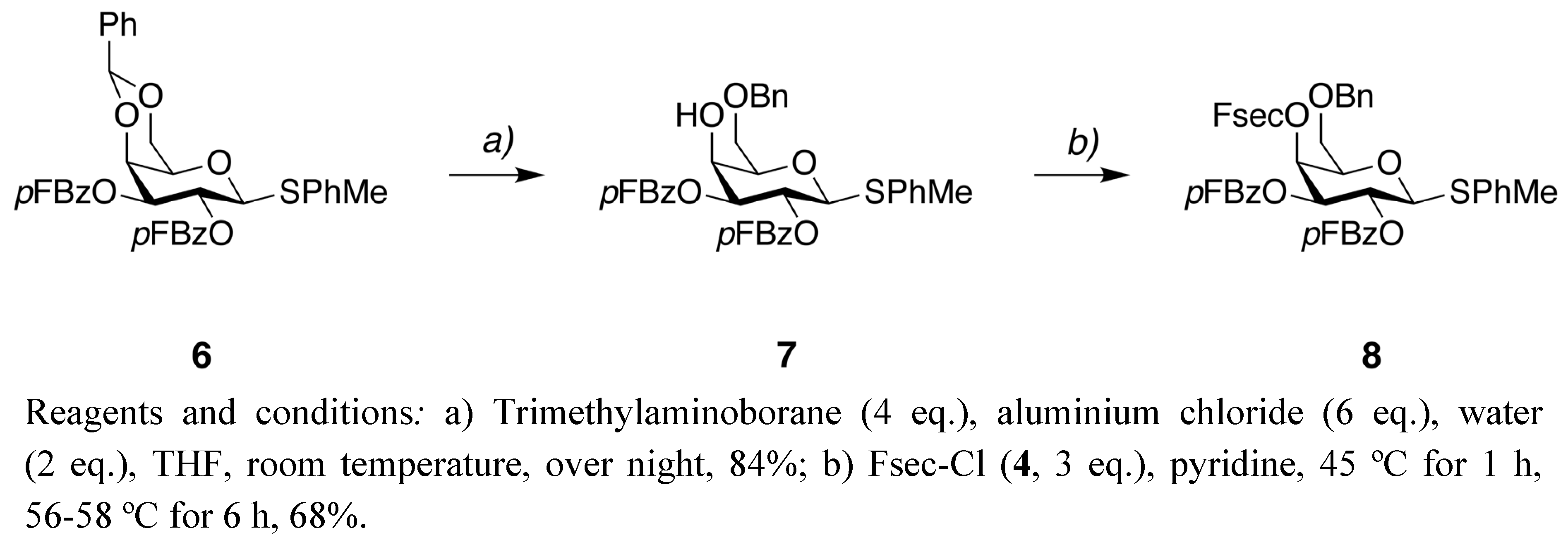
4-Methylphenyl 2,3-di-O-(4-fluorobenzoyl)-6-O-benzyl-1-thio-β-D-galactopyranoside (
7). Trimethyl-aminoborane (0.238 g, 3.26 mmol) was added to
6 [
20] (0.500 g, 0.808 mmol) in distilled THF (25 mL) followed by aluminium chloride (0.639 g, 4.79 mmol). After dissolution of the reagents water (0.029 mL, 1.61 mmol) was added and the mixture was stirred at room temperature overnight. Water and HCl (1M) was added to terminate the reaction and the mixture was extracted with EtOAc. The organic phase was washed with brine, dried with MgSO
4, and concentrated. Purification with flash column chromatography (Isolera, SNAP KP-SIL, 5–100% EtOAc in n-heptane, 22 min) gave
7 (0.419 g) in 84% yield.
