The Beneficial Effect of Hydrogen on CO Oxidation over Au Catalysts. A Computational Study

Abstract
:1. Introduction
2. Computational Details

3. Results
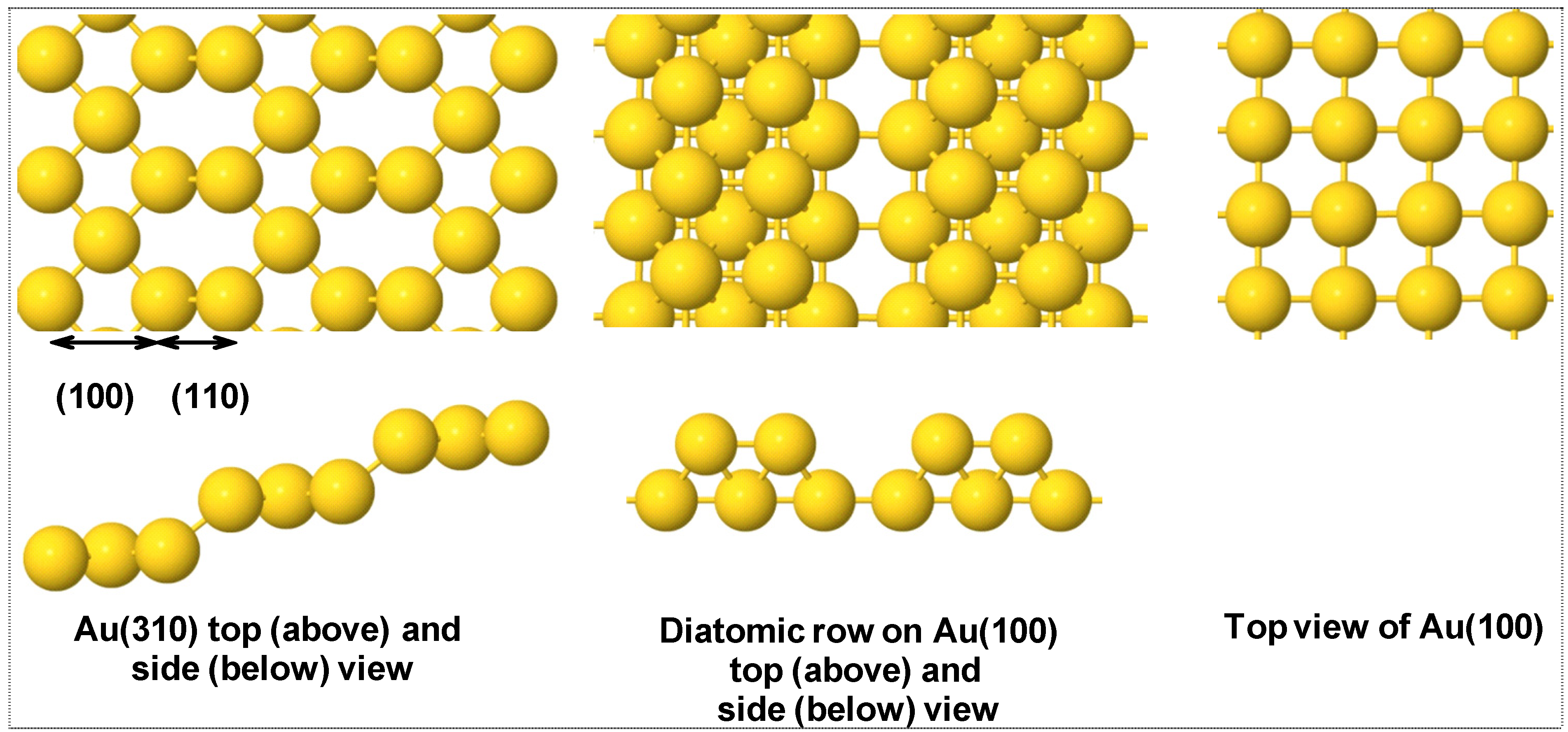
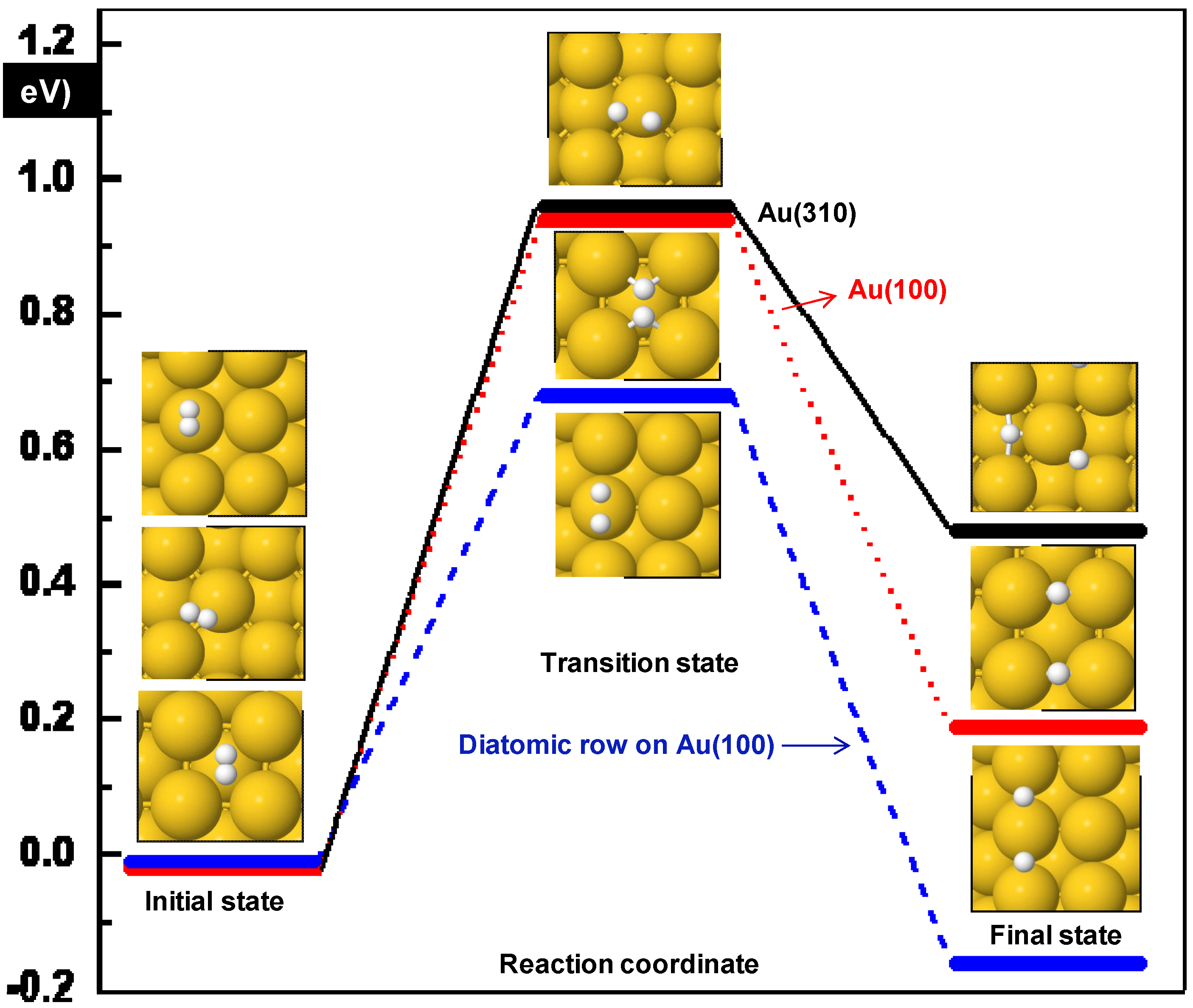
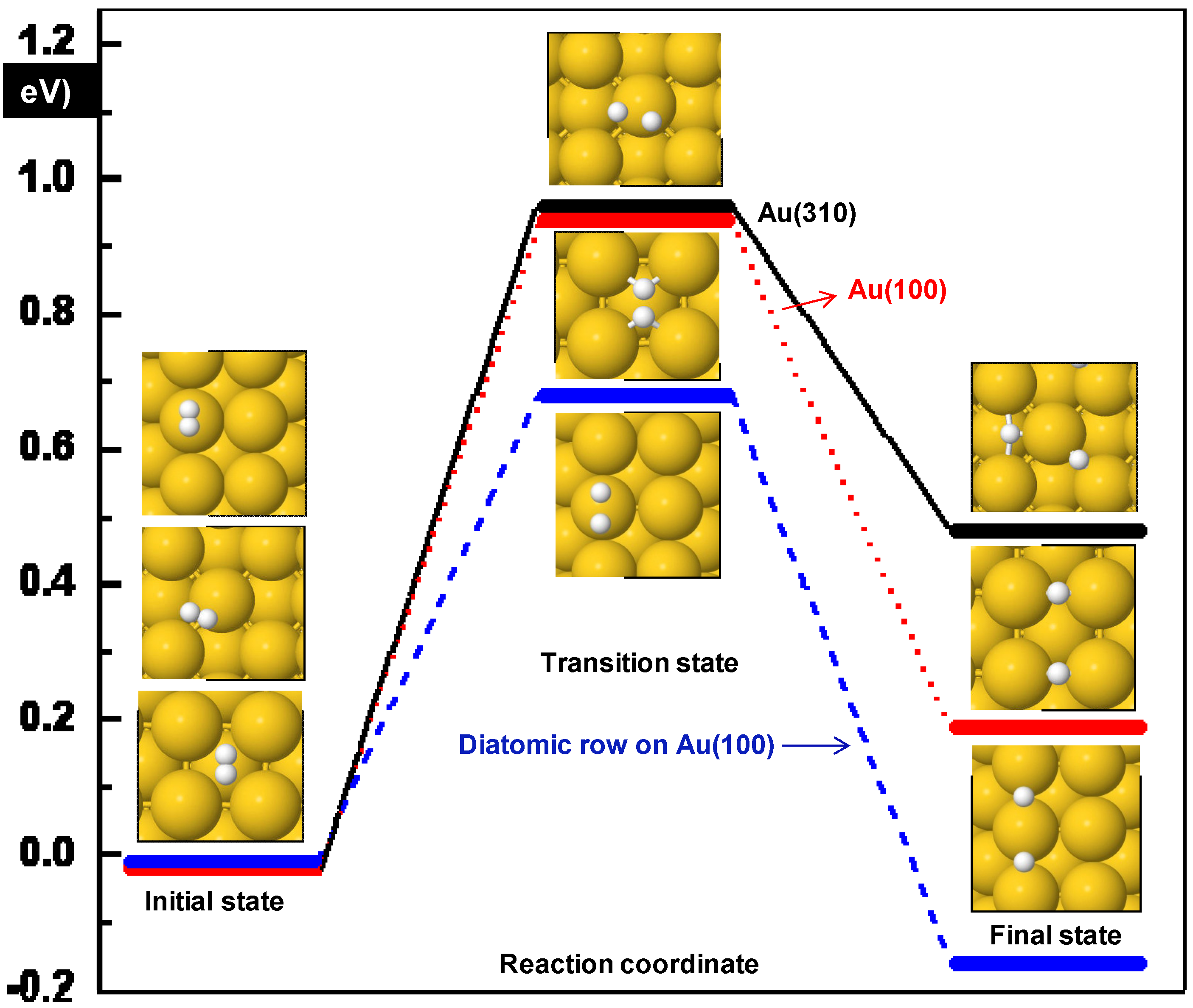
3.1. H2 Adsorption and Dissociation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Position | Eads (eV) |
