Recent Applications of Polymer Supported Organometallic Catalysts in Organic Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
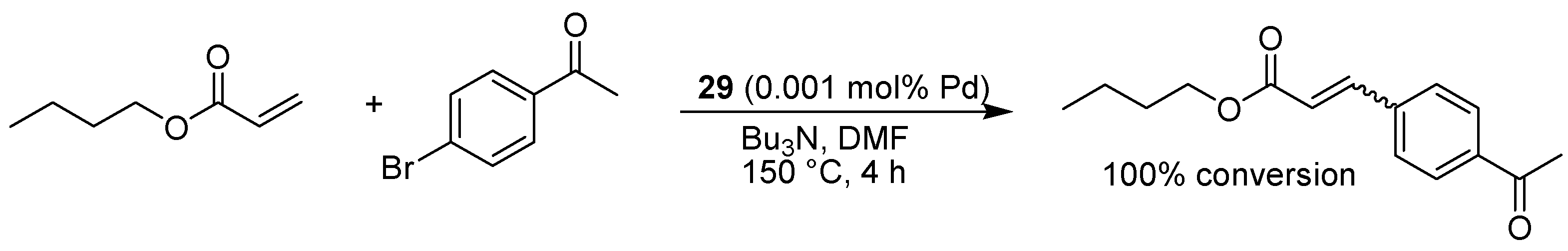{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Abbreviations
| acac | acetylacetonato |
| AIBN | azoisobutyronitrile |
| BSA | N,O-bis(tri-methyl silyl)-acetamide |
| DMA | N,N-dimethylacetamide |
| DPEN | 1,2-diphenyl-ethylenediamine |
| DVB | divinylbenzene |
| NHC | N-heterocyclic carbene |
| PEG | polyethylene glycol |
| PHOX | diphenylphosphinooxazoline |
| PS | polystyrene |
| TBAA | tetrabutyl-ammonium acetate |
| TEMPO | 2,2,6,6-tetramethyl-piperidin-1-oxyl |
| TOF | turnover frequency |
| TON | turnover number |
1. Introduction
2. Cobalt
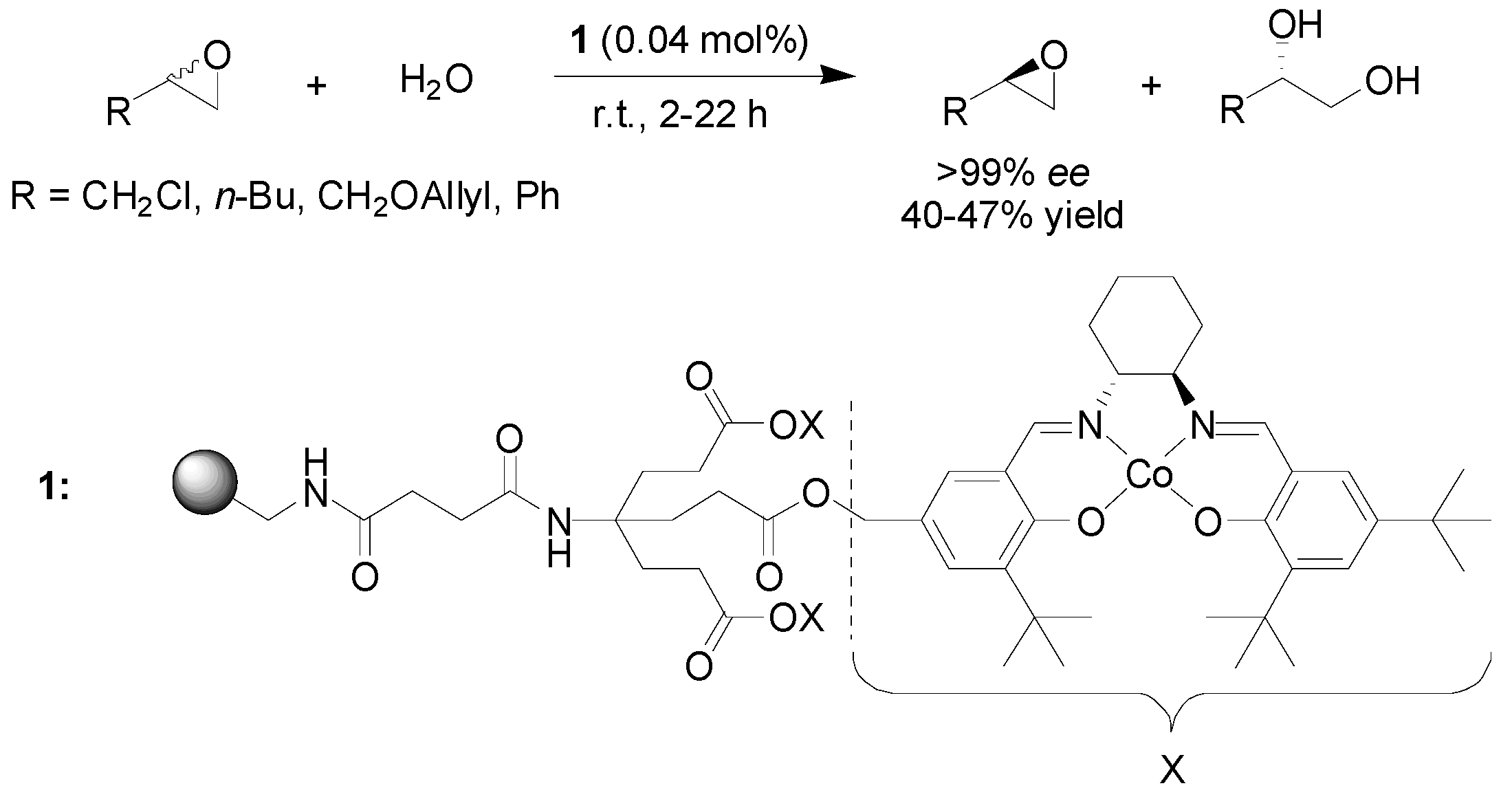
2.1. Ring Opening of Epoxides

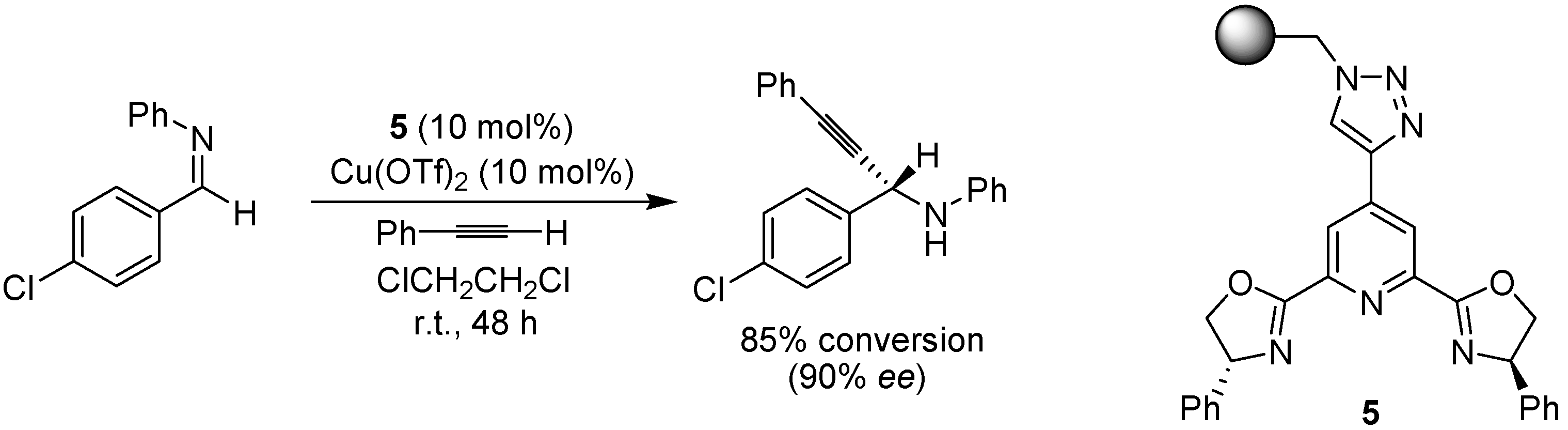
3. Copper
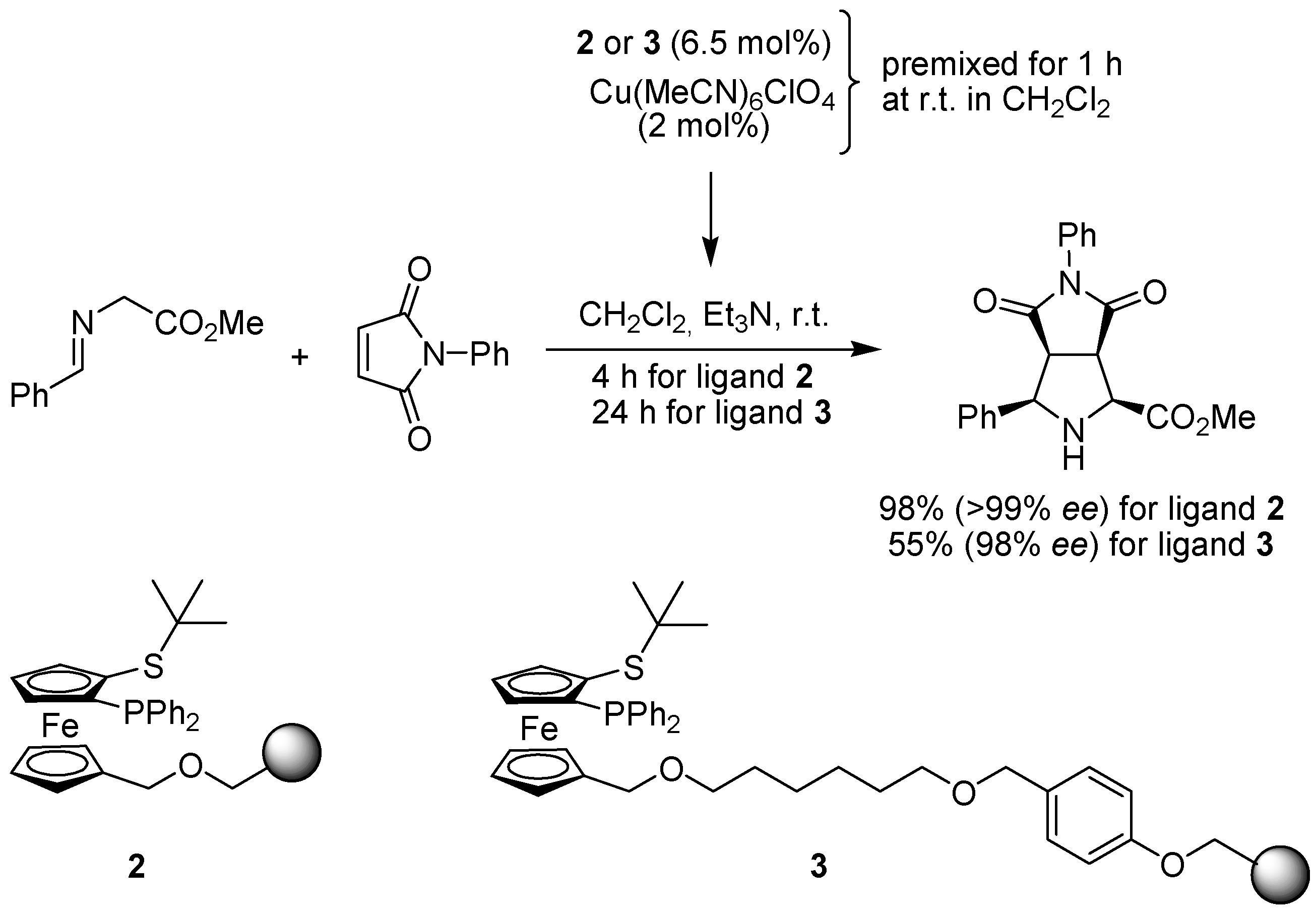
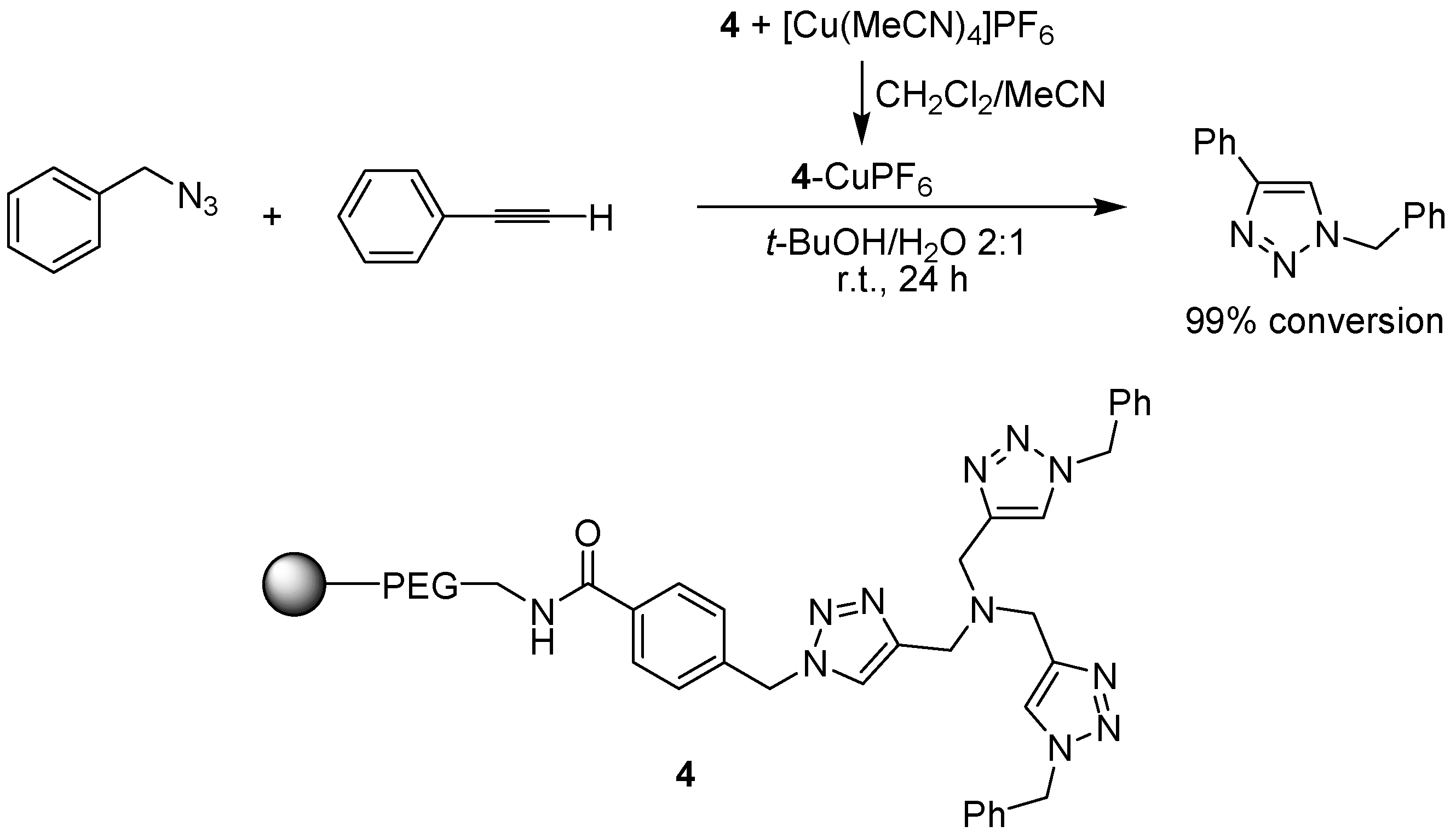
3.1. Cycloaddition Reactions


