3-Acetyloxy-2-cyano-2-(alkylaminocarbamoyl)propyl Groups as Biodegradable Protecting Groups of Nucleoside 5´-mono-Phosphates

Abstract
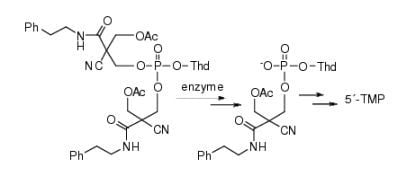
:
1. Introduction

2. Results and Discussion
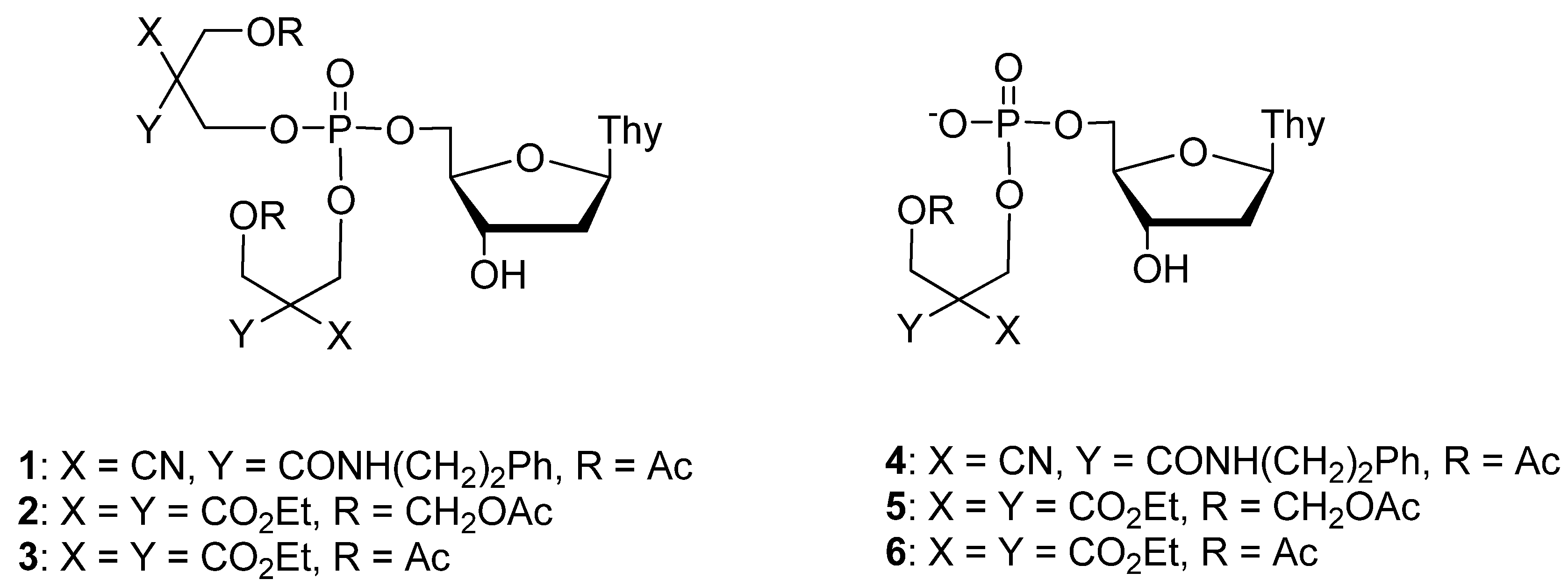
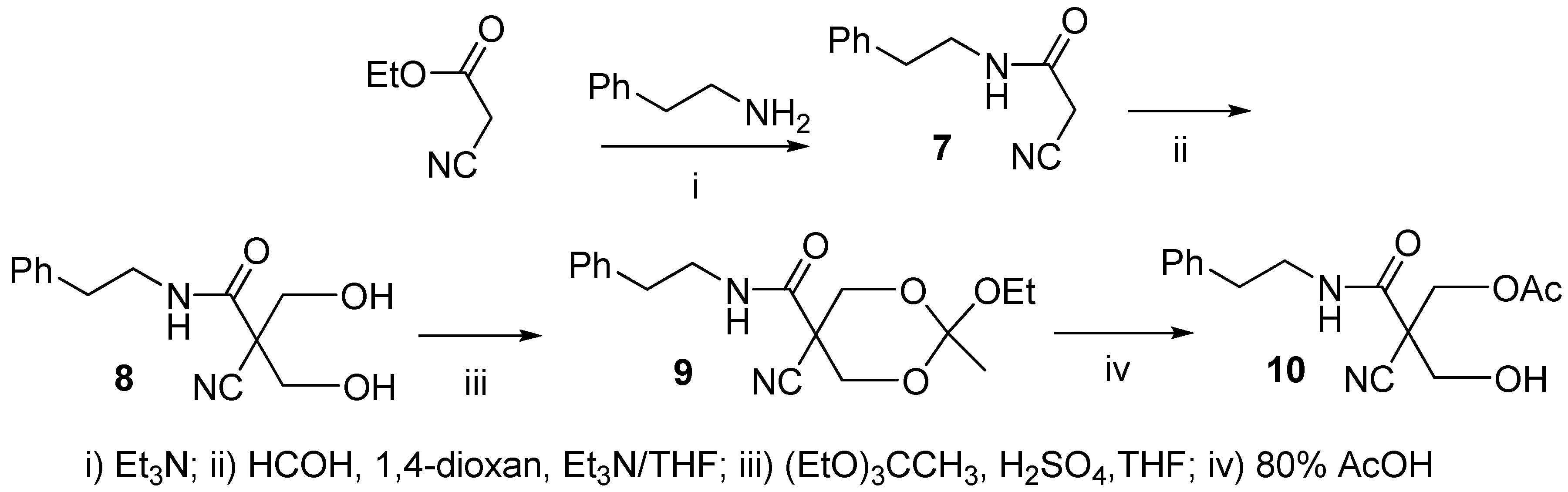
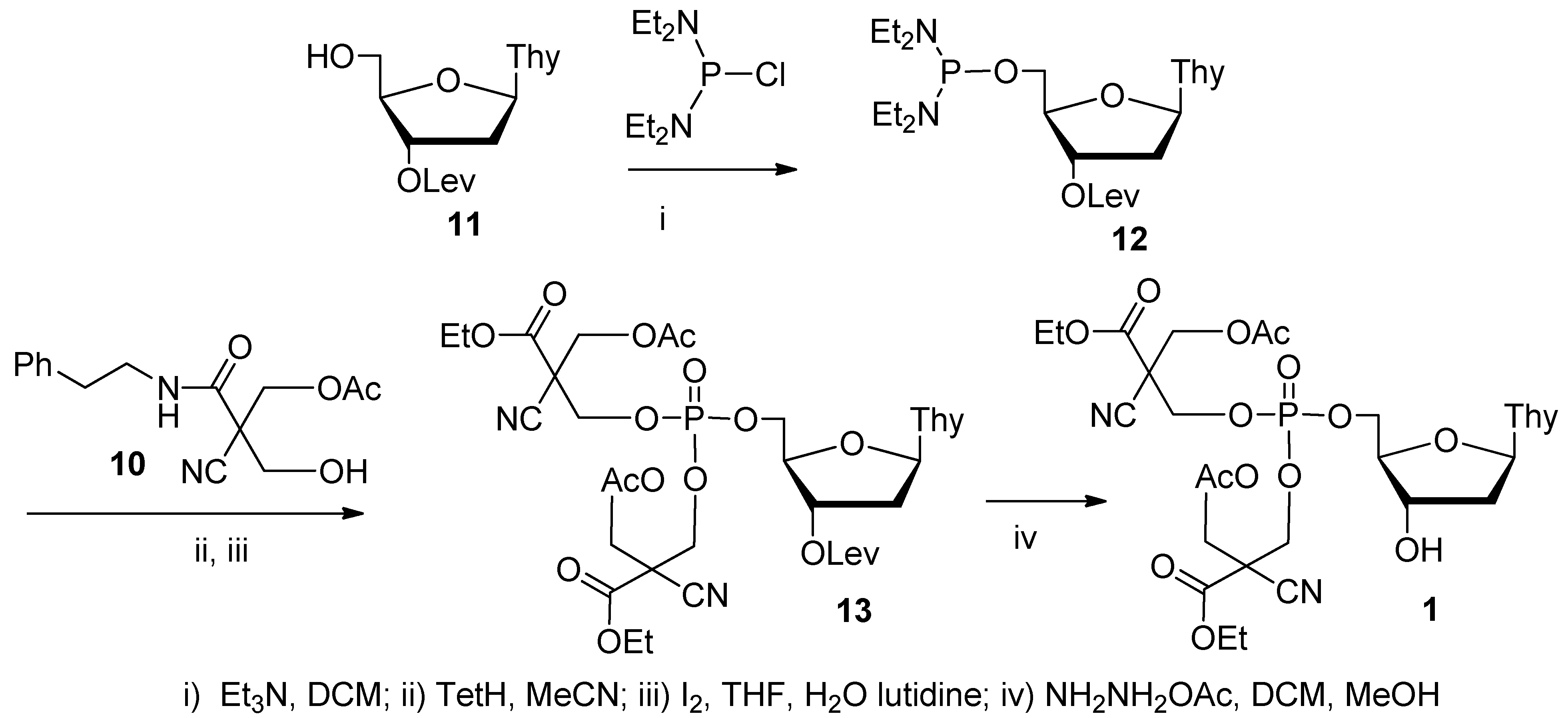
2.1. Synthesis


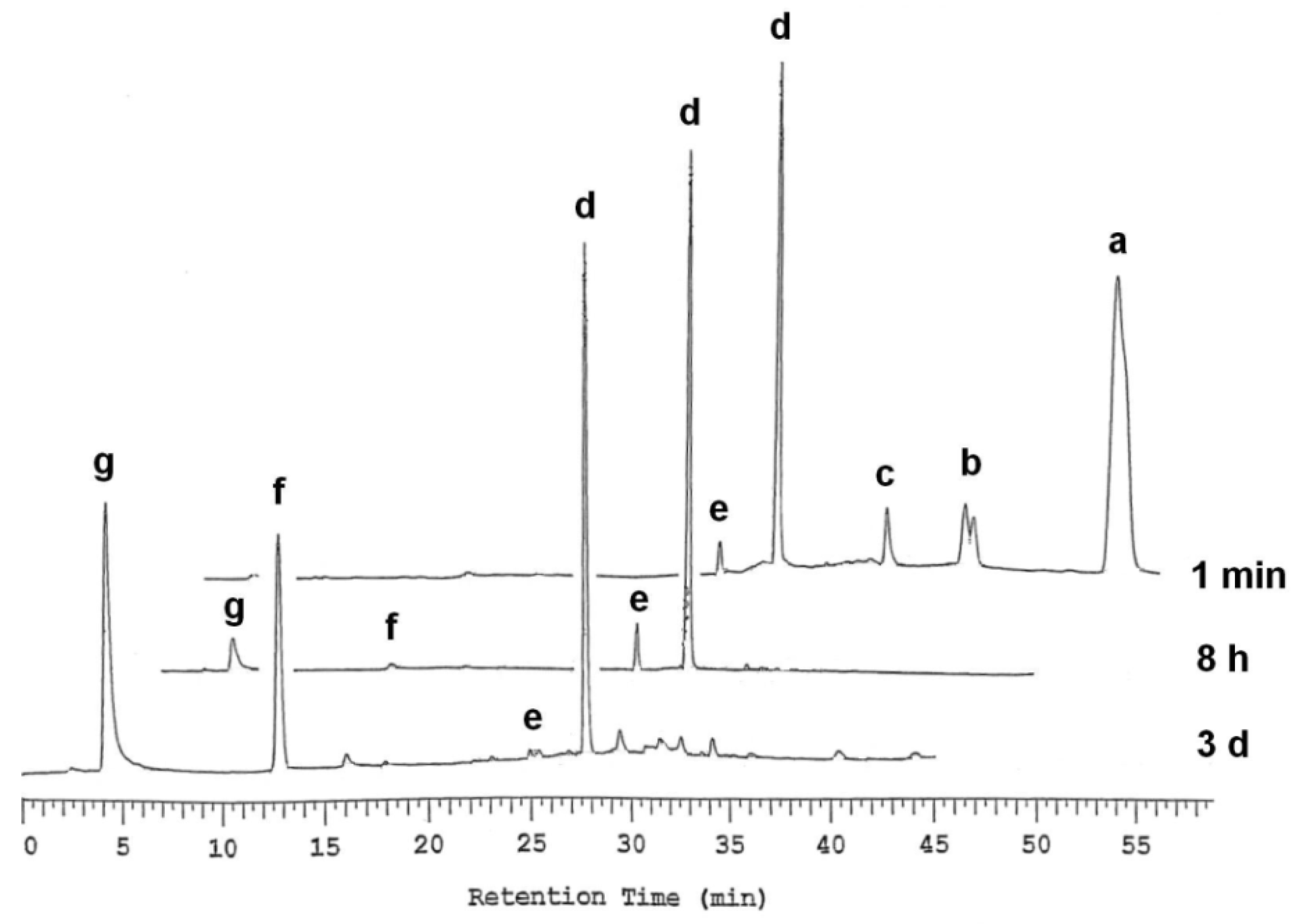
2.2. Hydrolytic Stability of Thymidine 5´-Bis[3-acetyloxy-2-cyano-2-(2-phenylethylcarbamoyl)propyl] phosphate (1)

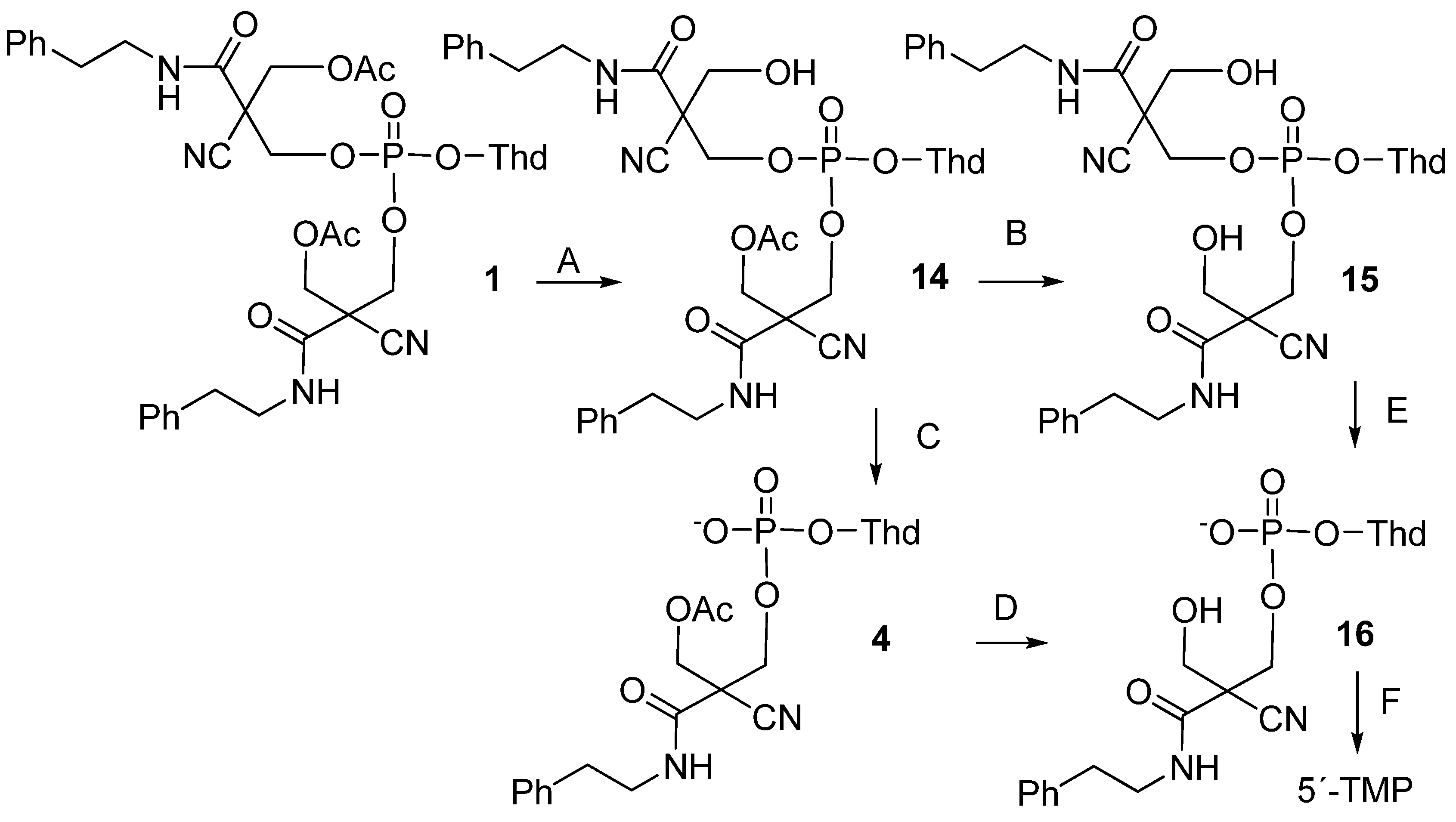
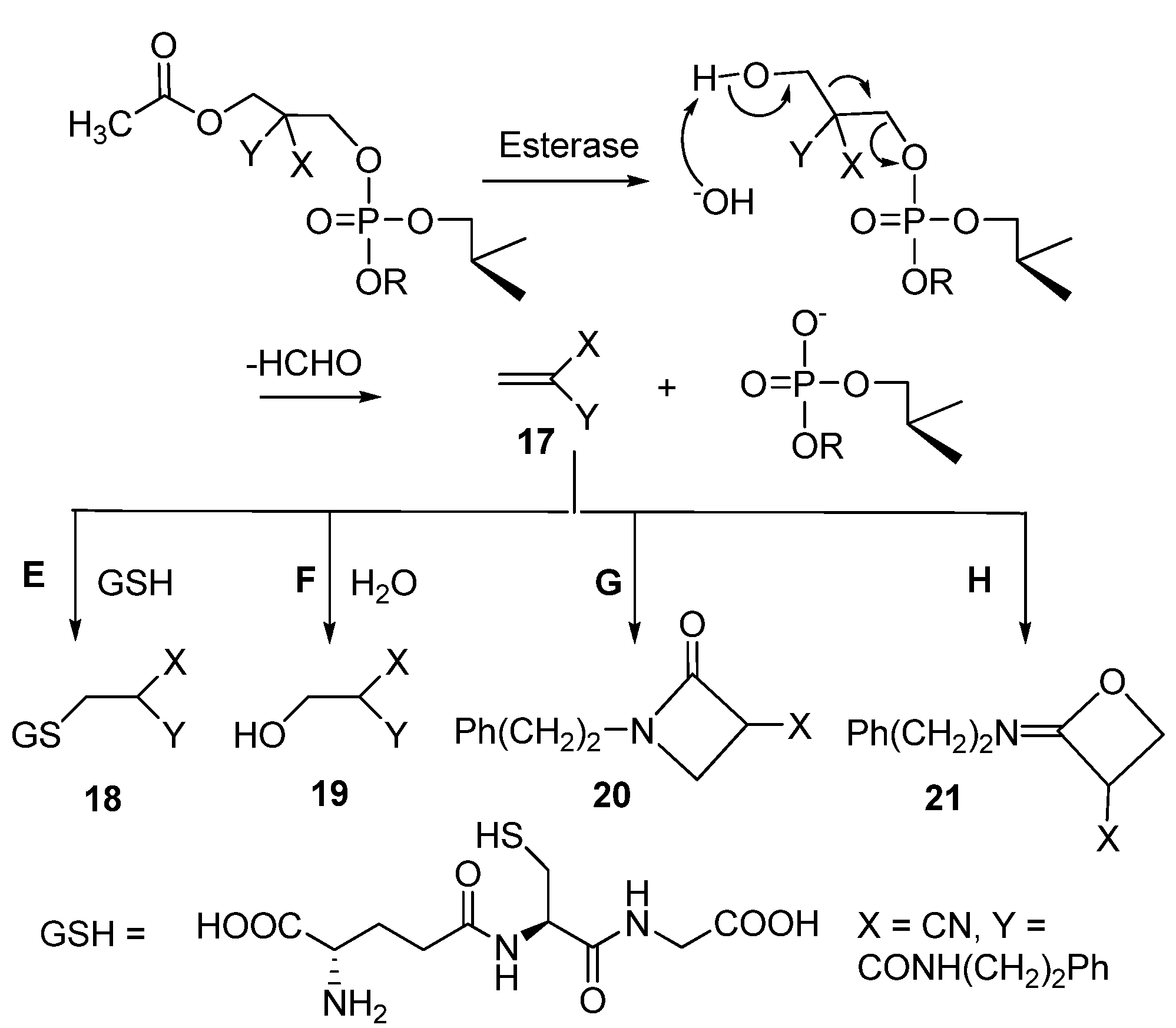
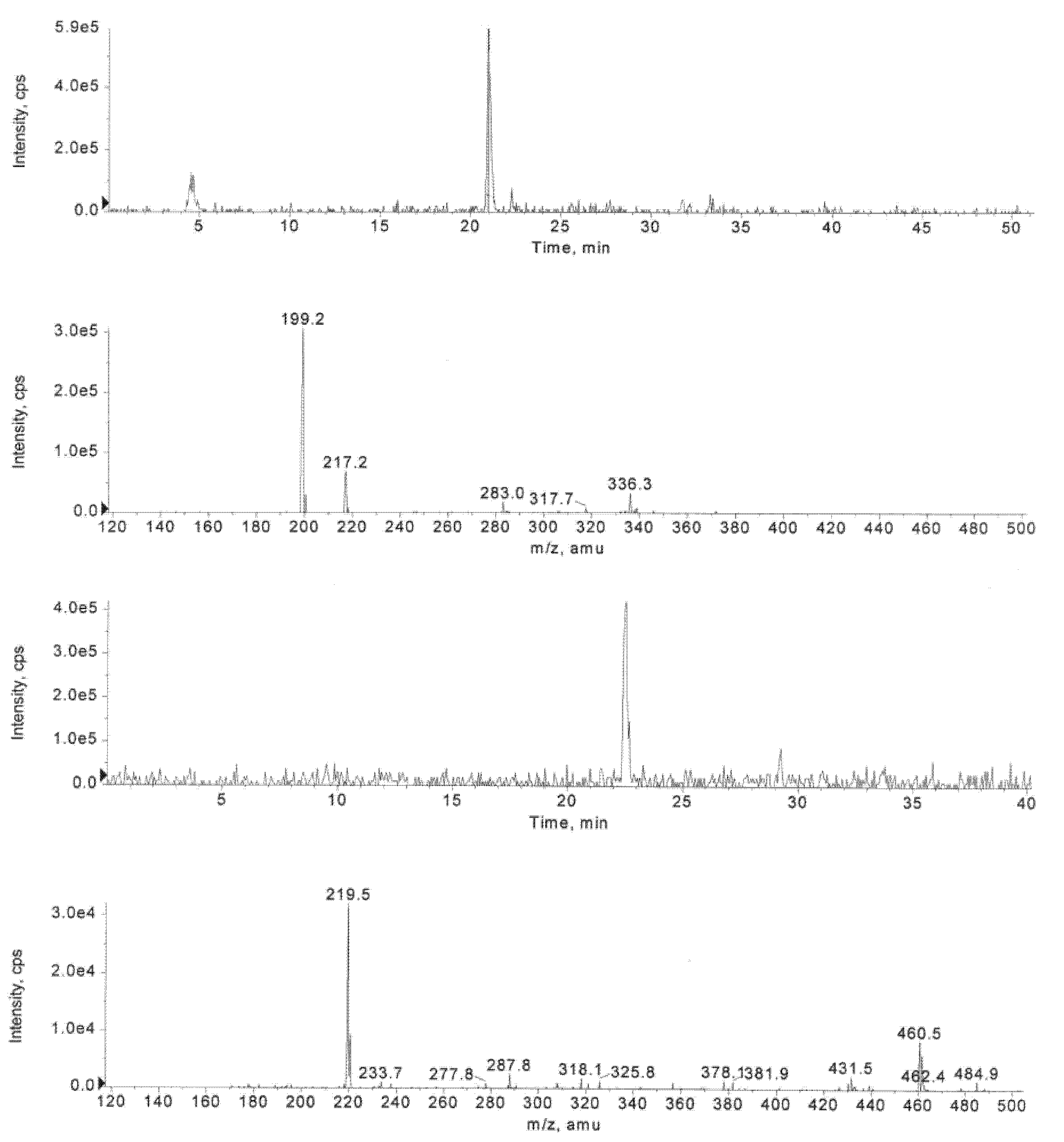
2.3. Enzymatic Deprotection of Thymidine 5´-Bis[3-acetyloxy-2-cyano-2-(2-phenylethylcarbamoyl)-propyl]phosphate (1)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| t1/2 / min | |||
|---|---|---|---|
| Reaction | 1 | 2 | 3 |
| A | 3.8 | 0.17 | 3.2 |
| D | 2838 | 210 | 9060 |
| B | ≈ 2 | 0.90 | 31a |
| C | ≈ 0.2 | 10.4 | 31a |
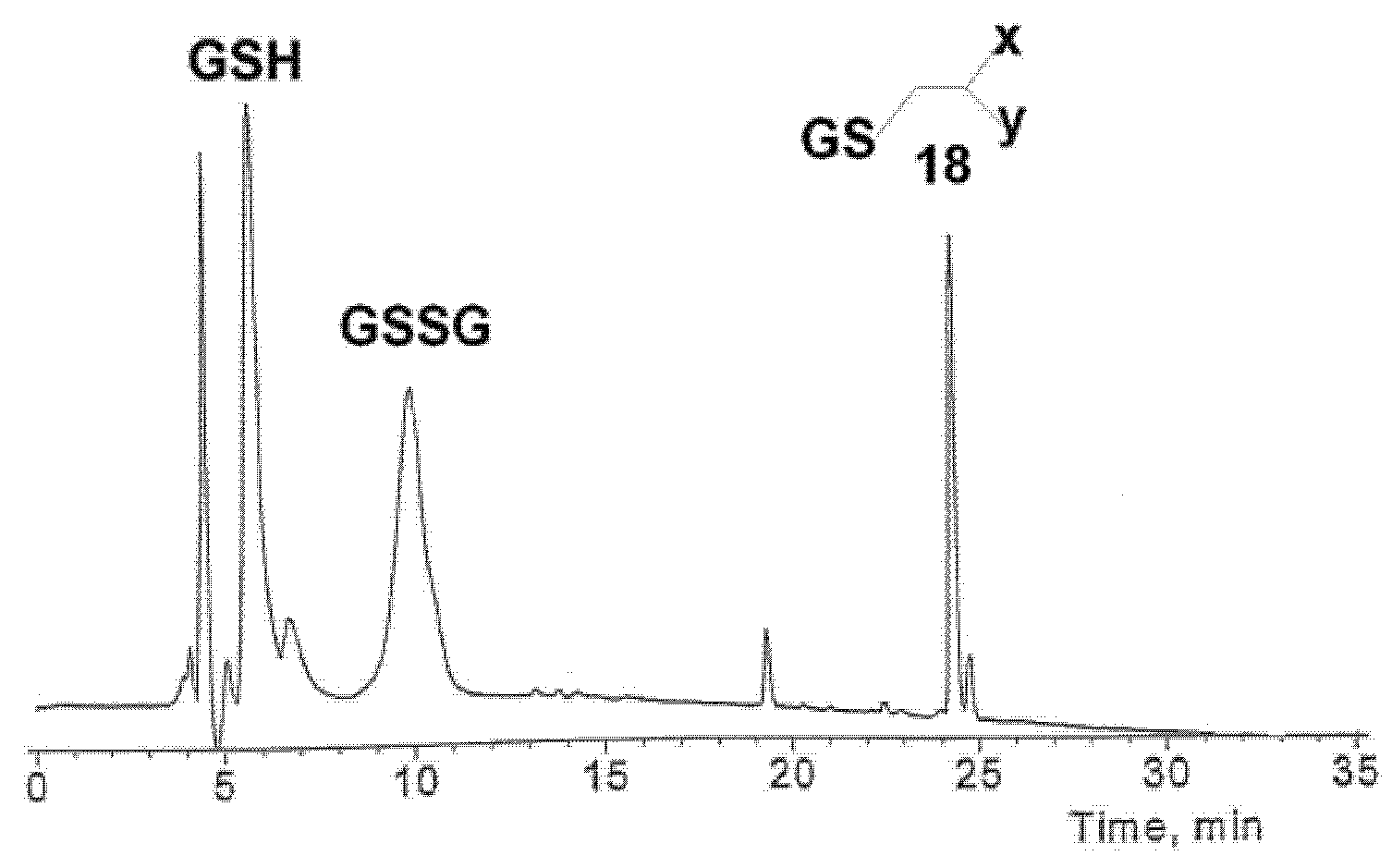
2.4. Reactions of 2-cyano-N-(2-phenyl)Ethylacrylamide with Glutathione




3. Experimental
3.1. General
3.2. Materials
3.3. Kinetic Measurements
4. Conclusions
Acknowledgements
References and Notes
- Hecker, S.J.; Erion, M.D. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008, 51, 2328–2345. [Google Scholar] [CrossRef]
- Poijärvi-Virta, P.; Lönnberg, H. Prodrug approaches of nucleotides and oligonucleotides. Curr. Med. Chem. 2006, 13, 3441–3465. [Google Scholar] [CrossRef]