1H-NMR (CDCl
3) δ 8.06–7.93 (m, 4H, ArH), 7.45 (d, 2H,
J = 8.0 Hz, ArH), 7.40–7.28 (m, 5H, ArH), 7.12–6.93 (m, 6H, ArH), 5.79 (t, 1H,
J = 9.9 Hz, H-2), 5.34 (dd, 1H,
J = 2.9 and 9.9 Hz, H-3), 4.93 (d, 1H,
J = 9.9 Hz, H-1), 4.61 (s, 2H, -OC
H2C-), 4.43 (s, 1H, H-4), 3.96–4.83 (m, 3H, H-5 and H-6), 2.32 (s, 3H, -C
H3);
13C-NMR (CDCl
3) δ 167.1 (
J = 6.7 Hz), 164.6 (
J = 6.5 Hz), 167.5 (
J = 54.9 Hz), 137.9 (
J = 68.0 Hz), 133.2, 132.3 (
J = 10.7 Hz), 129.6, 128.4, 128.3, 127.8, 127.7, 125.5 (
J = 37.6 Hz), 125.4 (
J = 37.6 Hz), 115.5 (
J = 20.3 Hz), 115.6 (
J = 20.3 Hz), 86.6, 77.2, 75.7, 73.7, 69.6, 68.3, 68.2, 21.1;
19F-NMR (CDCl
3) δ -105.0, -105.4; MS(ES+) calculated for C
34H
31F
2O
7S (M+H) 621.18, found 620.99.
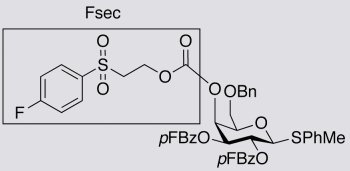
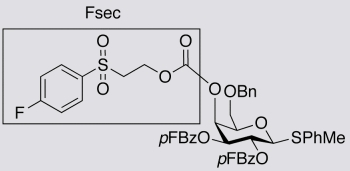
4-Methylphenyl6-O-benzyl-2,3-di-O-(4-fluorobenzoyl)-4-O-[2-(4-fluorophenyl)sulfonylethoxy-carbonyl]-1-thio-β-D-galactopyranoside (8). Compound 4 (0.08 g, 0.30 mmol) was added to 7 (0.062 g, 0.10 mmol) in pyridine (1.0 mL). The mixture was stirred at 45 °C for 1 h and then at 56–58 °C for 6 h. The pyridine was removed under reduced pressure and the residue was purified by column chromatography (toluene-EtOAc 3:1) to give 8 (0.058 g) as a white solid in 68% yield. 1H- NMR (CDCl3) δ 7.95–8.02 (m, 2H, Ar-H), 7.84–7.94 (m, 4H, Ar-H), 7.27–7.40 (m, 8H, Ar-H), 6.97–7.11 (m, 6H, Ar-H), 5.59 (t, 1H, J = 10.0 Hz, H-2), 5.48 (m, 1H, H-4), 5.39 (dd, 1H, J = 3.6 and 10.0 Hz, H-3), 4.87 (d, 1H, J = 10.0 Hz, H-1), 4.51 (dd, 2H, J = 11.6 and 26.2 Hz, -CH2O-), 4.28 (t, 1H, J = 6.4 Hz, -SO2CH2CH2O-), 3.99 (t, 1H, J = 6.4 Hz, H-5), 3.68 (dd, 1H, J = 6.0 and 9.8 Hz, H-6), 3.58 (dd, 1H, J = 6.8 and 9.8 Hz, H-6), 3.29–3.38 (m, 2H, -SO2CH2CH2O-), 2.32 (s, 3H, -CH3); 13C-NMR (CDCl3) δ 166.2 (J = 256.0 Hz), 166.1 (J = 254.1 Hz), 166.1 (J = 253.6 Hz), 164.4, 164.4, 154.1, 138.5, 137.7, 135.4 (J = 3.4 Hz), 133.1, 132.5 (J = 9.3 Hz), 132.4 (J = 9.4 Hz), 131.2 (J = 9.7 Hz), 129.9, 129.9, 128.9, 128.5, 128.0, 127.9, 125.5 (J = 3.1 Hz), 125.1 (J = 3.0 Hz), 117.0 (J = 22.7 Hz), 115.9 (J = 21.9 Hz), 115.8 (J = 21.9 Hz), 87.4, 75.9, 73.7, 73.1, 72.6, 68.4, 67.7, 61.4, 55.0, 21.3; 19F- NMR (CDCl3) δ -105.1, -104.6, -102.8 (Fsec); MS(ES+) calculated for C43H39F3O12S2 (M+H2O) 868.18, found 868.43; [α]D25 = 4.6° (c 1.0, CHCl3).