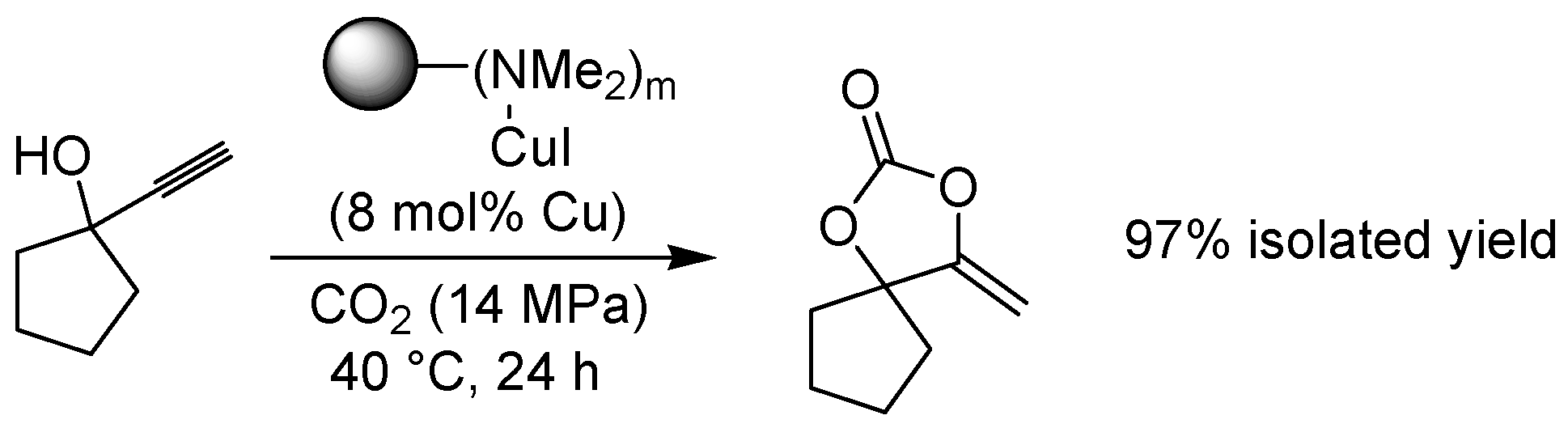
3.2. Other Copper-Catalyzed Reactions


4. Ruthenium
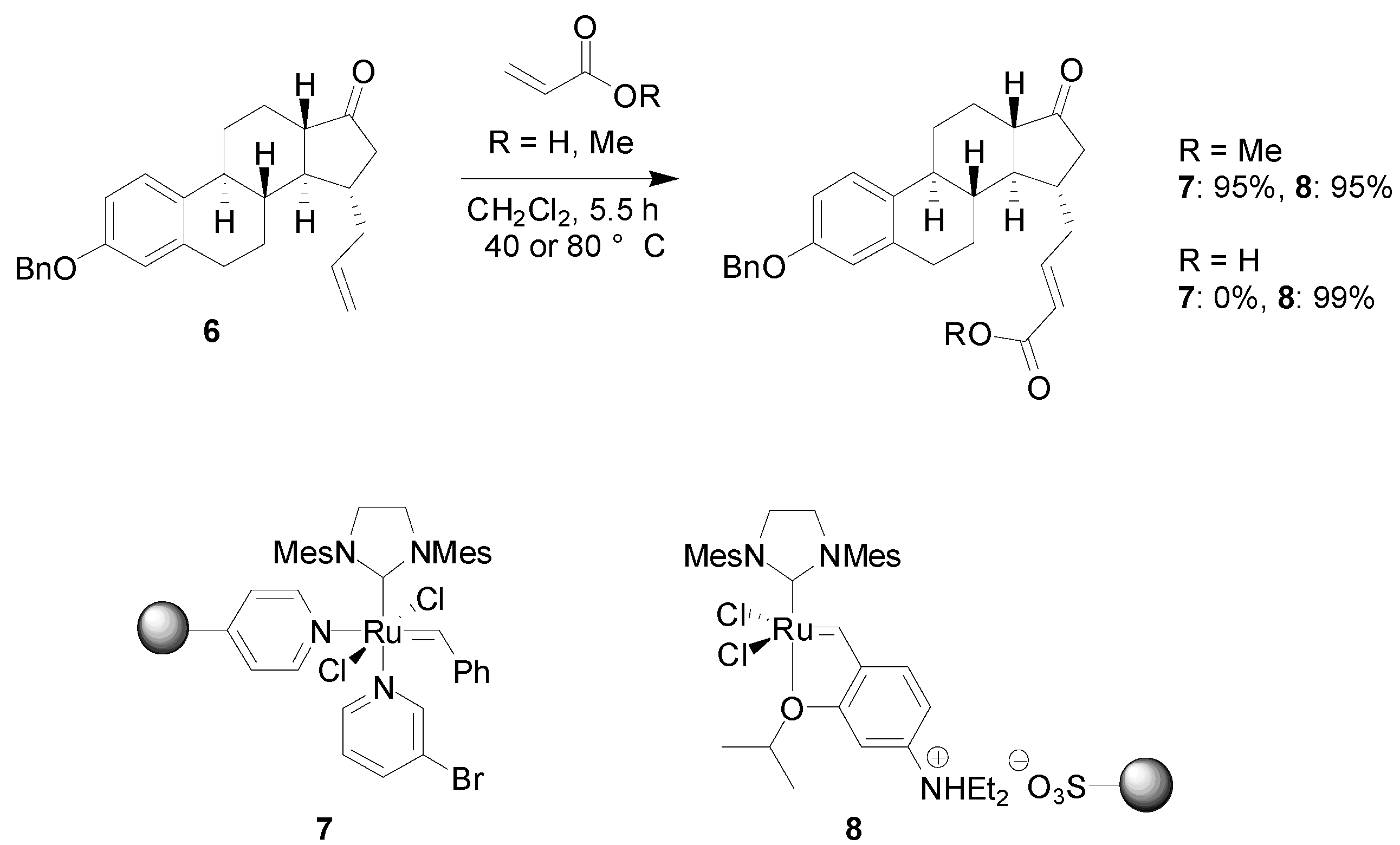
4.1. Metathesis

4.2. Asymmetric Transfer Hydrogenation

4.3. Halogenation

4.4. Cyclopropanation

4.5. Ring Opening of Epoxides

5. Rhodium
5.1. Conjugate Addition

5.2. Rhodium-Catalyzed Carbonylation, Hydroformylation and Hydrogenation



6. Palladium