- Mackman, R.L.; Cihlar, T. Prodrug strategies in the design of nucleoside and nucleotide antiviral therapeutics. Annu. Rep. Med. Chem. 2004, 39, 305–321. [Google Scholar] [CrossRef]
- Schultz, C. Prodrugs of biologically active phosphate esters. Bioorg. Med. Chem. 2003, 11, 885–898. [Google Scholar] [CrossRef]
- Anastasi, C.; Quelever, G.; Burlet, S.; Garino, C.; Souard, F.; Kraus, J.L. New antiviral nucleoside prodrugs await application. Curr. Med. Chem. 2003, 10, 1825–1843. [Google Scholar] [CrossRef]
- Wagner, C.R.; Iyer, V.V.; McIntee, E.J. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med. Res. Rev. 2000, 20, 417–451. [Google Scholar] [CrossRef]
- Krecmerova, M.; Holy, A.; Pohl, R.; Masojidkova, M.; Andrei, G.; Naesens, L.; Neyts, J.; Balzarini, J.; De Clercq, E.; Snoeck, R. Ester prodrugs of cyclic 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine: Synthesis and antiviral activity. J. Med. Chem. 2007, 50, 5765–5772. [Google Scholar]
- Padmanabhan, S.; Coughlin, J.E.; Zhang, G.; Kirk, C.J.; Iyer, R.P. Anti-HBV nucleotide prodruganalogs: Synthesis, bio-reversibility, and cytotoxicity studies. Bioorg. Med. Chem. Lett. 2006, 16, 1491–1494. [Google Scholar]
- Khan, S.R.; Nowak, B.; Plunkett, W.; Farquhar, D. Bis(pivaloyloxymethyl) thymidine 5'-phosphate is a cell membrane-permeable precursor of thymidine 5'-phosphate in thymidine kinase deficient CCRF CEM cells. Biochem. Pharmacol. 2005, 69, 1307–1313. [Google Scholar] [CrossRef]