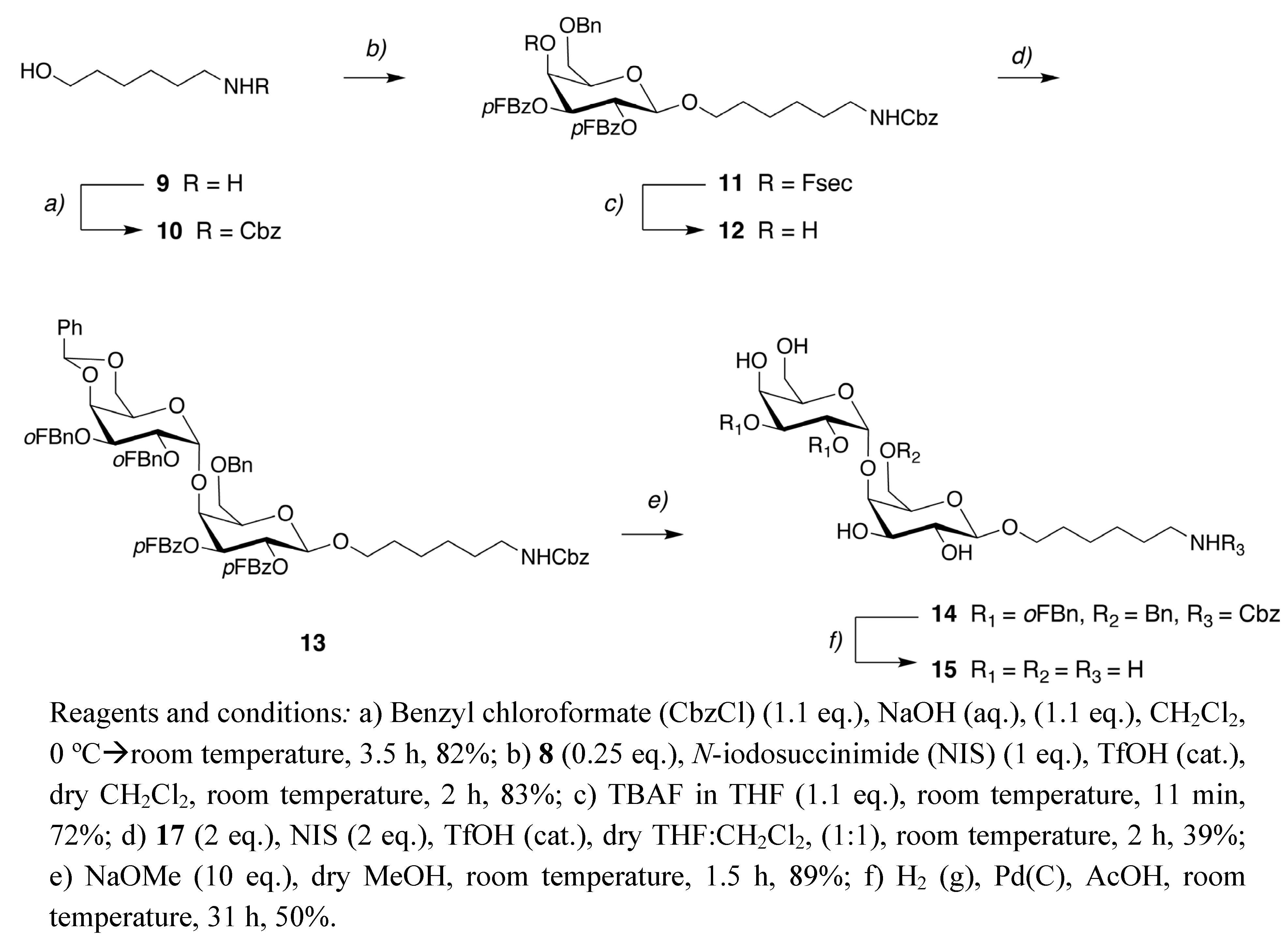
6-Benzyloxycarbonylamino-1-hexanol (10). Benzyl chloroformate (CbzCl, 0.390 mL, 2.73 mmol) was added dropwise to a solution of 6-amino-1-hexanol (9, 0.301 g, 2.57 mmol) in NaOH (aq.) (2.81 mL, 2.81 mmol) at 0 °C. The solution was allowed to reach room temperature, stirred for 30 min and then CH2Cl2 was added (2 mL). After stirring at room temperature for 3 h the mixture was concentrated in vacuo. The residue was purified with flash column chromatography (Isolera, SNAP KP-SIL, 5–95% EtOAc in n-heptane, 22 min) to give 10 (0.528 g) in 82% yield.1H-NMR (CDCl3) δ 7.38–7.27 (m, 5H, ArH), 5.08 (s, 2H, -COOCH2C-), 4.87 (s, 1H, -NH-), 3.60 (t, 2H, J = 6.5 Hz, -CH2OH), 3.23–3.09 (m, 2H, -CH2NH-), 1.59–1.45 (m, 4H, -CH2CH2OH, -CH2CH2NH-), 1.42–1.23 (m, 4H, 2 × -CH2CH2CH2-); 13C-NMR (CDCl3) δ 156.6, 136.7, 128.6, 128.2, 66.7, 62.2, 41.0, 32.6, 30.0, 26.4, 25.4; MS(ES+) calculated for C14H22NO (M+H) 252.16, found 252.42.