6.1. Allylic Substitution


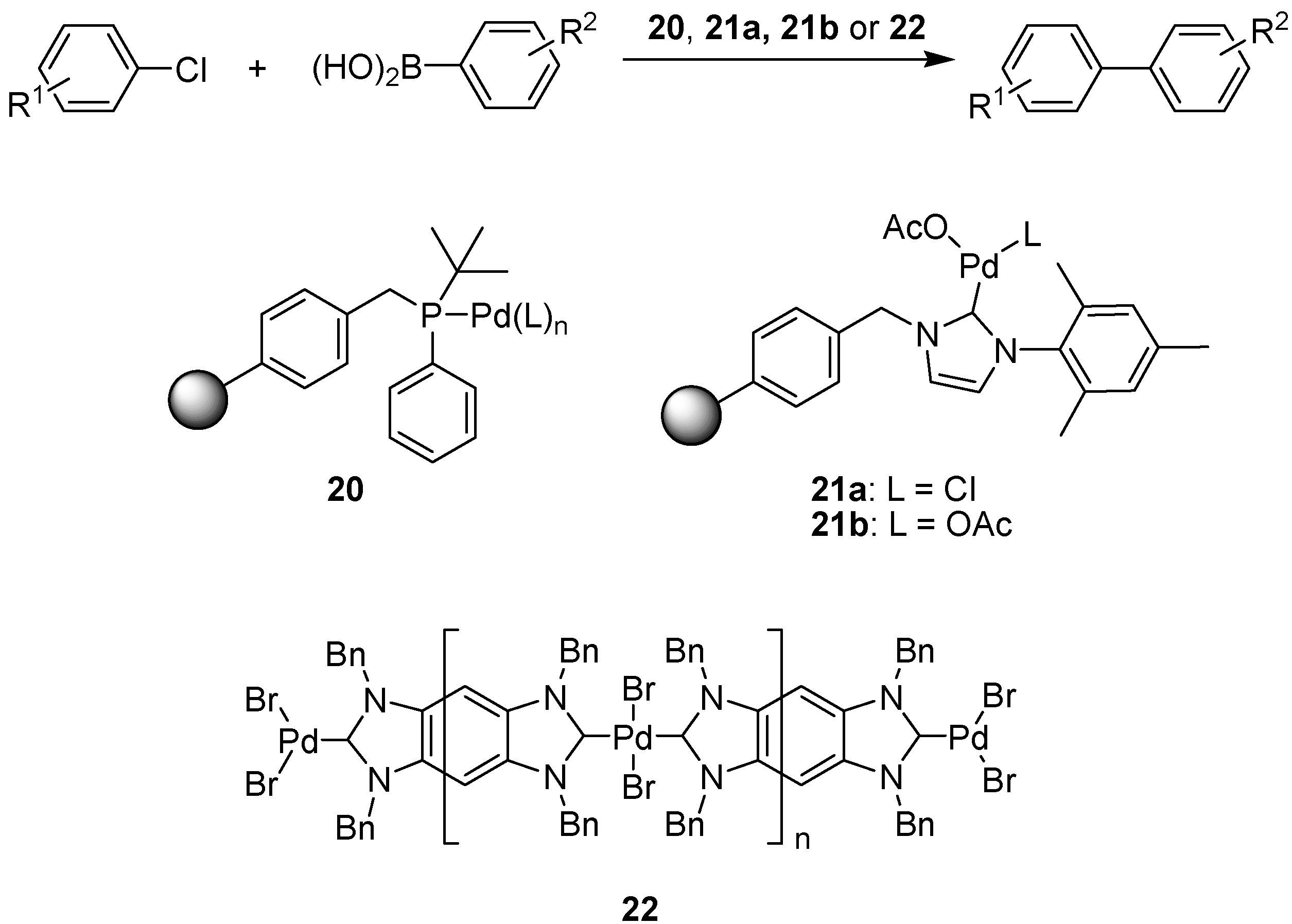
6.2. Suzuki Coupling


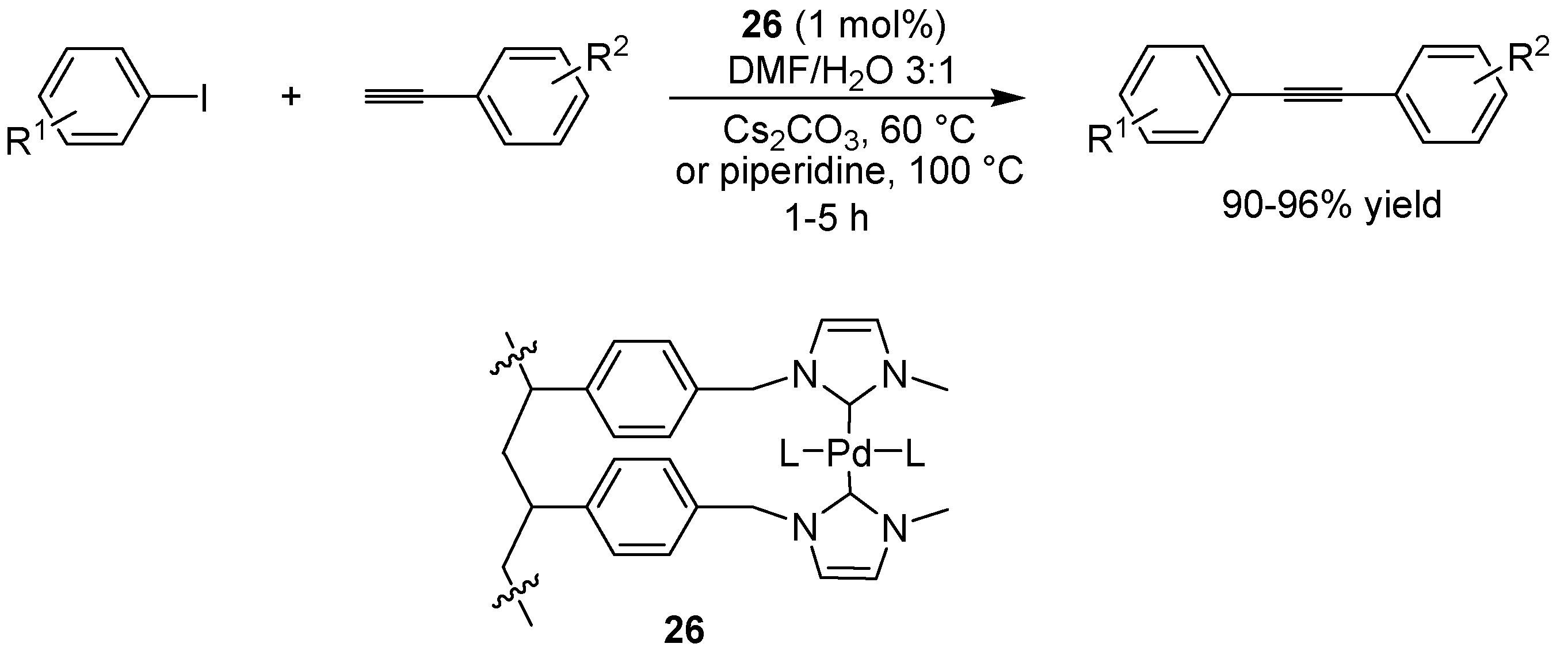
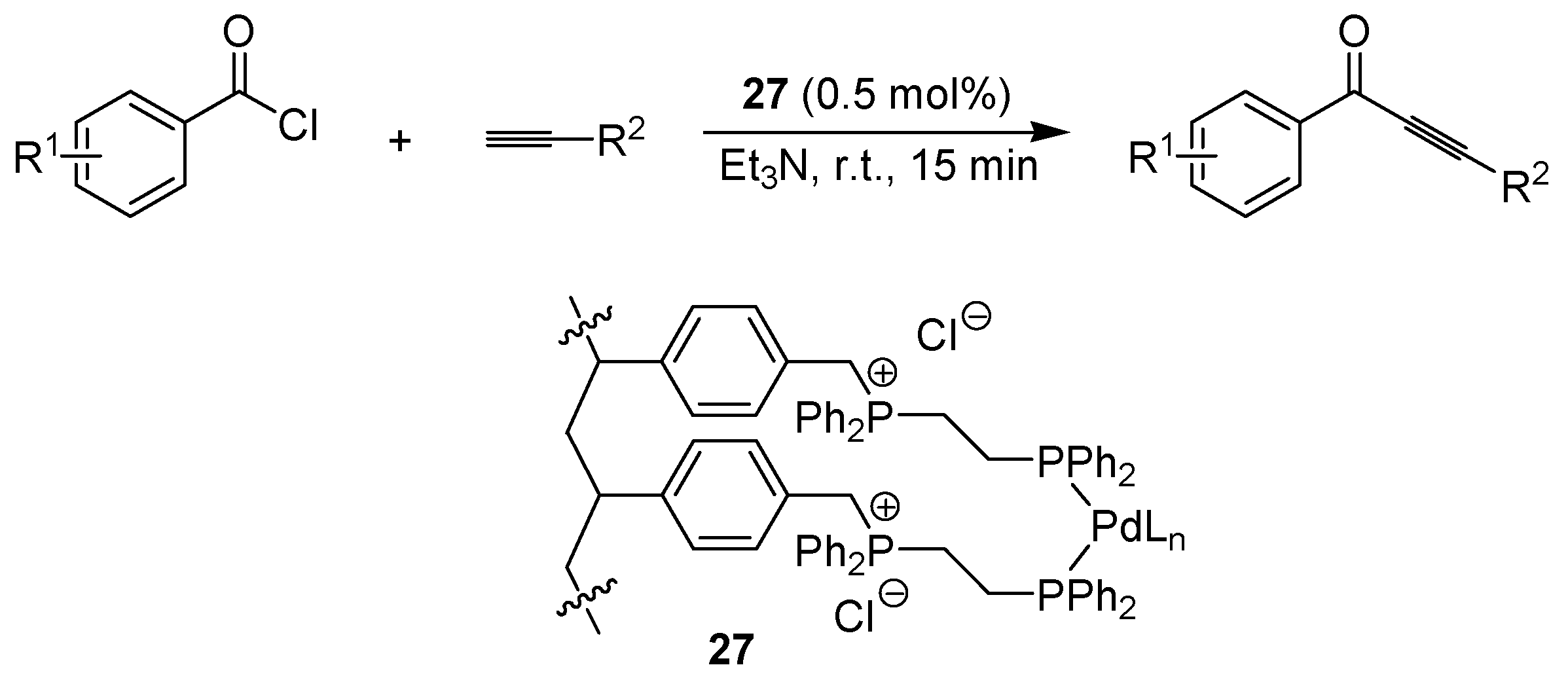
6.3. Sonogashira Coupling


6.4. The Heck Reaction



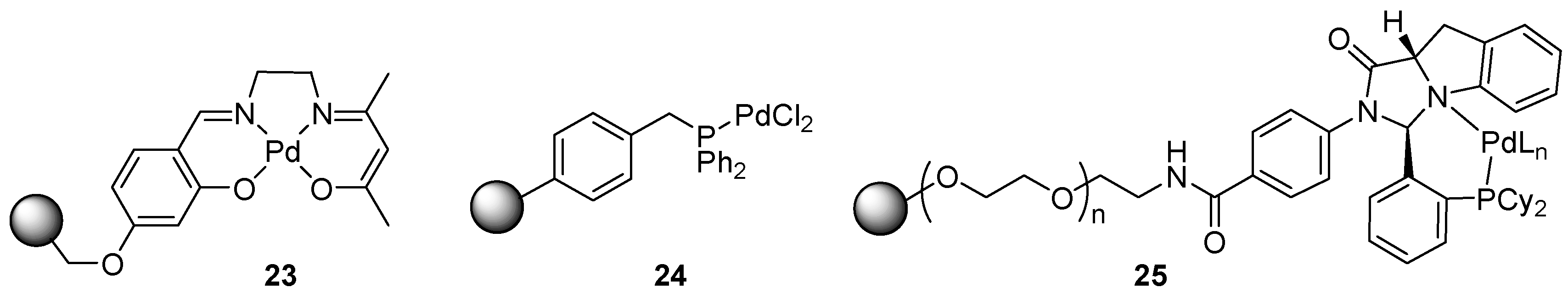
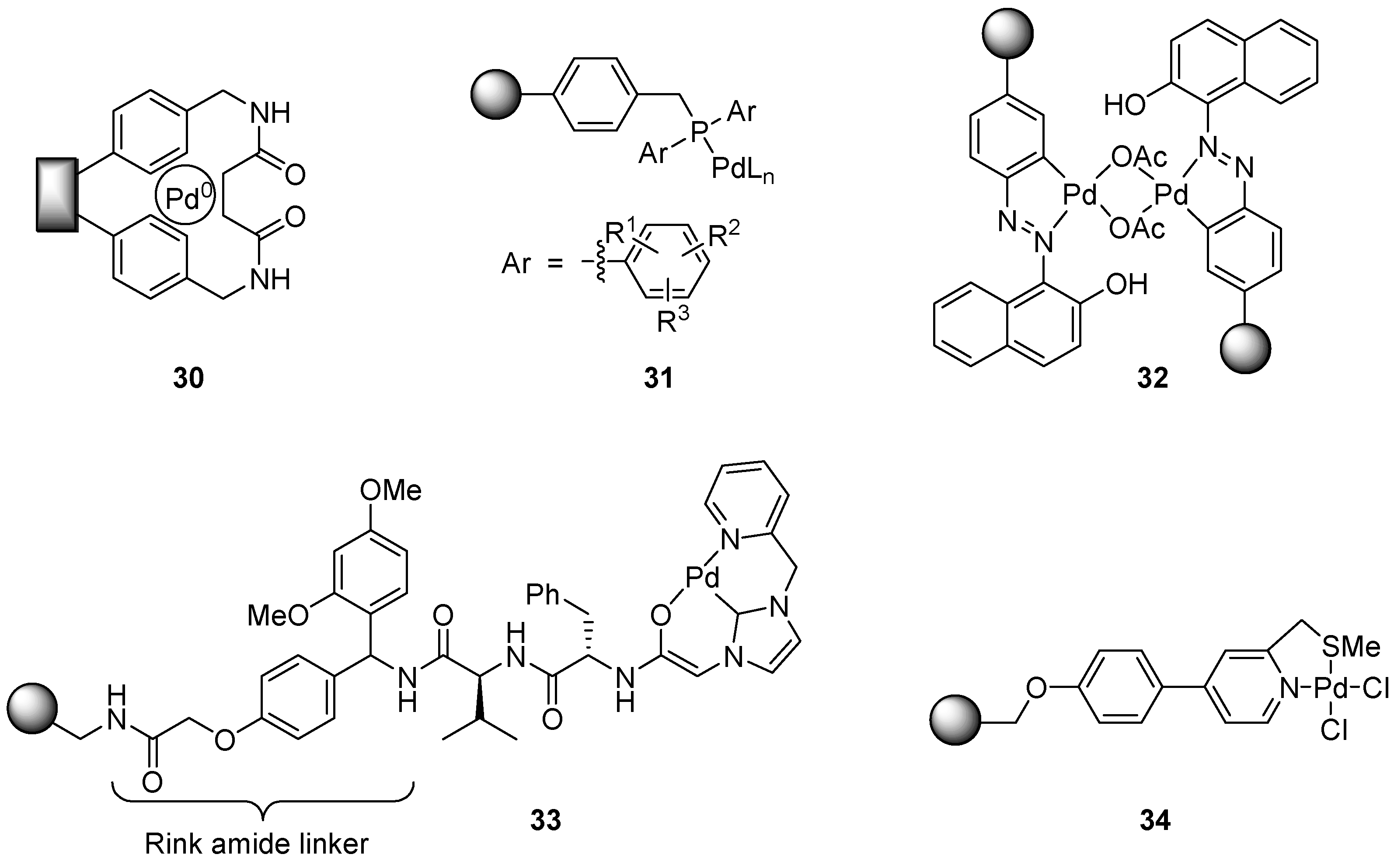
6.5. Catalysts Applicable Towards Several Cross-Coupling Reaction Types