- Choi, J.R.; Cho, D.G.; Roh, K.Y.; Hwang, J.T.; Ahn, S.; Jang, H.S.; Cho, W.Y.; Kim, K.W.; Cho, Y.G.; Kim, J.; Kim, Y.Z. A novel class of phosphonate nucleosides 9-((1-phosphonomethoxycycloproyl)-methyl)-guanine as a potent and selective anti-Hbv agent. J. Med. Chem. 2004, 47, 2864–2869. [Google Scholar]
- Rose, J.D.; Parker, W.B.; Someya, H.; Shaddix, S.G.; Montgomery, J.; Secrist III, J.A. Enhancement of nucleoside cytotoxicity through nucleotide prodrugs. J. Med. Chem. 2002, 45, 4505–4512. [Google Scholar] [CrossRef]
- Shaw, J.-P.; Louie, M.S.; Krishnamurthy, V.V.; Arimilli, M.N.; Jones, R.J.; Bidgood, A.M.; Lee, W.A.; Cundy, K.C. Pharmacokinetics and metabolism of selected prodrugs of PMEA in rats. Drug Metab. Dispos. 1997, 25, 362–366. [Google Scholar]
- Starrett, J.E., Jr.; Tortolani, D.R.; Russell, J.; Hitchcock, M.J.M.; Whiterock, V.; Martin, J.C.; Mansuri, M.M. Synthesis, oral bioavailability determination, and in vitro evaluation of prodrugs of the antiviral agent 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA). J. Med. Chem. 1994, 37, 1857–1864. [Google Scholar] [CrossRef]
- Farquhar, D.; Khan, S.; Srivastava, D.N.; Saunders, P.P. Synthesis and antitumor evaluation of bis[(pivaloyloxy)methyl] 2’-deoxy-5-fluorouridine 5’-monophosphate (FdUMP): a strategy to introduce nucleotides into hells. J. Med. Chem. 1994, 37, 3902–3909. [Google Scholar] [CrossRef]