6-(Benzyloxycarbonylamino)-hexyl 6-O-benzyl-2,3-di-O-(4-fluorobenzoyl)-4-O-[2-(4-fluorophenyl-sulfonyl)ethoxycarbonyl]-β-D-galactopyranoside (11). Compound 10 (0.118 g, 0.470 mmol), 8 (0.100 g, 0.118 mmol), and NIS (0.106g, 0.471 mmol) were dried under vacuum and in the absence of light for 2 h. Distilled CH2Cl2 (5 mL) and TfOH (0.1 M in CH2Cl2, 0.001 mmol) were added and the solution was stirred in room temperature in the absence of light for 2 h. The reaction mixture was concentrated and the residue was purified with flash column chromatography (Isolera, SNAP KP-SIL, 5–95% EtOAc in n-heptane, 22 min) giving 11 (0.064 g) in 83% yield.1H-NMR (CDCl3) δ 8.02–7.86 (m, 6H, ArH), 7.39–7.24 (m, 12H, ArH), 7.11–6.99 (m, 4H, ArH), 5.56 (dd, 1H, J = 8.0 and 10.3 Hz, H-2), 5.44 (d, 1H, J = 3.3 Hz, H-4), 5.37 (dd, 1H, J = 3.3 and 10.3 Hz, H-3), 5.09 (s, 2H, -COOCH2Ph), 4.65 (d, 1H, J = 8.0 Hz, H-1), 4.51 (q, 2H, J = 11.9 and 26.7 Hz, -OCH2Ph), 4.29 (t, 2H, J = 5.7 Hz, -COOCH2CH2-), 3.96 (t, 1H, J = 6.5 Hz, H-5), 3.92–3.85 (m, 1H, -OCHHCH2CH2-), 3.67–3.62 (m, 1H, H-6), 3.60–3.53 (m, 1H, H-6), 3.52–3.44 (m, 1H, -OCHHCH2CH2-), 3.39–3.30 (m, 2H, -COOCH2CH2-), 3.10–3.01 (m, 2H, -NHCH2-), 1.58–1.44 (m, 2H, -OCH2CH2CH2-), 1.35–1.13 (m, 6H, -CH2CH2CH2NH-); 13C-NMR (CDCl3) δ 167.4 (J = 8.6 Hz), 167.2, 164.9 (J = 6.5 Hz), 164.7, 164.4, 164.3, 156.4, 154.0, 137.6, 136.7, 135.3 (J = 5.3 Hz), 132.3 (J = 4.2 and 9.2 Hz), 131.2 (J = 9.8 Hz), 128.5 (J = 7.2 Hz), 128.2, 127.9, 127.8, 125.6 (J = 4.7 Hz), 125.1 (J = 4.3 Hz), 117.0, 116.8, 115.7 (J = 15.9 and 22.1 Hz), 101.2 (C-1), 77.2, 73.6, 72.3, 71.9, 71.8, 69.9, 69.7, 67.4, 66.7, 61.3, 54.9, 40.9, 29.7, 29.3, 26.8, 25.5; 19F-NMR (CDCl3) δ -103.0, -104.6, -105.2; MS(ES+) calculated for C50H51F3NO14S (M+H) 978.30, found 978.46.