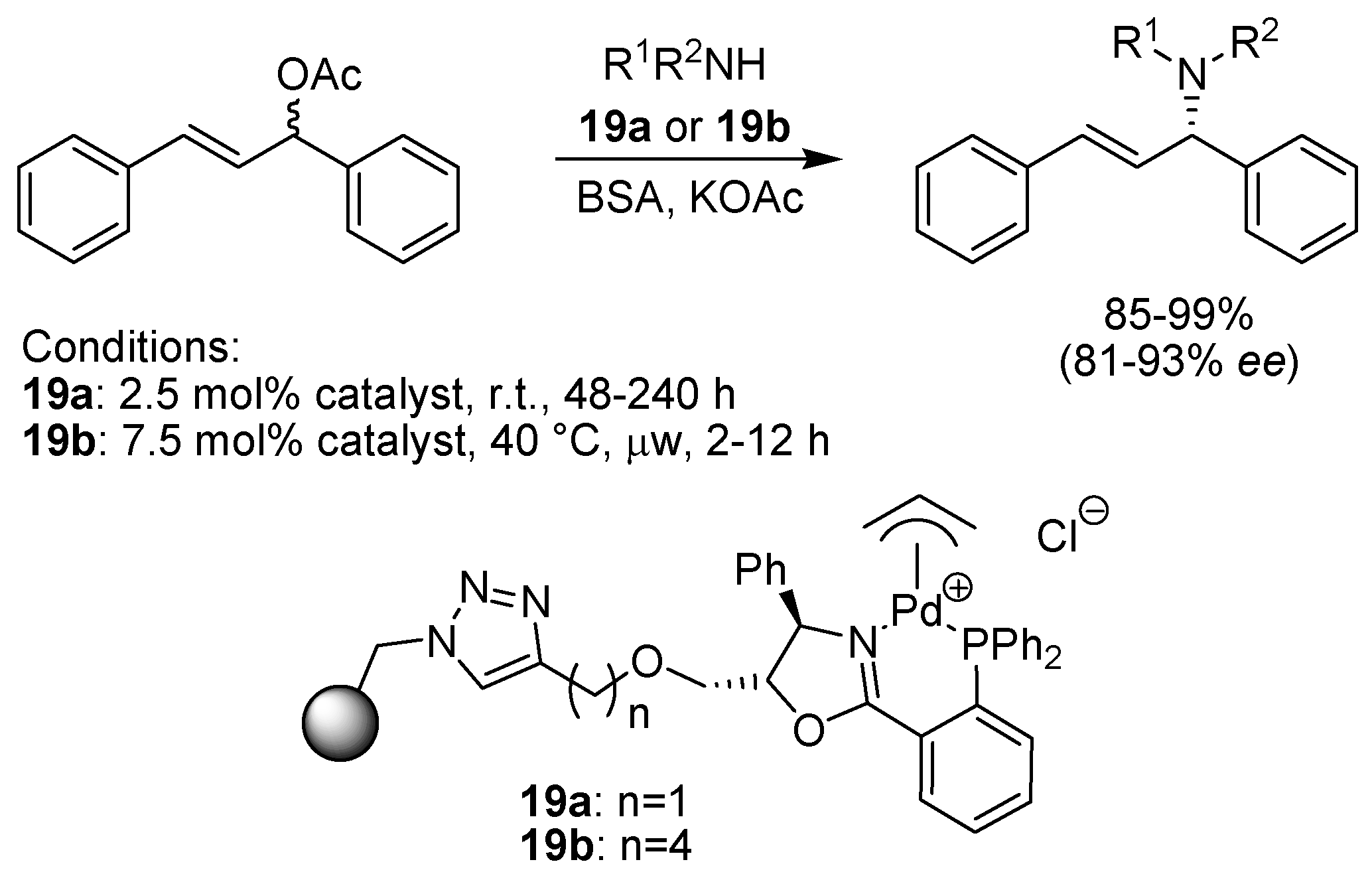
6.6. Aryl Amination (C-N Coupling)