- Tang, Y.-b.; Peng, Z.-g.; Liu, Z.-y.; Li, Y.-p.; Jiang, J.-d.; Li, Z.-r. Some new acyclic nucleotide analogues as antiviral prodrugs: Synthesis and bioactivities in vitro. Bioorg. Med. Chem. Lett. 2007, 17, 6350–6353. [Google Scholar] [CrossRef]
- Mackman, R.L.; Zhang, L.; Prasad, V.; Boojamra, C.G.; Douglas, J.; Grant, D.; Hui, H.; Kim, C.U.; Laflamme, G.; Parrish, J.; Stoycheva, A.D.; Swaminathan, S.; Wang, K.; Cihlar, T. Synthesis, anti-HIV activity, and resistance profile of thymidine phosphonomethoxy nucleosides and their bis-isopropyloxymethylcarbonyl (bisPOC) prodrugs. Bioorg. Med. Chem. 2007, 15, 5519–5528. [Google Scholar] [CrossRef]
- De Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef]
- Arimrilli, M.N.; Kim, C.U.; Dougherty, J.; Mulato, A.; Oliyai, R.; Shaw, J.-P.; Cundy, K.C.; Bischofberger, N. Synthesis, in vitro biological evaluation and oral bioavailability of 9-[2-(phosphonomethoxy)propyl]adenine (PMPA) prodrugs. Antivir. Chemother. 1997, 8, 557–564. [Google Scholar]
- Shaw, J.-P.; Sueoka, C.M.; Oliyai, R.; Lee, W.A.; Arimilli, M.N.; Kim, C.U.; Cundy, K.C. Metabolism and pharmacokinetics of novel oral prodrugs of 9-[(R)-2-(phosphonomethoxy)propyl]adenine (PMPA) in dogs. Pharm. Res. 1997, 14, 1824–1829. [Google Scholar] [CrossRef]
- Villard, A.-L.; Coussot, G.; Lefebvre, I.; Augustijns, P.; Aubertin, A.-M.; Gosselin, G.; Peyrottes, S.; Perigaud, C. Phenyl phosphotriester derivatives of AZT: Variations upon the SATE moiety. Bioorg. Med. Chem. 2008, 16, 7321–7329. [Google Scholar] [CrossRef]