|---|---|---|
| H | (100)bridge at outer step | −2.18 |
| (100)bridge at inside step | −2.14 | |
| bridge at step | −1.96 | |
| (100) hollow | −1.9 | |
| top (100) | −1.89 | |
| top at step (110) | −1.98 | |
| O2 + H | bridge at step + (100) | −2.21 |
| bridge at inside step | ||
| OOH | bridge at step | −1.00 |
| OH + O | bridge at step + (100) hollow | −4.97 |
| OOH + CO | bridge at step + (100) bridge at inside step | −1.31 |
| OH + CO2 | bridge at step + (100) bridge at outer step (across) | −2.2 |
| O2 + H2 | bridge at step | −0.19 |
| OH+ OH | bridge at step + (100) hollow | −4.00 |
| O + H | bridge at step + (100) bridge at inside step- | −5.49 |
| OH | bridge at step | −2.31 |
| OH + H | bridge at step + (100) bridge at inside step | −4.44 |
| H2O | top at step (flat) | −0.23 |
| H2O + O | bridge at step + (100) bridge at outer step | −3.46 |
| CO + O | top at step + (100) hollow | −3.61 |
| Surface | Initial position | Ea (eV) | ΔE (eV) | dH-H (Å) | ν (cm−1) |
|---|---|---|---|---|---|
| Diatomic rows Au(100) | Top | 0.69 | −0.16 | 1.41 | 1044i |
| Au(100) | hollow | 0.96 | 0.21 | 1.04 | 241i |