6.7. Allylic Imidate Rearrangement

7. Iridium
7.1. Transfer Dehydrogenation

8. Concluding Remarks
Acknowledgements
References and Notes
- Ley, S.V.; Baxendale, I.R.; Bream, R.N.; Jackson, P.S.; Leach, A.G.; Longbottom, D.A.; Nesi, M.; Scott, J.S.; Storer, R.I.; Taylor, S.J. Multi-step organic synthesis using solid-supported reagents and scavengers: a new paradigm in chemical library generation. J. Chem. Soc. Perkin Trans. 1 2000, 3815–4195. [Google Scholar]
- Clapham, B.; Reger, T.S.; Janda, K.D. Polymer-supported catalysis in synthetic organic chemistry. Tetrahedron 2001, 57, 4637–4662. [Google Scholar] [CrossRef]
- Bergbreiter, D.E. Using soluble polymers to recover catalysts and ligands. Chem. Rev. 2002, 102, 3345–3383. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Marco, M. Preparation of polymer-supported ligands and metal complexes for use in catalysis. Chem. Rev. 2002, 102, 3217–3273. [Google Scholar] [CrossRef]
- McNamara, C.A.; Dixon, M.J.; Bradley, M. Recoverable catalysts and reagents using recyclable polystyrene-based supports. Chem. Rev. 2002, 102, 3275–3299. [Google Scholar] [CrossRef]
- Bräse, S.; Lauterwasser, F.; Ziegert, R.E. Recent advances in asymmetric C-C and C-heteroatom bond forming reactions using polymer-bound catalysts. Adv. Synth. Catal. 2003, 345, 869–929. [Google Scholar] [CrossRef]
- Guino, M.; Hii, K.K.M. Applications of phosphine-functionalised polymers in organic synthesis. Chem. Soc. Rev. 2007, 36, 608–617. [Google Scholar] [CrossRef]
- Bergbreiter, D.E.; Tian, J.H.; Hongfa, C. Using Soluble Polymer Supports To Facilitate Homogeneous Catalysis. Chem. Rev. 2009, 109, 530–582. [Google Scholar] [CrossRef]
- Bräse, S.; Kirchhoff, J.H.; Kobberling, J. Palladium-catalysed reactions in solid phase organic synthesis. Tetrahedron 2003, 59, 885–939. [Google Scholar] [CrossRef]
- Graden, H.; Kann, N. Solid phase synthesis using organometallic reagents. Curr. Org. Chem. 2005, 9, 733–763. [Google Scholar] [CrossRef]
- Ljungdahl, N.; Bromfield, K.; Kann, N. Solid phase organometallic chemistry. Top. Curr. Chem. 2007, 278, 89–134. [Google Scholar] [CrossRef]
- Beligny, S.; Rademann, J. Oxidizing and reducing agents. In The Power of Functional Resins in Organic Synthesis; Tulla-Puche, J., Albericio, F., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 83–99. [Google Scholar]
- Goyal, P.; Zheng, X.L.; Weck, M. Enhanced cooperativity in hydrolytic kinetic resolution of epoxides using poly(styrene) resin-supported dendronized Co-(salen) catalysts. Adv. Synth. Catal. 2008, 350, 1816–1822. [Google Scholar] [CrossRef]
- Priego, J.; Mancheno, O.G.; Cabrera, S.; Arrayas, R.G.; Llamas, T.; Carretero, J.C. 1-Phosphino-2-sulfenylferrocenes: efficient ligands in enantioselective palladium-catalyzed allylic substitutions and ring opening of 7-oxabenzonorbornadienes. Chem. Commun. 2002, 2512–2513. [Google Scholar]
- Martin-Matute, B.; Pereira, S.I.; Pena-Cabrera, E.; Adrio, J.; Silva, A.M.S.; Carretero, J.C. Synthesis of polymer-supported Fesulphos ligands and their application in asymmetric catalysis. Adv. Synth. Catal. 2007, 349, 1714–1724. [Google Scholar]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef]
- Chan, T.R.; Fokin, V.V. Polymer-supported copper(I) catalysts for the experimentally simplified azide-alkyne cycloaddition. QSAR Comb. Sci. 2007, 26, 1274–1279. [Google Scholar] [CrossRef]
- Tilliet, M.; Lundgren, S.; Moberg, C.; Levacher, V. Polymer-bound pyridine-bis(oxazoline). Preparation through click chemistry and evaluation in asymmetric catalysis. Adv. Synth. Catal. 2007, 349, 2079–2084. [Google Scholar] [CrossRef]
- Weissberg, A.; Halak, B.; Portnoy, M. The first solid-phase synthesis of bis(oxazolinyl)pyridine ligands. J. Org. Chem. 2005, 70, 4556–4559. [Google Scholar] [CrossRef]
- Jiang, H.F.; Wang, A.Z.; Liu, H.L.; Qi, C.R. Reusable polymer-supported amine-copper catalyst for the formation of alpha-alkylidene cyclic carbonates in Supercritical carbon dioxide. Eur. J. Org. Chem. 2008, 2309–2312. [Google Scholar]
- Wang, L.; Huang, C.Y.; Cai, C. Polymer-supported copper complex for the direct synthesis of O-aryloxime ethers via cross-coupling of oximes and arylboronic acids. Catal. Commun. 2010, 11, 532–536. [Google Scholar] [CrossRef]
- Kirschning, A.; Harmrolfs, K.; Mennecke, K.; Messinger, J.; Schon, U.; Grela, K. Homo- and heterogeneous Ru-based metathesis catalysts in cross-metathesis of 15-allylestrone - Towards 17 beta-hydroxysteroid dehydrogenase type 1 inhibitors. Tetrahedron Lett. 2008, 49, 3019–3022. [Google Scholar]