6-(Benzyloxycarbonylamino)hexyl 6-O-benzyl-2,3-di-O-(4-fluorobenzoyl)-β-D-galactopyranoside (12). Compound 11 (0.013 g, 0.013 mmol) was dissolved in THF (13 mL) and TBAF in THF (0.015 mL, 0.015 mmol) was added. The solution was stirred at room temperature for 11 min and purification by preparative HPLC (30–100% CH3CN in H2O, 50 min) afforded 12 (7.2 mg) in 72% yield.1H-NMR (CDCl3) δ 8.07–7.96 (m, 4H, ArH), 7.38–7.28 (m, 10H, ArH), 7.08–6.99 (m, 4H, ArH), 5.72 (dd, 1H, J = 8.0 and 10.3 Hz, H-2), 5.25 (dd, 1H, J = 3.1 and 10.3 Hz, H-3), 5.09 (s, 2H, -COOCH2), 4.72 (t, 1H, J = 5.8 Hz), 4.63 (d, 1H, J = 8.0 Hz, H-1), 4.61 (d, 2H, J = 3.1 Hz, -OCH2Ph), 4.38 (d, 1H, J = 2.9 Hz, H-4), 3.96–3.87 (m, 1H, -OCHHCH2-), 3.86–3.78 (m, 3H), 3.54–3.46 (m, 1H, -OCHHCH2-), 3.05 (s, 2H, -CH2CH2NH-), 1.60–1.41 (m, 2H, -OCH2CH2CH2-), 1.36–1.10 (m, 6H, -OCH2CH2CH2CH2CH2CH2NH-); 13C-NMR (CDCl3) δ 167.4 (J = 16.5 Hz), 164.8 (J = 16.4 Hz), 164.8 (J = 53.8 Hz), 137.5, 132.6 (J = 9.5 Hz), 132.4 (J = 9.4 Hz), 128.7, 128.3, 128.1, 125.9 (J = 2.8 Hz), 125.5 (J = 2.8 Hz), 115.8 (J = 22.1 Hz), 115.7 (J = 22.1 Hz), 101.6, 74.5, 74.0, 73.3, 70.1, 70.0, 69.4, 68.4, 66.9, 41.0, 29.7, 29.3, 26.3, 25.6; 19F-NMR (CDCl3) δ -105.1, -105.5; MS(ES+) calculated for C41H44F2NO10 (M+H) 748.29, found 748.40.
6-(Benzyloxycarbonylamino)hexyl 6-O-benzyl-[2,3-di-O-(4-fluorobenzoyl)-]-4-O-{4,6-O-benzylidene-2,3-di-O-(2-fluorobenzyl)-α-D-galactopyranosyl}-β-D-galactopyranoside (13). Compound 12 (0.159 g, 0.213 mmol), 17 (donor) (0.252 g, 0.427 mmol) and NIS (0.097 g, 0.429 mmol) were dried under vacuum and in the absence of light for 2 h. Distilled THF-CH2Cl2 (1:1, 8 mL) and TfOH (0.1 M in CH2Cl2, 0.001 mmol) was added. The reaction mixture was stirred at room temperature in absence of light for 2 h. The mixture was then concentrated and the residue was purified with preparative HPLC (20–100% CH3CN in H2O, 50 min) to give 13 (0.100 g) in 39% yield. 1H-NMR (CDCl3) δ 8.05–7.91 (m, 4H, ArH), 7.52 (t, 1H, J = 7.5 Hz, ArH), 7.49–7.40 (m, 3H, ArH), 7.39–7.19 (m, 15H, ArH), 7.12–6.94 (m, 8H, ArH), 5.64 (dd, 1H, J = 7.8 and 10.8 Hz, H-2), 5.37 (s, 1H, -CH2-), 5.14 (dd, 1H, J = 2.6 and 10.8 Hz, H-3), 5.09 (s, 2H, -COOCH2-), 5.06 (d, 1H, J = 3.3 Hz, H-1´), 4.72–4.92 (m, 4H), 4.65 (d, 1H, J = 7.8 Hz), 4.41 (d, 1H, J = 2.6 Hz, H-4), 4.33 (d, 1H, J = 2.6 Hz), 4.27 (d, 2H, J = 2.6 Hz), 4.23 (dd, 1H, J = 3.2 and 10.2), 4.12 (dd, 1H, J = 3.3 