- Peyrottes, S.; Egron, D.; Lefebvre, I.; Gosselin, G.; Imbach, J.L.; Perigaud, C. SATE pronucleotide approaches: an overview. Mini-Rev. Med. Chem. 2004, 4, 395–408, and references therein. [Google Scholar]
- Perigaud, C.; Gosselin, G.; Lefebvre, I.; Girardet, J.L.; Benzaria, S.; Barber, I.; Imbach, J.L. Rational design for cytosolic delivery of nucleoside monophosphates: "SATE" and "DTE" as enzyme-labile transient phosphate protecting groups. Bioorg. Med. Chem. Lett. 1993, 3, 2521–2526. [Google Scholar] [CrossRef]
- Routledge, A.; Walker, I.; Freeman, S.; Hay, A.; Mahmood, N. Synthesis, bioactivation and anti-HIV activity of 4-acyloxybenzyl bis(nucleoside-5'-yl) phosphates. Nucleos. Nucleot. 1995, 14, 1545–1558. [Google Scholar] [CrossRef]
- Srivastva, D.N.; Farquhar, D. Bioreversible phosphate protective groups: Synthesis and stability of model acyloxymethyl phosphates. Bioorg. Chem. 1984, 12, 118–129. [Google Scholar] [CrossRef]
- Farquhar, D.; Srivastva, D.N.; Kuttesch, N.J.; Saunders, P.P. Biologically reversible phosphate-protective groups. J. Pharm. Sci. 1983, 72, 324–325. [Google Scholar] [CrossRef]
- Mittchell, A.G.; Nicholls, D.; Walker, I.; Irwin, W.J.; Freeman, S. Prodrugs of phosphonoformate: Products, kinetics and mechanisms of hydrolysis of dibenzyl (methoxycarbonyl)phosphonate. J. Chem. Soc., Perkin Trans. 2 1991, 1297–1303. [Google Scholar]