| Au(310) | (100) top | 0.98 | 0.50 | 1.45 | 322i |
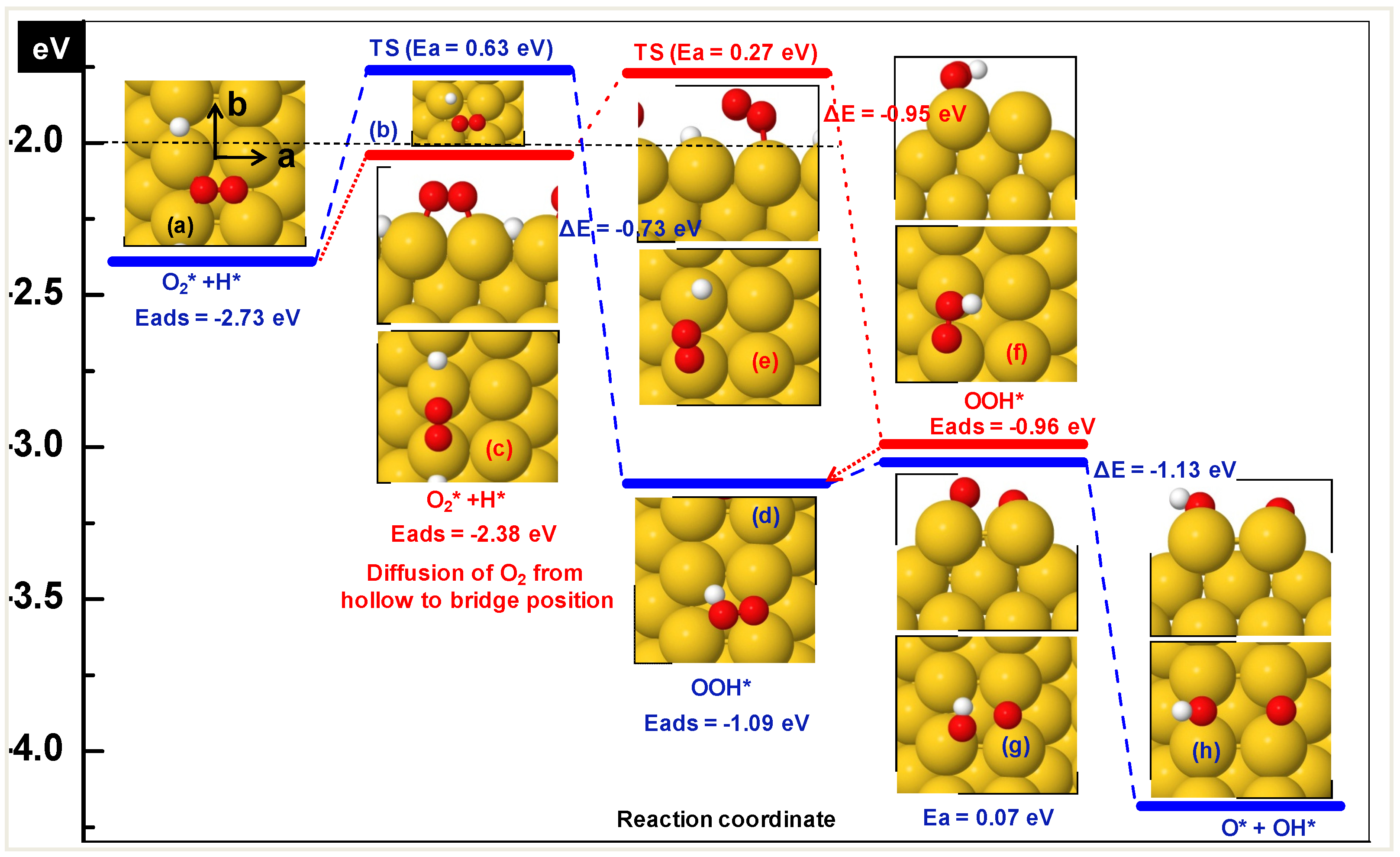
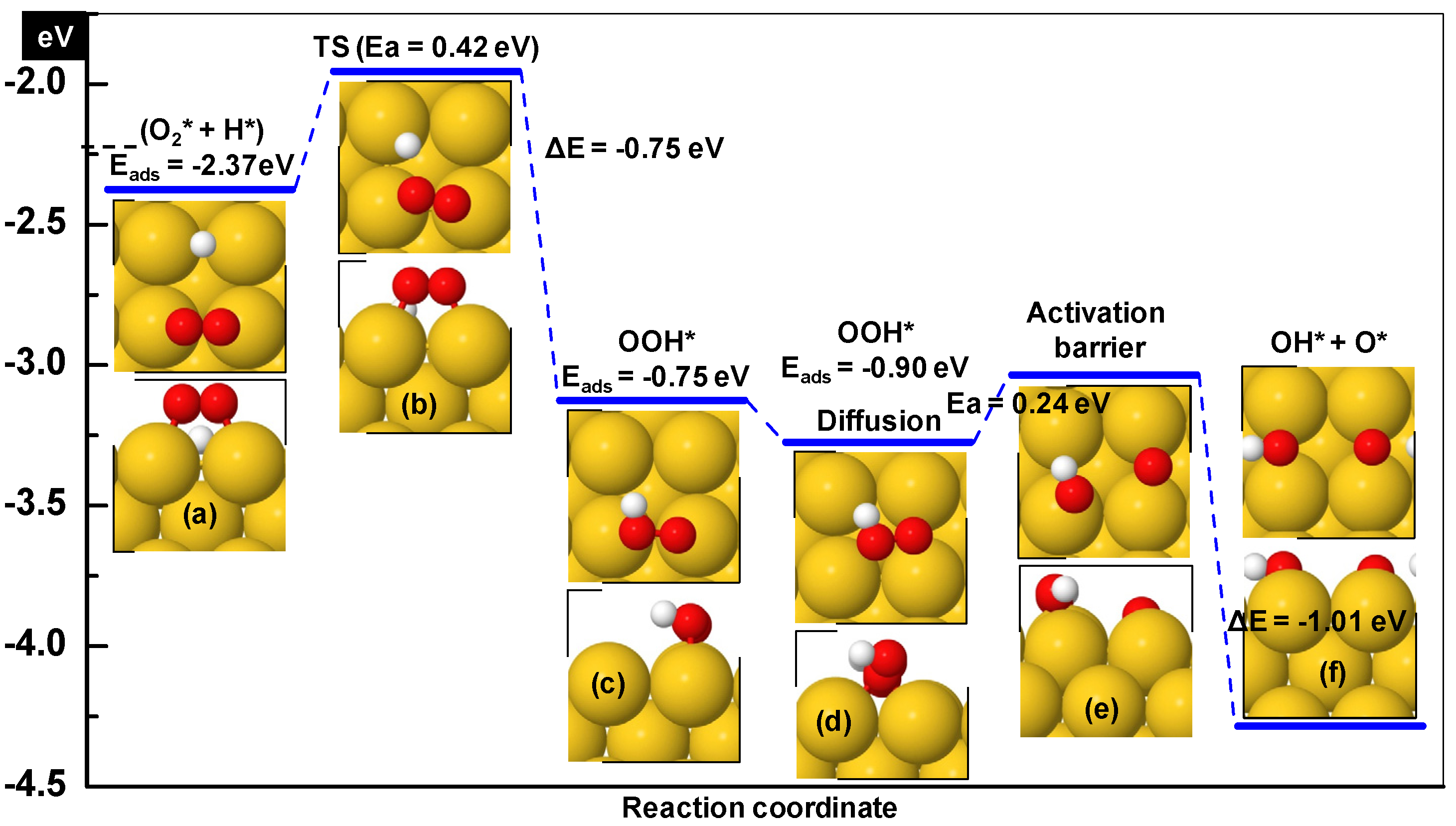
3.1.1. Hydroperoxy Formation and Decomposition on Au (O2 + H ➔ OOH ➔ OH + O)

| Step | Ea (eV) | ΔE (eV) | dTS (Å) | ν (cm−1) |
|---|---|---|---|---|
| O2* + H* ➔ OOH* | 0.33 | −1.2 | 2.53 | 112i |
| OOH* ➔ OH* + O* | 0.52 | −0.21 | 2.07 | 388i |