and 10.2 Hz, H-2´), 4.02–3.85 (m, 3H, -OCHHCH2-), 3.84 (t, 1H, J = 6.4 Hz), 3.65 (dd, 1H, J = 6.1 and 9.7 Hz), 3.55–3.46 (m, 1H, -OCHHCH2-), 3.37 (dd, 2H, J = 12.6 and 61.2 Hz, H-6´), 3.10–3.00 (m, 2H, -CH2CH2NH-), 1.62–1.44 (m, 2H, -OCH2CH2-), 1.35–1.12 (m, 6H, -OCH2CH2CH2CH2CH2CH2NH-); 13C-NMR (CDCl3) δ 167.3 (J = 14.2 Hz), 165.0 (J = 25.0 Hz), 164.8 (J = 21.0 Hz), 161.9 (J = 27.2 Hz), 159.4 (J =27.0 Hz), 156.5, 138.2, 138.0, 136.8, 132.4 (J = 9.3 and 15.3 Hz), 130.7 (J = 4.10 Hz), 130.1 (J = 3.4 Hz), 129.2 (J = 4.7 Hz), 128.9, 128.6, 128.5, 128.2, 128.1, 127.7, 126.4, 126.1, 125.9, 125.8, 125.7, 125.6 (J = 3.7 Hz), 124.1 (J = 3.5 Hz), 124.0 (J = 10.0 Hz), 116.0 (J = 23.4 Hz), 115.8 (J = 22.7 Hz), 115.2 (J =28.5 Hz), 101.4, 100.8, 77.3, 76.7, 76.0, 75.9, 74.6, 74.3, 74.0, 73.2, 70.1, 70.0, 69.1, 68.0, 67.9, 66.8, 64.9 (J = 3.9 Hz), 63.4, 40.9, 29.9, 29.4, 26.5, 25.6; 19F-NMR (CDCl3) δ -104.4, -105.5, -118.7, -119.3; [α]D25 +87.1° (c 1.0, CH2Cl2); MS(ES+) calculated for C68H68F4NO15 (M+H) 1214.45, found 1214.85.
6-Aminohexyl 4-O-α-D-galactopyranosyl-β-D-galactopyranoside (15). Compound 13 (0.048 g, 0.040 mmol) was dissolved in dry MeOH (12 mL) and NaOMe in MeOH (0.40 mL, 0.40 mmol) was slowly added. The solution was stirred for 1.5 h in room temperature and AcOH was then added until the pH was neutral. The mixture was concentrated in vacuo and subsequent purification by preparative HPLC (5–100% CH3CN in H2O, 50 min) gave 14 (0.034 g) in 89% yield. Compound 14 (0.032 g, 0.033 mmol) was then dissolved in AcOH (5 mL) and Pd(C) (0.044 mg) was added. The slurry was stirred under H2 (g) for 31 h. The Pd catalyst was removed by filtration through Celite and the filtrate was concentrated in vacuo. The residue was purified by preparative HPLC (0–100% CH3CN in H2O, 50 min) to give 15 (7.2 mg) in 50% yield.1H-NMR (D2O) δ 4.97 (d, 1H, J = 3.7 Hz, H-1´), 4.49 (d, 1H, J = 7.7 Hz, H-1), 4.41–4.35 (m, 1H), 4.04 (d, 2H, J = 2.6 Hz), 3.97–3.81 (m, 5H), 3.80–3.58 (m, 5H), 3.57–3.50 (m, 1H), 3.01 (t, 2H, J = 7.5 Hz, -CH2NH2), 1.73–1.61 (m, 4H, -OCH2CH2-, -CH2CH2NH2), 1.48–1.36 (m, 4H, -OCH2CH2CH2CH2-); 13C-NMR (D2O) δ 103.5 (C-1), 100.8 (C-1´), 77.8, 75.7, 73.1, 71.7, 71.4, 71.0, 69.8, 69.7, 69.4, 61.2, 60.8, 40.1 (-CH2NH2), 29.2 (-CH2CH2NH2), 27.2 (-OCH2CH2-), 25.9 (-CH2C H2CH2NH2), 25.3 (-OCH2CH2CH2-); [α]D25 +38.8° (c 1.0, H2O); MS(ES+) calculated for C18H36NO (M+H) 442.23, found 442.25.