- Mitchell, A.G.; Thompson, W.; Nicholls, D.; Irwin, W.J.; Freeman, S. Bioreversible protection for the phospho group: bioactivation of the di(4-acyloxybenzyl) and mono(4-acyloxybenzyl) phosphoesters of methylphosphonate and phosphonoacetate. J. Chem. Soc. Perkin Trans. 1 1992, 2345–2353. [Google Scholar]
- Thomson, W.; Nicholls, D.; Irwin, W.J.; Al-Mushadani, J.S.; Freeman, S.; Karpas, A.; Petrik, J.; Mahmood, N.; Hay, A.J. Synthesis, bioactivation and anti-HIV activity of the bis(4-acyloxybenzyl) and mono(4-acyloxybenzyl) esters of the 5'-monophosphate of AZT. J. Chem. Soc., Perkin Trans. 1 1993, 1239–1245. [Google Scholar]
- Ora, M.; Taherpour, S.; Linna, R.; Leisvuori, A.; Hietamaki, E.; Poijärvi-Virta, P.; Beigelman, L.; Lönnberg, H. Biodegradable protections for nucleoside 5'-monophosphates: Comparative study on the removal of O-acetyl and O-acetyloxymethyl protected 3-hydroxy-2,2-bis(ethoxycarbonyl)propyl groups. J. Org. Chem. 2009, 74, 4992–5001. [Google Scholar]
- Ora, M.; Mäki, E.; Poijärvi, P.; Neuvonen, K.; Oivanen, M.; Lönnberg, H. Hydrolytic stability of nucleoside phosphotriesters derived from bis(hydroxymethyl)-1,3-dicarbonyl compounds and their congeners: towards a novel pro-drug strategy for antisense oligonucleotides. J. Chem. Soc., Perkin Trans. 2 2001, 881–885. [Google Scholar]