- Haraguchi, N.; Tsuru, K.; Arakawa, Y.; Itsuno, S. Asymmetric transfer hydrogenation of imines catalyzed by a polymer-immobilized chiral catalyst. Org. Biomol. Chem. 2009, 7, 69–75. [Google Scholar] [CrossRef]
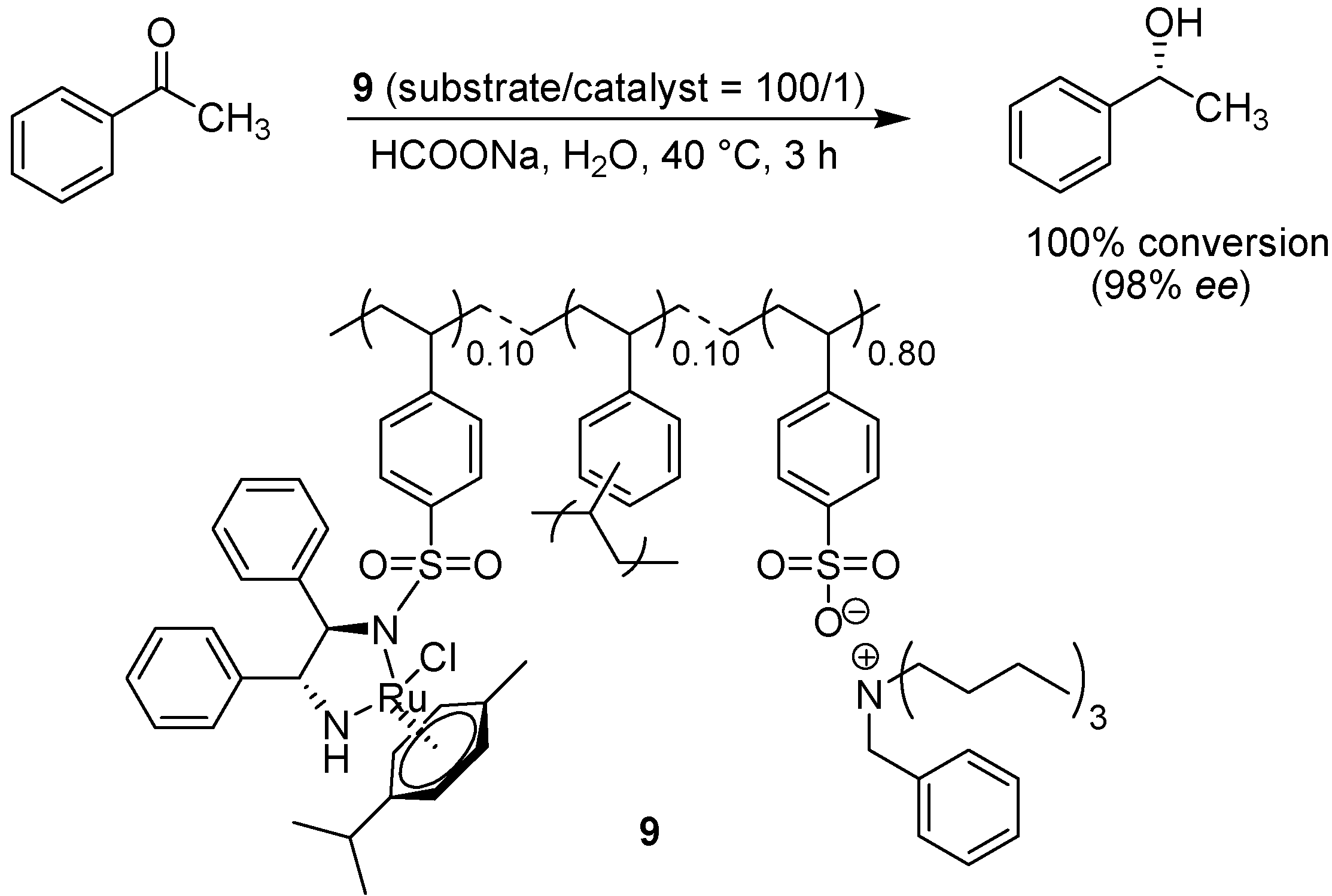
- Arakawa, Y.; Chiba, A.; Haraguchi, N.; Itsuno, S. Asymmetric transfer hydrogenation of aromatic ketones in water using a polymer-supported chiral catalyst containing a hydrophilic pendant group. Adv. Synth. Catal. 2008, 350, 2295–2304. [Google Scholar] [CrossRef]
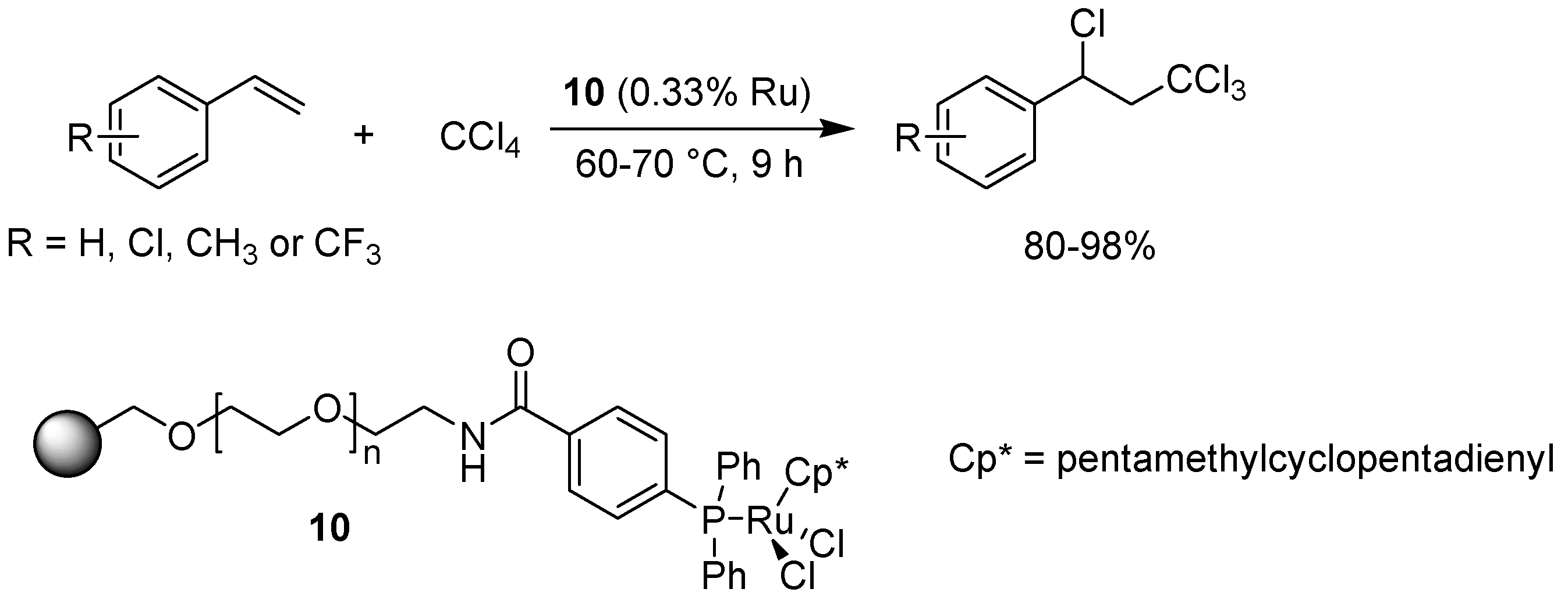
- Severin, K. Ruthenium catalysts for the Kharasch reaction. Curr. Org. Chem. 2006, 10, 217–224. [Google Scholar] [CrossRef]
- Oe, Y.; Uozumi, Y. Highly efficient heterogeneous aqueous Kharasch reaction with an amphiphilic resin-supported ruthenium catalyst. Adv. Synth. Catal. 2008, 350, 1771–1775. [Google Scholar] [CrossRef]
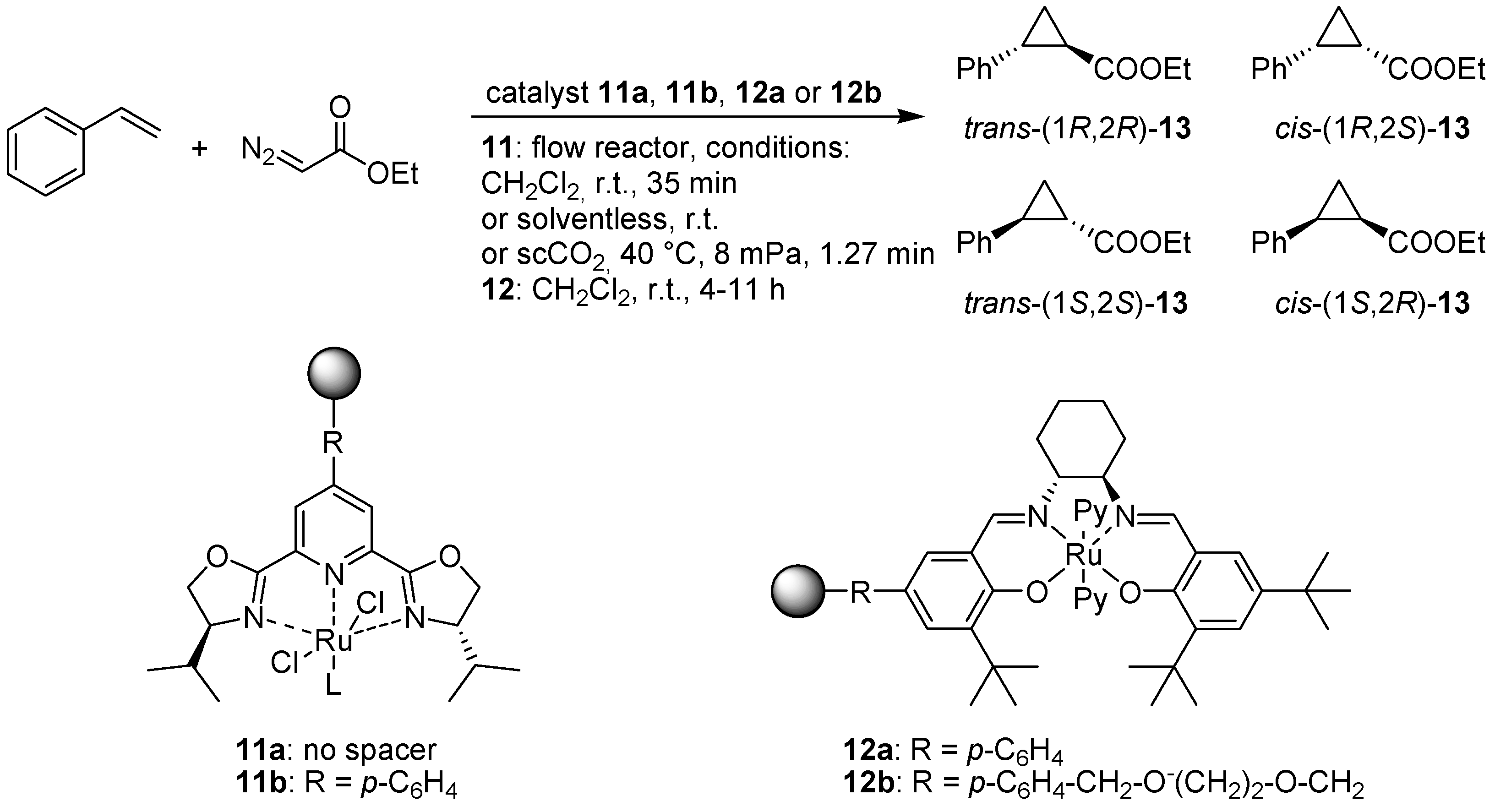
- Burguete, M.I.; Cornejo, A.; Garcia-Verdugo, E.; Gil, M.J.; Luis, S.V.; Mayoral, J.A.; Martinez-Merino, V.; Sokolova, M. Pybox monolithic miniflow reactors for continuous asymmetric cyclopropanation reaction under conventional and supercritical conditions. J. Org. Chem. 2007, 72, 4344–4350. [Google Scholar] [CrossRef]
- Gill, C.S.; Venkatasubbaiah, K.; Jones, C.W. Recyclable polymer- and silica-supported ruthenium(II)-salen bis-pyridine catalysts for the asymmetric cyclopropanation of olefins. Adv. Synth. Catal. 2009, 351, 1344–1354. [Google Scholar] [CrossRef]
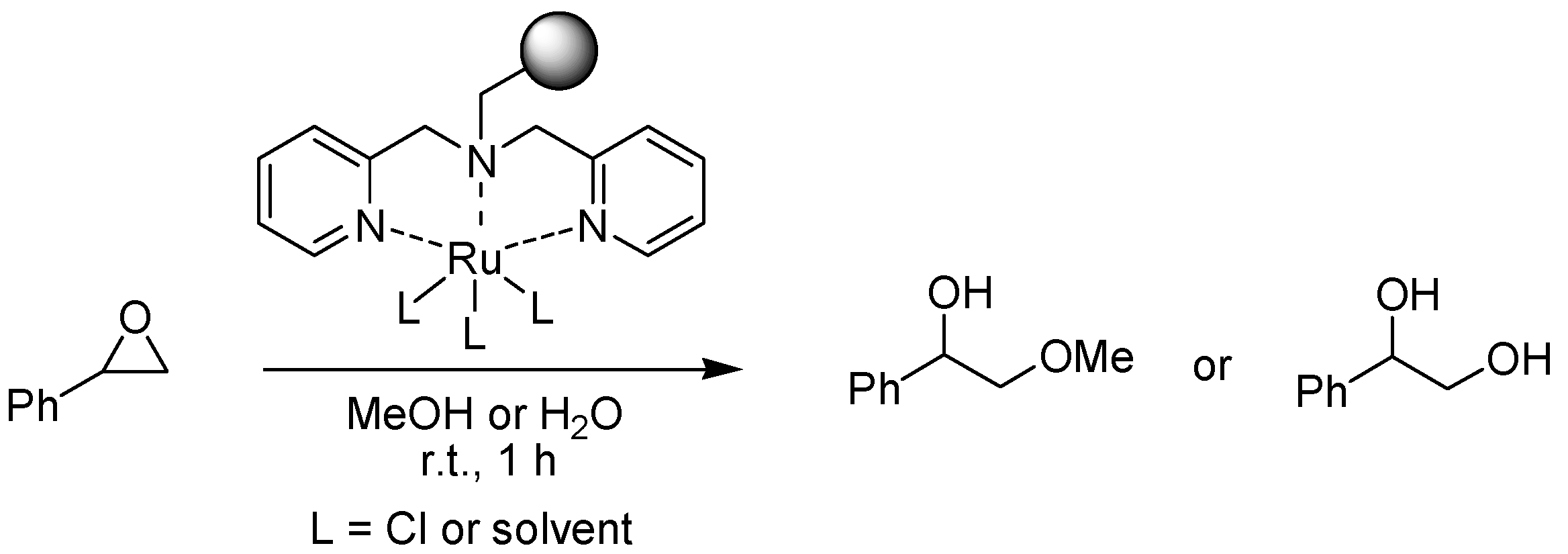
- Lee, S.H.; Lee, E.Y.; Yoo, D.W.; Hong, S.J.; Lee, J.H.; Kwak, H.; Lee, Y.M.; Kim, J.; Cheal, K.A.; Lee, J.K. Novel polymer-supported ruthenium and iron complexes that catalyze the conversion of epoxides into diols or diol mono-ethers: clean and recyclable catalysts. New J. Chem. 2007, 31, 1579–1582. [Google Scholar] [CrossRef]
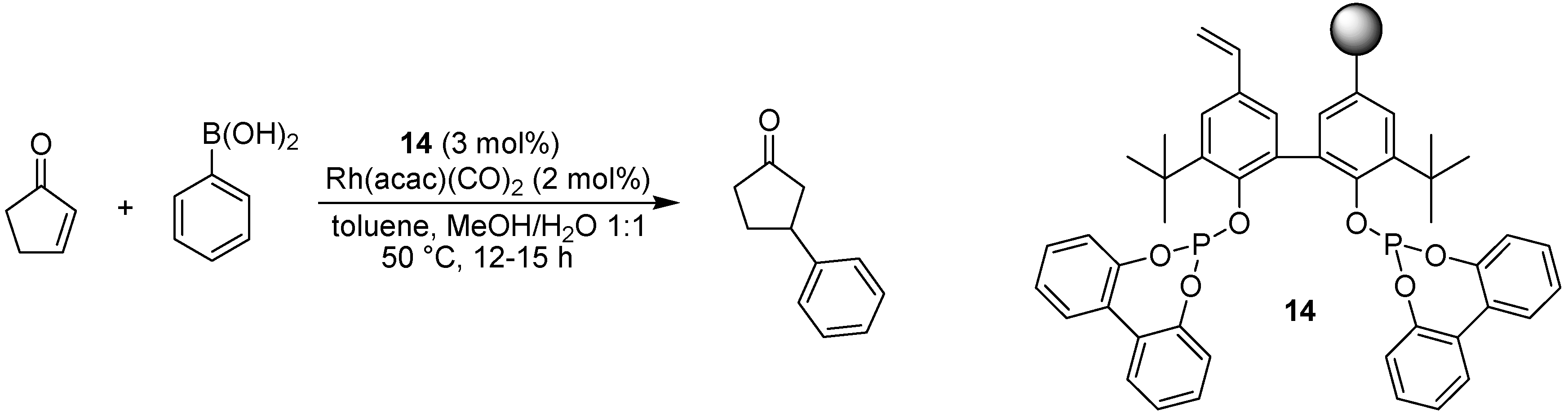
- Jana, R.; Tunge, J.A. A Homogeneous, Recyclable Rhodium(I) Catalyst for the Hydroarylation of Michael Acceptors. Org. Lett. 2009, 11, 971–974. [Google Scholar] [CrossRef]
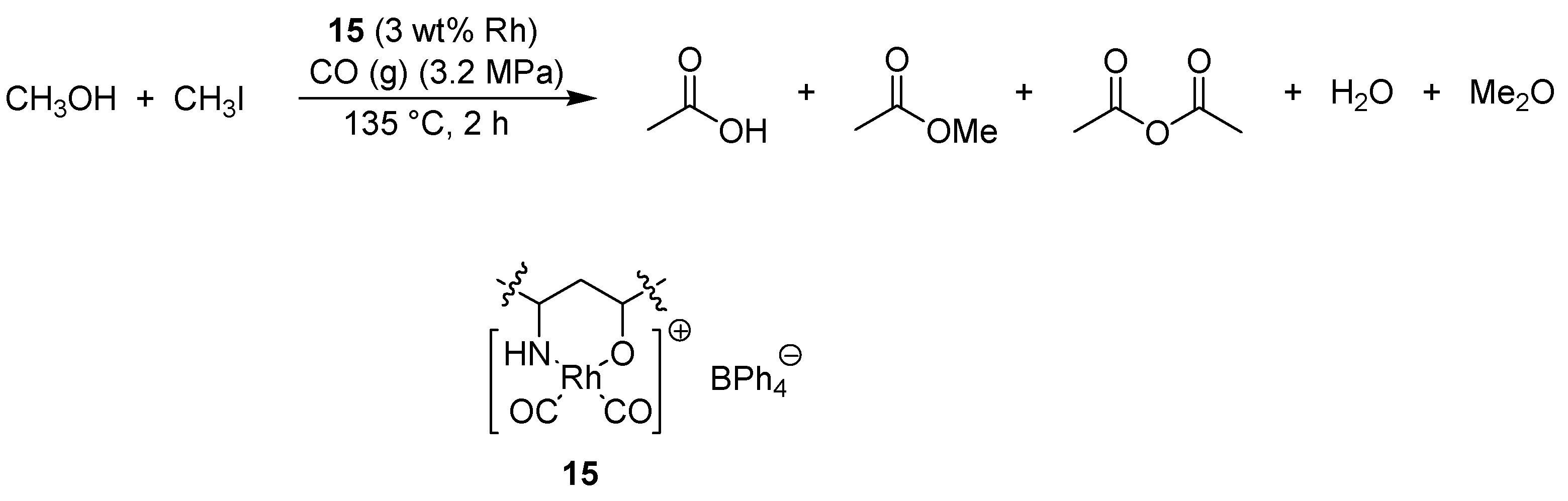
- Zhang, S.; Guo, C.; Qian, Q.; Yuan, G. Synthesis of acetic acid and acetic anhydride from methanol carbonylation with polymer supported rhodium catalyst. Catal. Commun. 2008, 9, 853–858. [Google Scholar] [CrossRef]
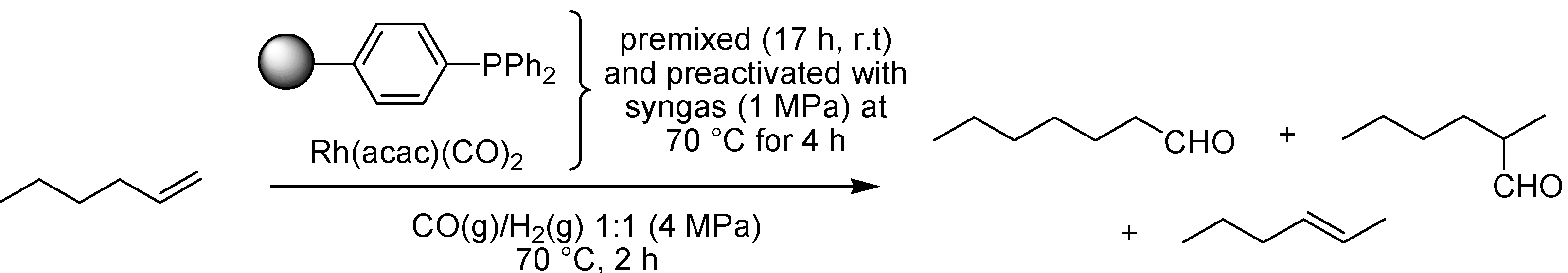
- Fujita, S.I.; Akihara, S.; Fujisawa, S.; Arai, M. Hydroformylation of 1-hexene using polymer-supported rhodium catalysts in supercritical carbon dioxide. J. Mol. Catal. A: Chem. 2007, 268, 244–250. [Google Scholar] [CrossRef]
- Pawar, G.M.; Weckesser, J.; Blechert, S.; Buchmeiser, M.R. Ring opening metathesis polymerization-derived block copolymers bearing chelating ligands: synthesis, metal immobilization and use in hydroformylation under micellar conditions. Beilstein J. Org. Chem. 2010, 6, 10–3762. [Google Scholar]
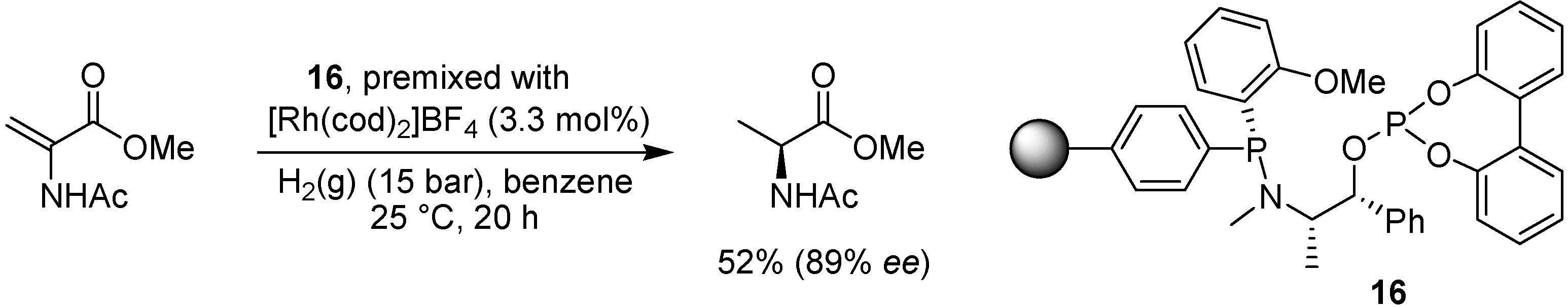
- den Heeten, R.; Swennenhuis, B.H.G.; van Leeuwen, P.; de Vries, J.G.; Kamer, P.C.J. Parallel synthesis and screening of polymer-supported phosphorus-stereogenic aminophosphane-phosphite and -phosphinite ligands. Angew. Chem. Int. Ed. 2008, 47, 6602–6605. [Google Scholar]
- Uozumi, Y.; Suzuka, T. π-Allylic sulfonylation in water with amphiphilic resin-supported palladium-phosphine complexes. Synthesis 2008, 1960–1964. [Google Scholar] [CrossRef]
- Uozumi, Y.; Takenaka, H.; Suzuka, T. Allylic substitution of meso-1,4-diacetoxycycloalkenes in water with an amphiphilic resin-supported chiral palladium complex. Synlett 2008, 1557–1561. [Google Scholar]
- Swennenhuis, B.H.G.; Chen, R.F.; van Leeuwen, P.; de Vries, J.G.; Kamer, P.C.J. Supported chiral monodentate ligands in rhodium-catalysed asymmetric hydrogenation and palladium-catalysed asymmetric allylic alkylation. Eur. J. Org. Chem. 2009, 5796–5803. [Google Scholar]
- Popa, D.; Marcos, R.; Sayalero, S.; Vidal-Ferran, A.; Pericas, M.A. Towards continuous flow, highly enantioselective allylic amination: ligand design, optimization and supporting. Adv. Synth. Catal. 2009, 351, 1539–1556. [Google Scholar] [CrossRef]
- Schweizer, S.; Becht, J.M.; Le Drian, C. Highly efficient and reusable supported Pd catalysts for Suzuki-Miyaura reactions of aryl chlorides. Org. Lett. 2007, 9, 3777–3780. [Google Scholar] [CrossRef]
- Schweizer, S.; Becht, J.M.; Le Drian, C. Highly efficient reusable polymer-supported Pd catalysts of general use for the Suzuki reaction. Tetrahedron 2010, 66, 765–772. [Google Scholar]