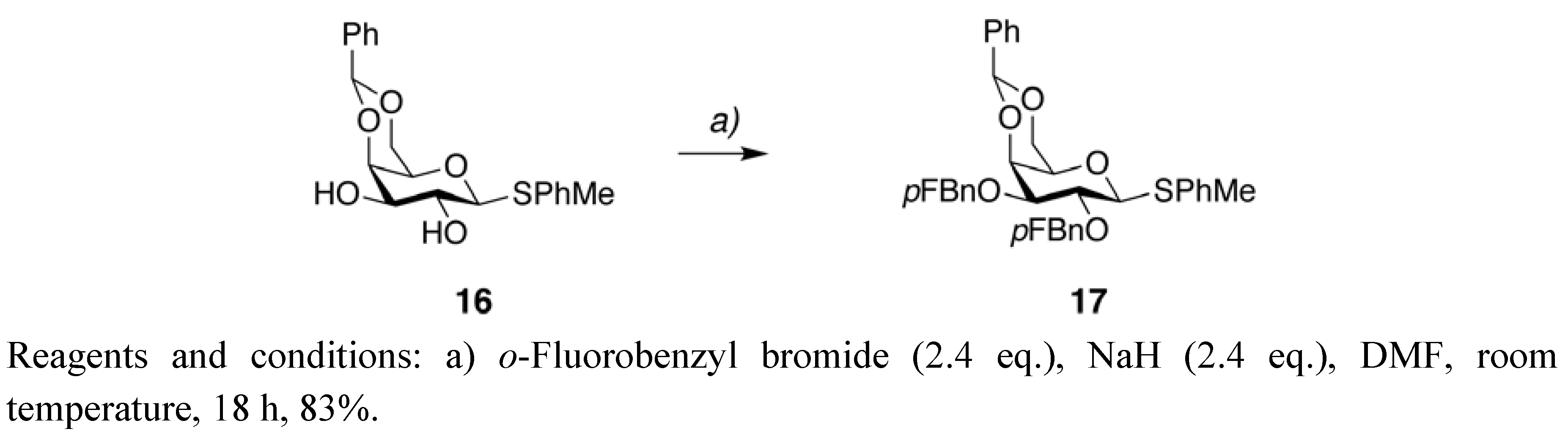
4-Methylphenyl 4,6-O-benzylidene-2,3-di-O-(2-fluorobenzyl)-1-thio-β-D-galactopyranoside (
17). Compound
16 [
15] (0.401 g, 1.071 mmol) was dissolved in DMF (6 mL) and stirred at 0 °C for 30 min before NaH (60% oil dispersion, 2.58 mmol) was added. After 10 min 2-fluorobenzyl bromide (0.311 mL, 2.58 mmol) was added dropwise and the mixture stirred at room temperature for 18 h. The reaction was quenched with MeOH and the mixture was diluted with CH
2Cl
2 and washed with H
2O. The crude was concentrated and purified with flash column chromatography (n-heptane-EtOAc 5:1) to give
17 (0.522 g) as a white solid in 83%.
1H-NMR (CDCl
3) δ 7.75–7.68 (m, 2H, ArH), 7.66–7.56 (m, 2H, ArH), 7.55–7.40 (m, 4H, ArH), 7.37–7.26 (m, 2H, ArH), 7.20 (t, 1H,
J = 7.4 Hz, ArH), 7.16–7.02 (m, 5H, ArH), 5.57 (s, 1H, PhC
H-), 4.84 (s, 2H,
oFPhC
H2O-), 4.80 (dd, 2H,
J = 10.8 and 126.8 Hz,
oFPhC
H2O-), 4.65 (d, 1H,
J = 9.3 Hz, H-1), 4.44 (d, 1H,
J = 9.3 Hz, H-6), 4.31 (d, 1H,
J = 2.7 Hz, H-4), 4.06 (d, 1H,
J = 12.3 Hz, H-6), 3.92 (t, 1H,
J = 9.3 Hz, H-2), 3.71 (dd, 1H,
J = 2.7 and 9.3 Hz, H-3), 4.46 (s, 1H, H-5), 2.28 (s, 3H, -PhC
H3);
13C-NMR (CDCl
3) δ 161.7 (
J = 16.2 Hz), 159.3 (
J = 16.9 Hz), 138.0, 137.7, 133.4, 130.6 (
J = 4.2 Hz), 130.3 (
J = 4.1 Hz), 129.7, 129.5 (
J = 8.2 Hz), 129.3 (
J = 8.0 Hz), 129.0, 128.7, 128.1, 126.7, 125.7 (
J = 14.6 Hz), 125.1 (
J = 14.6 Hz), 124.1 (
J = 3.4 and 15.2 Hz), 115.1 (
J = 21.5 Hz), 101.2, 86.5, 81.7, 75.3, 73.3, 69.8, 69.4, 68.4 (
J = 4.0 Hz), 64.9 (
J = 3.8 Hz), 21.2;
19F-NMR (CDCl
3) δ -119.1, -119.6; MS(ES+) calculated for C
34H
33F
2O
5S 591.20 (M+H), found 591.25.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