- Poijärvi, P.; Mäki, E.; Tomperi, J.; Ora, M.; Oivanen, M.; Lönnberg, H. Towards nucleotide prodrugs derived from 2,2-bis(hydroxymethyl)malonate and its congeners: hydrolytic cleavage of 2-cyano-2-(hydroxymethyl)-3-methoxy-3-oxopropyl and 3-(alkylamino)-2-cyano-2-(hydroxymethyl)-3-oxopropyl protections from the internucleosidicphosphodiester and phosphorothioate linkages. Helv. Chim. Acta 2002, 85, 1869–1876. [Google Scholar] [CrossRef]
- Poijärvi, P.; Oivanen, M.; Lönnberg, H. Towards oligonucleotide prodrugs: 2,2-bis(ethoxycarbonyl) and 2-(alkylaminocarbonyl)-2-cyano substituted 3-(pivaloyloxy)propyl groups as biodegradable protecting groups for internucleosidicphosphoromonothioate linkages. Lett. Org. Chem. 2004, 1, 183–188. [Google Scholar] [CrossRef]
- Poijärvi, P.; Heinonen, P.; Virta, P.; Lönnberg, H. 2,2-Bis(ethoxycarbonyl)- and 2-(alkylaminocarbonyl)-2-cyano-substituted 3-(pivaloyloxy)propyl groups as biodegradable phosphate protections of oligonucleotides. Bioconjugate Chem. 2005, 16, 1564–1571. [Google Scholar] [CrossRef]
- Erion, M.D.; van Poelje, P.D.; Mackenna, D.A.; Colby, T.J.; Montag, A.C.; Fujitaki, J.M.; Linemeyer, D.L.; Bullough, D.A. Liver-targeted drug delivery using HepDirectprodrugs. J. Pharmacol. Exp. Ther. 2005, 312, 554–560. [Google Scholar]
- Casanova, M.; Heck, H. Further studies of the metabolic incorporation and covalent binding of inhaled [3H]- and [14C]formaldehyde in Fischer-344 rats: effects of glutathione depletion. Toxicol. Appl. Pharmacol. 1987, 89, 105–121. [Google Scholar] [CrossRef]