- Lee, D.H.; Kim, J.H.; Jun, B.H.; Kang, H.; Park, J.; Lee, Y.S. Macroporous polystyrene-supported palladium catalyst containing a bulky N-heterocyclic carbene ligand for Suzuki reaction of aryl chlorides. Org. Lett. 2008, 10, 1609–1612. [Google Scholar]
- Zeng, X.M.; Zhang, T.X.; Qin, Y.C.; Wei, Z.J.; Luo, M.M. Synthesis of a carbene transfer organometallic polymer and application to forming a recyclable heterogeneous catalyst for the Suzuki reactions of aryl chlorides. Dalton Trans. 2009, 8341–8348. [Google Scholar]
- Karimi, B.; Akhavan, P.F. Main-chain NHC-palladium polymer as a recyclable self-supported catalyst in the Suzuki-Miyaura coupling of aryl chlorides in water. Chem. Commun. 2009, 3750–3752. [Google Scholar] [CrossRef]
- Phan, N.T.S.; Styring, P. Supported phosphine-free palladium catalysts for the Suzuki-Miyaura reaction in aqueous media. Green Chem. 2008, 10, 1055–1060. [Google Scholar] [CrossRef]
- Bai, L.; Wang, J.X. Reusable, polymer-supported, palladium-catalyzed, atom-efficient coupling reaction of aryl halides with sodium tetraphenylborate in water by focused microwave irradiation. Adv. Synth. Catal. 2008, 350, 315–320. [Google Scholar] [CrossRef]
- Zhou, W.J.; Wang, K.H.; Wang, J.X.; Huang, D.F. Reusable, polystyrene-resin-supported, palladium-catalyzed, atom-efficient cross-coupling reaction of aryl halides with triarylbismuths. Eur. J. Org. Chem. 2010, 416–419. [Google Scholar]
- Uozumi, Y.; Matsuura, Y.; Arakawa, T.; Yamada, Y.M.A. Asymmetric Suzuki-Miyaura coupling in water with a chiral palladium catalyst supported on an amphiphilic resin. Angew. Chem. Int. Ed. 2009, 48, 2708–2710. [Google Scholar] [CrossRef]
- Glaser, C. Beiträge zur kenntniss des acetenylbenzols. Ber. Dtsch. Chem. Ges. 1869, 2, 422–424. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, D.H.; Jun, B.H.; Lee, Y.S. Copper-free Sonogashira cross-coupling reaction catalyzed by polymer-supported N-heterocyclic carbene palladium complex. Tetrahedron Lett. 2007, 48, 7079–7084. [Google Scholar] [CrossRef]
- Qin, Y.C.; Wei, W.; Luo, M.M. Suzuki-Miyaura cross-coupling of arenediazonium salts with arylboronic acids catalyzed by a recyclable polymer-supported N-heterocyclic carbene-palladium catalyst. Synlett 2007, 2410–2414. [Google Scholar]
- Suzuka, T.; Okada, Y.; Ooshiro, K.; Uozumi, Y. Copper-Free Sonogashira coupling in water with an amphiphilic resin-supported palladium complex. Tetrahedron 2010, 66, 1064–1069. [Google Scholar]
- Bakherad, M.; Keivanloo, A.; Bahramian, B.; Mihanparast, S. A diphenylphosphinoethane-functionalized polystyrene resin-supported Pd(0) complex as an effective catalyst for copper-free Sonogashira coupling reactions under aerobic conditions. Tetrahedron Lett. 2009, 50, 6418–6420. [Google Scholar] [CrossRef]
- Bakherad, M.; Keivanloo, A.; Bahramian, B.; Rajaie, M. A copper- and solvent-free coupling of acid chlorides with terminal alkynes catalyzed by a polystyrene-supported palladium(0) complex under aerobic conditions. Tetrahedron Lett. 2010, 51, 33–35. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Cacchi, S.; Fabrizi, G. An efficient palladium-catalyzed synthesis of cinnamaldehydes from acrolein diethyl acetal and aryl iodides and bromides. Org. Lett. 2003, 5, 777–780. [Google Scholar] [CrossRef]
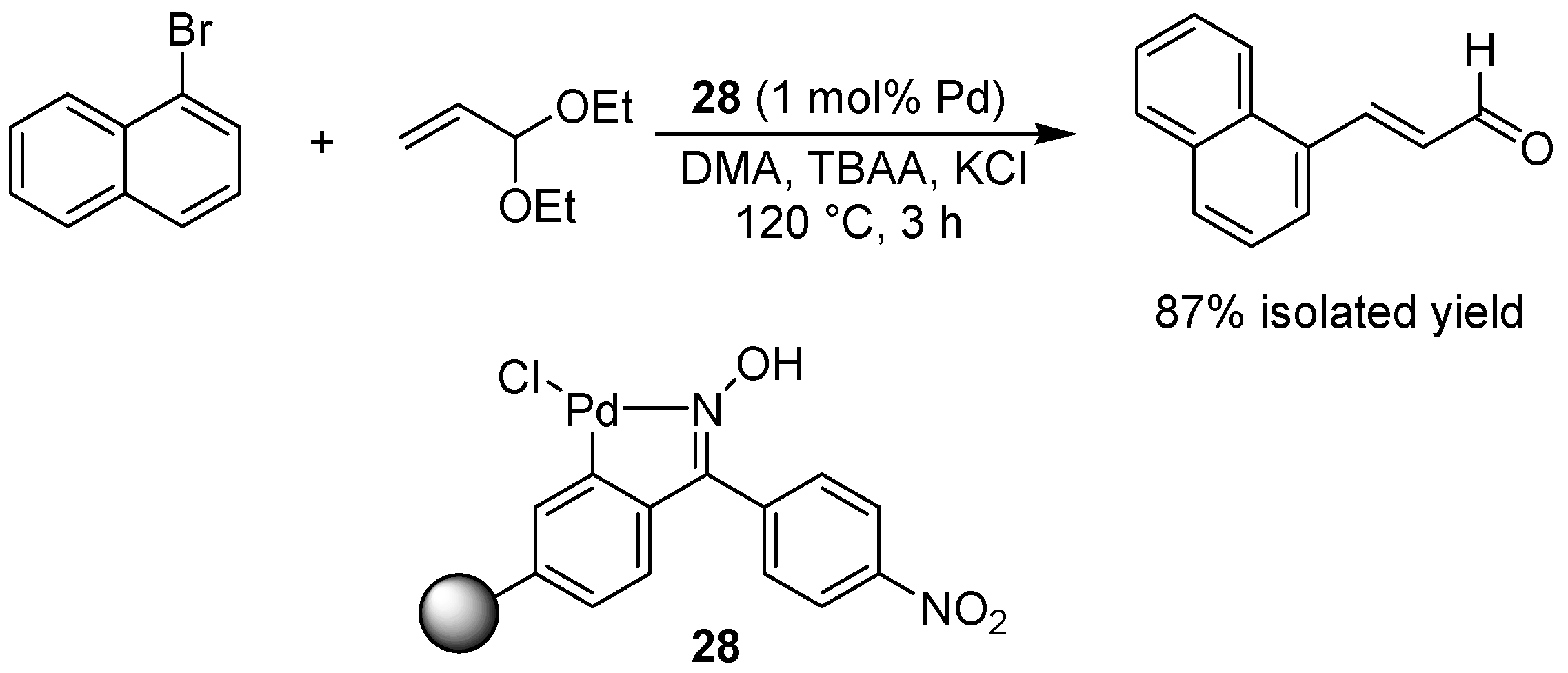
- Alacid, E.; Najera, C. Acrolein diethyl acetal: A three-carbon homologating reagent for the synthesis of beta-arylpropanoates and cinnamaldehydes by Heck reaction catalyzed by a Kaiser oxime resin derived palladacycle. Eur. J. Org. Chem. 2008, 3102–3106. [Google Scholar] [CrossRef]
- Alacid, E.; Najera, C. The Mizoroki-Heck reaction in organic and aqueous solvents promoted by a polymer-supported Kaiser oxime-derived palladacycle. ARKIVOC 2008, 50–67. [Google Scholar]
- Beletskaya, I.P.; Khokhlov, A.R.; Tarasenko, E.A.; Tyurin, V.S. Palladium supported on poly(N-vinylimidazole) or poly(N-vinylimidazole-co-N-vinylcaprolactam) as a new recyclable catalyst for the Mizoroki-Heck reaction. J. Organomet. Chem. 2007, 692, 4402–4406. [Google Scholar] [CrossRef]
- Yao, C.F.; Li, H.G.; Wu, H.H.; Liu, Y.M.; Wu, P. Mesostructured polymer-supported diphenylphosphine-palladium complex: An efficient and recyclable catalyst for Heck reactions. Catal. Commun. 2009, 10, 1099–1102. [Google Scholar] [CrossRef]
- Duong, H.A.; Tekavec, T.N.; Arif, A.M.; Louie, J. Reversible carboxylation of N-heterocyclic carbenes. Chem. Commun. 2004, 112–113. [Google Scholar]
- Bantu, B.; Pawar, G.M.; Decker, U.; Wurst, K.; Schmidt, A.M.; Buchmeiser, M.R. CO2 and Sn-II adducts of N-heterocyclic carbenes as delayed-action catalysts for polyurethane synthesis. Chem. Eur. J. 2009, 15, 3103–3109. [Google Scholar] [CrossRef]