- Lu, K.; Ye, W.; Ball, A.G.L.M.; Swenberg, J.A. Formation of S-[1-(N2-Deoxyguanosinyl)methyl]glutathione between Glutathione and DNA Induced by Formaldehyde. J. Am. Chem. Soc. 2009, 131, 3414–3415. [Google Scholar]
- Guzaev, A.; Lönnberg, H. Bis(hydroxymethylation) of the active methylene group of 1,3-dicarbonyl and related compounds. Synthesis 1997, 1281–1284. [Google Scholar] [CrossRef]
- Guzaev, A.; Salo, H.; Azhayev, A.; Lönnberg, H. Novel non-nucleosidic building blocks for the preparation of multilabeled oligonucleotides. Bioconjugate Chem. 1996, 7, 240–248. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1, 8 and 10 are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ora, M.; Mäntyvaara, A.; Lönnberg, H. 3-Acetyloxy-2-cyano-2-(alkylaminocarbamoyl)propyl Groups as Biodegradable Protecting Groups of Nucleoside 5´-mono-Phosphates. Molecules 2011, 16, 552-566. https://doi.org/10.3390/molecules16010552
Ora M, Mäntyvaara A, Lönnberg H. 3-Acetyloxy-2-cyano-2-(alkylaminocarbamoyl)propyl Groups as Biodegradable Protecting Groups of Nucleoside 5´-mono-Phosphates. Molecules. 2011; 16(1):552-566. https://doi.org/10.3390/molecules16010552
Chicago/Turabian StyleOra, Mikko, Anne Mäntyvaara, and Harri Lönnberg. 2011. "3-Acetyloxy-2-cyano-2-(alkylaminocarbamoyl)propyl Groups as Biodegradable Protecting Groups of Nucleoside 5´-mono-Phosphates" Molecules 16, no. 1: 552-566. https://doi.org/10.3390/molecules16010552