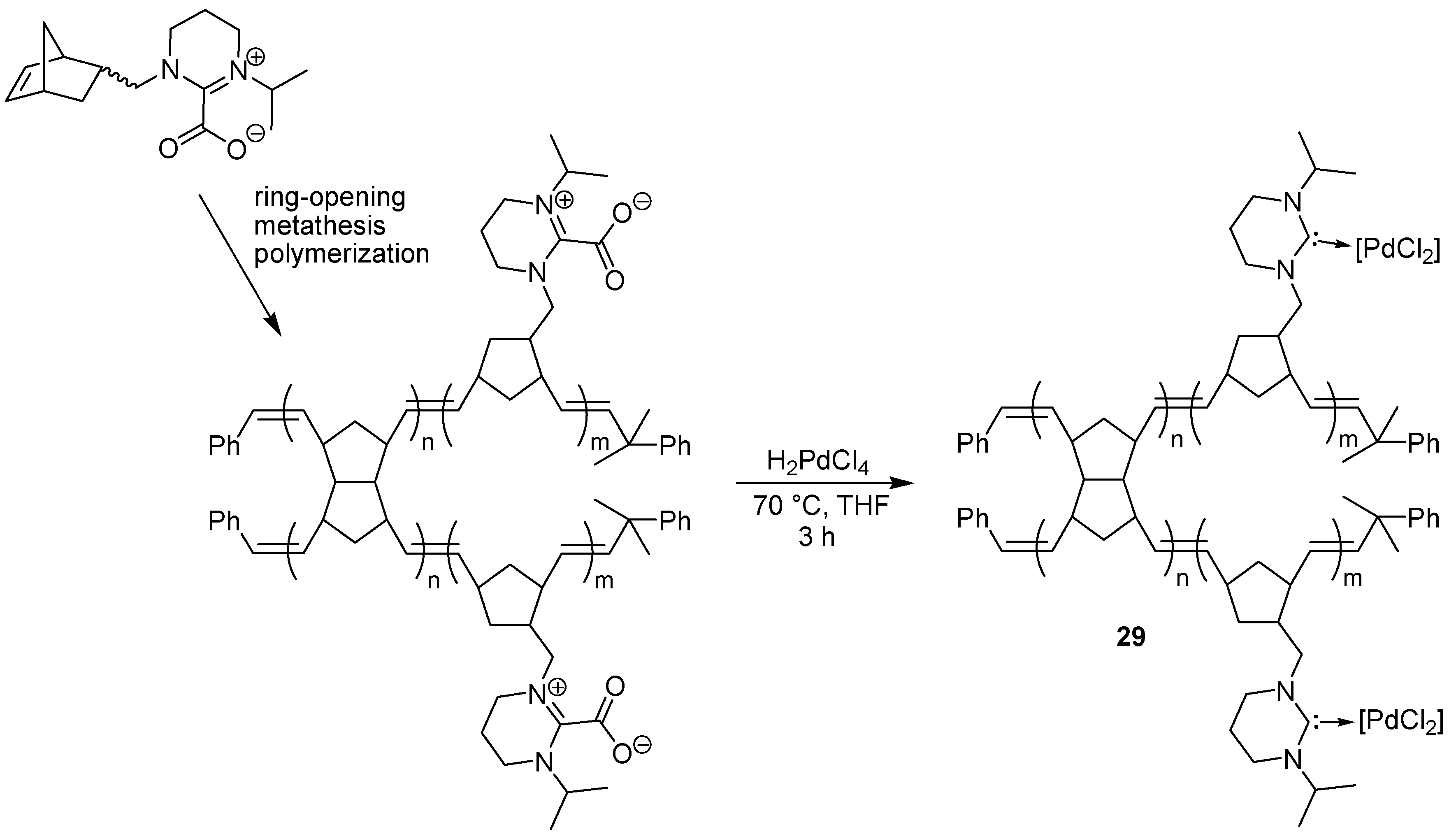
- Pawar, G.M.; Buchmeiser, M.R. Polymer-supported, carbon dioxide-protected N-heterocyclic carbenes: synthesis and application in organo- and organometallic catalysis. Adv. Synth. Catal. 2010, 352, 917–928. [Google Scholar] [CrossRef]
- Atrash, B.; Bradley, M.; Kobylecki, R.; Cowell, D.; Reader, J. Revolutionizing resin handling for combinatorial synthesis. Angew. Chem. Int. Ed. 2001, 40, 938–941. [Google Scholar] [CrossRef]
- Najman, R.; Cho, J.K.; Coffey, A.F.; Davies, J.W.; Bradley, M. Entangled palladium nanoparticles in resin plugs. Chem. Commun. 2007, 5031–5033. [Google Scholar]
- Schweizer, S.; Becht, J.M.; Le Drian, C. Development of efficient and reusable diarylphosphinopolystyrene-supported palladium catalysts for C-C bond forming cross-coupling reactions. Adv. Synth. Catal. 2007, 349, 1150–1158. [Google Scholar] [CrossRef]
- Islam, S.M.; Mondal, P.; Roy, A.S.; Mondal, S.; Hossain, D. Heterogeneous Suzuki and copper-free Sonogashira cross-coupling reactions catalyzed by a reusable palladium(II) complex in water medium. Tetrahedron Lett. 2010, 51, 2067–2070. [Google Scholar] [CrossRef]
- Worm-Leonhard, K.; Meldal, M. Green Catalysts: Solid-Phase Peptide Carbene Ligands in Aqueous Transition-Metal Catalysis. Eur. J. Org. Chem. 2008, 5244–5253. [Google Scholar] [CrossRef]
- Jones, R.C.; Canty, A.J.; Deverell, J.A.; Gardiner, M.G.; Guijt, R.M.; Rodemann, T.; Smith, J.A.; Tolhurst, V.A. Supported palladium catalysis using a heteroleptic 2-methylthiomethylpyridine-N,S-donor motif for Mizoroki-Heck and Suzuki-Miyaura coupling, including continuous organic monolith in capillary microscale flow-through mode. Tetrahedron 2009, 65, 7474–7481. [Google Scholar] [CrossRef]
- Basu, B.; Das, S.; Das, P.; Mandal, B.; Banerjee, D.; Almqvist, F. Palladium Supported on a Polyionic Resin as an Efficient, Ligand-Free, and Recyclable Catalyst for Heck, Suzuki-Miyaura, and Sonogashira Reactions. Synthesis 2009, 1137–1146. [Google Scholar]
- Dahan, A.; Portnoy, M. Pd catalysis on dendronized solid support: Generation effects and the influence of the backbone structure. J. Am. Chem. Soc. 2007, 129, 5860–5869. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Zhang, W.Q.; Wang, Y.; Zhang, M.C. Palladium-iminodiacetic acid immobilized on pH-responsive polymeric microspheres: Efficient quasi-homogeneous catalyst for Suzuki and Heck reactions in aqueous solution. Adv. Synth. Catal. 2008, 350, 2065–2076. [Google Scholar] [CrossRef]
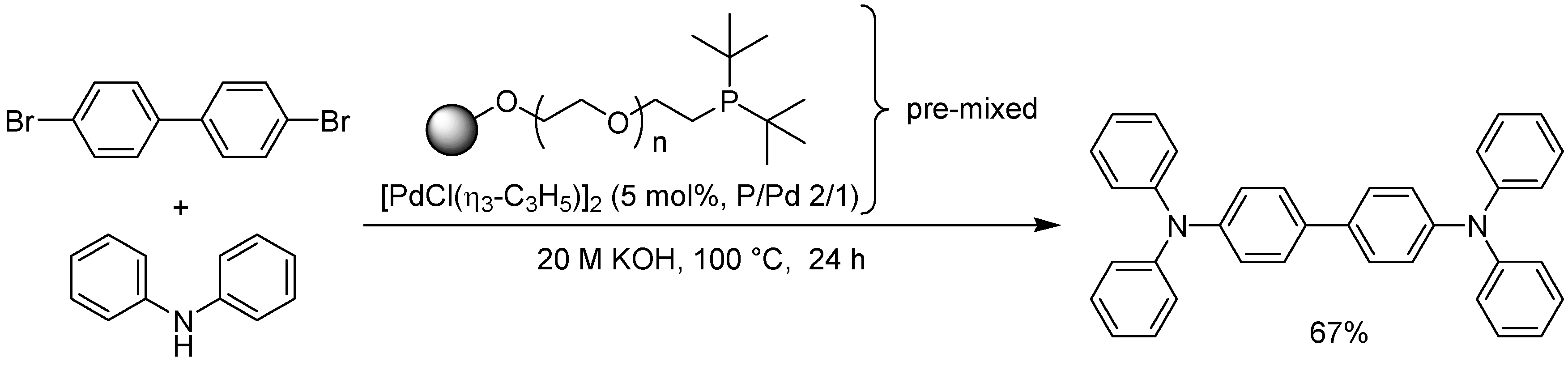
- Hirai, Y.; Uozumi, Y. Clean synthesis of triarylamines: Buchwald-Hartwig reaction in water with amphiphilic resin-supported palladium complexes. Chem. Commun. 2010, 46, 1103–1105. [Google Scholar] [CrossRef]
- Inasaki, T.; Ueno, M.; Miyamoto, S.; Kobayashi, S. Polymer-incarcerated palladium with active phosphine as recoverable and reusable Pd catalyst for the amination of aryl chlorides. Synlett 2007, 3209–3213. [Google Scholar]
- Nishio, R.; Sugiura, M.; Kobayashi, S. Preparation of phosphinated polymer-incarcerated palladium and its application to C-N and C-C bond-forming reactions. Chem. Asian J. 2007, 2, 983–995. [Google Scholar] [CrossRef]
- Christensen, H.; Kiil, S.; Dam-Johansen, K. Applicability of a fiber-supported catalyst on a Buchwald-Hartwig amination reaction. Org. Process Res. Dev. 2007, 11, 956–965. [Google Scholar] [CrossRef]
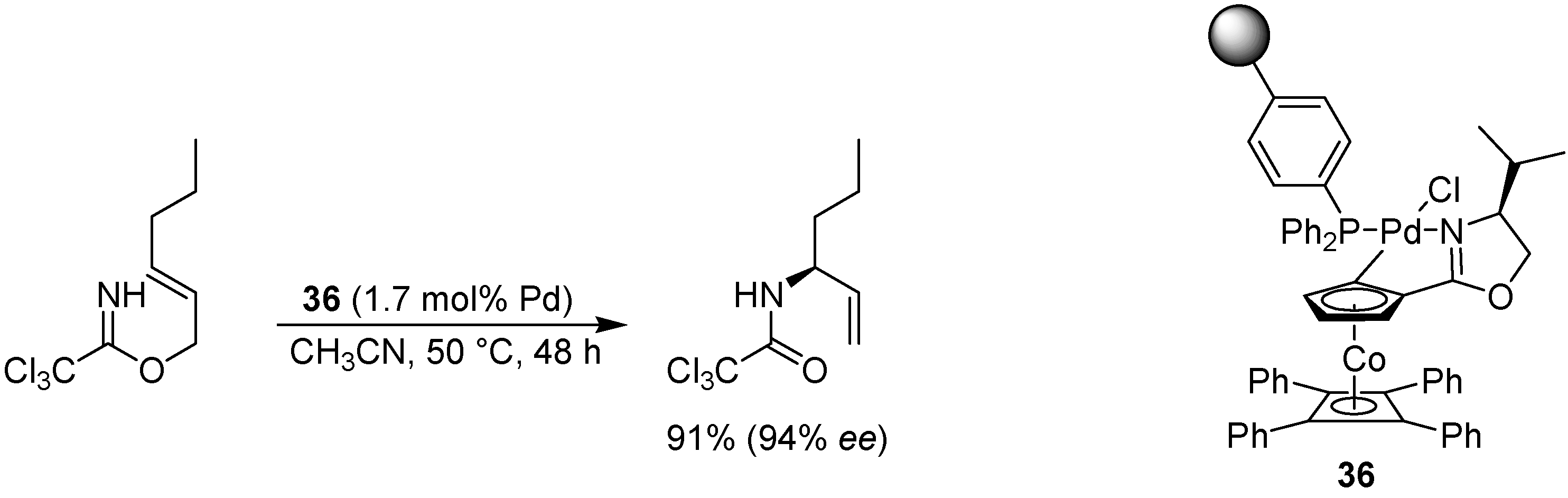
- Nomura, H.; Richards, C.J. An investigation into the allylic imidate rearrangement of trichloroacetimidates catalysed by cobalt oxazoline palladacycles. Chem. Eur. J. 2007, 13, 10216–10224. [Google Scholar] [CrossRef]
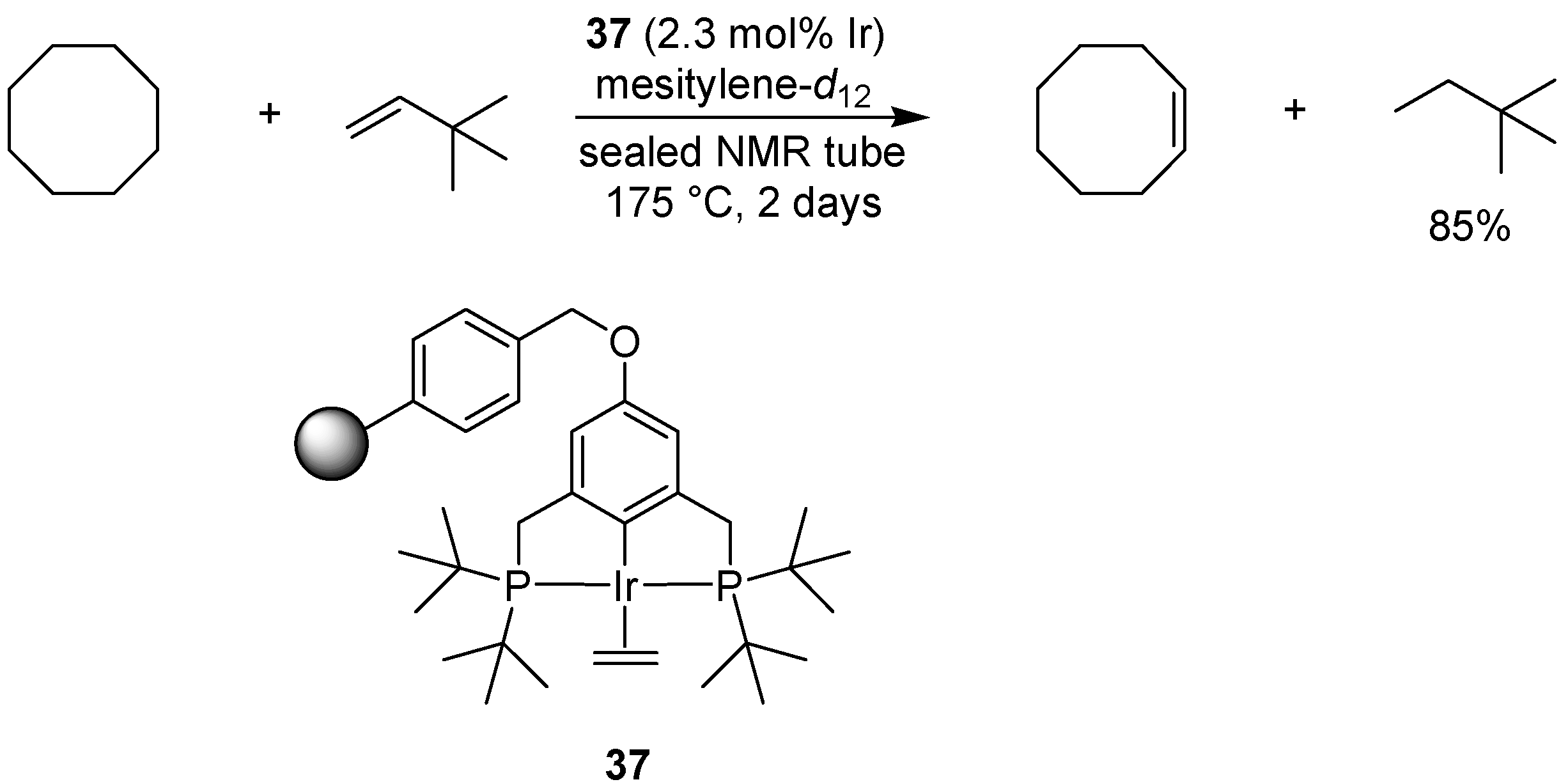
- Huang, Z.; Brookhart, M.; Goldman, A.S.; Kundu, S.; Ray, A.; Scott, S.L.; Vicente, B.C. Highly active and recyclable heterogeneous iridium pincer catalysts for transfer dehydrogenation of alkanes. Adv. Synth. Catal. 2009, 351, 188–206. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kann, N. Recent Applications of Polymer Supported Organometallic Catalysts in Organic Synthesis. Molecules 2010, 15, 6306-6331. https://doi.org/10.3390/molecules15096306
Kann N. Recent Applications of Polymer Supported Organometallic Catalysts in Organic Synthesis. Molecules. 2010; 15(9):6306-6331. https://doi.org/10.3390/molecules15096306
Chicago/Turabian StyleKann, Nina. 2010. "Recent Applications of Polymer Supported Organometallic Catalysts in Organic Synthesis" Molecules 15, no. 9: 6306-6331. https://doi.org/10.3390/molecules15096306