| CO* + OOH* ➔ CO2* + OH* | 0.50 | −3.71 | - | |
| O2* + H2* ➔ OH* + OH* | 1.95 | −2.83 | 1.46 | |
| OH* + OH* ➔ H2O* + O* | 0.1 | −0.06 | 1.85 | |
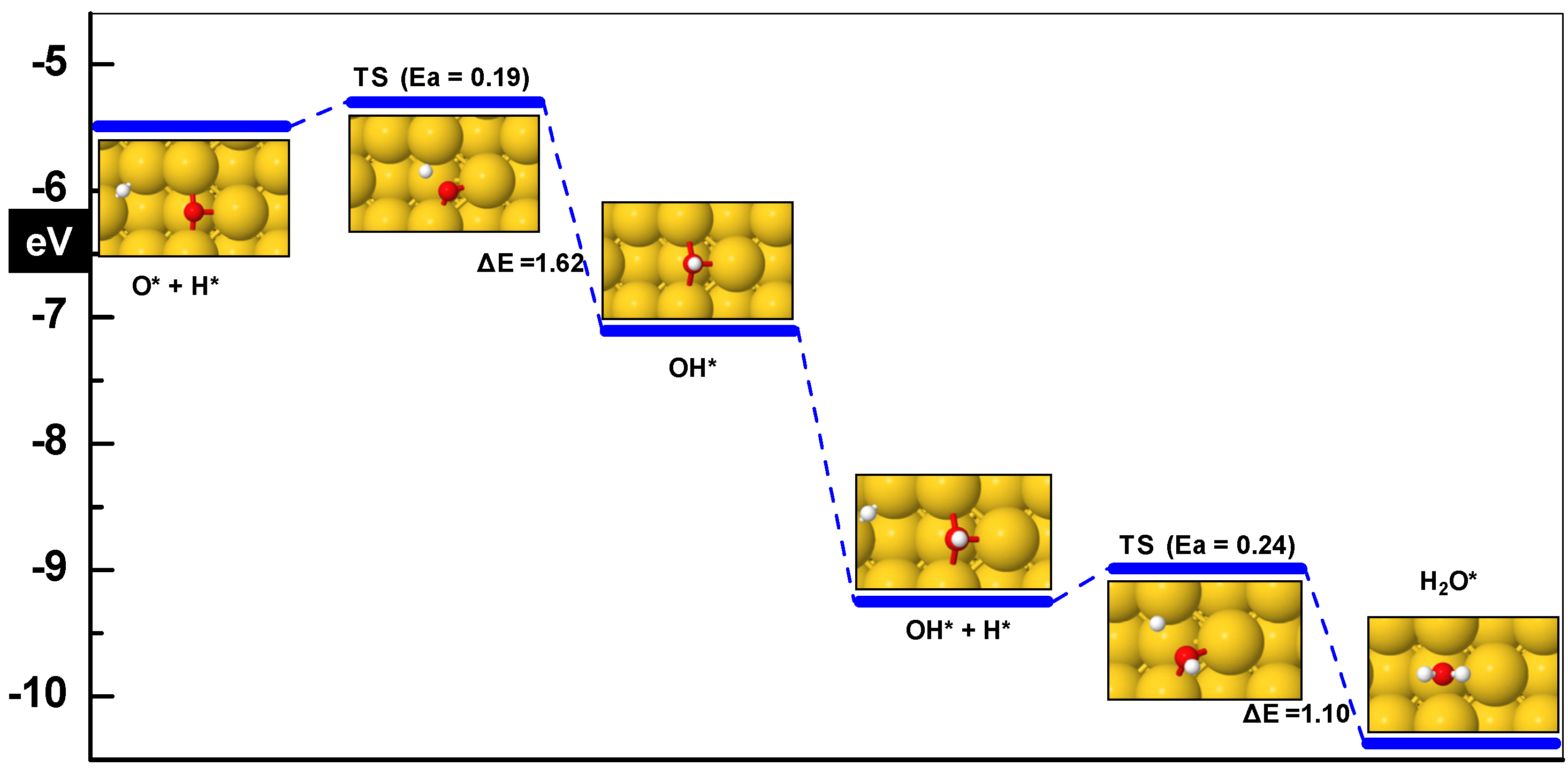
| O* + H* ➔ OH* | 0.19 | −1.62 | 1.87 | 533i |
| OH* + H* ➔ H2O* | 0.23 | −1.1 | 1.86 | 277i |
| CO* + O* ➔ CO2 | 0.01 | −2.85 | 3.29 | 53i |
| Overall Reaction | ||||
| CO + O2 + H2 ➔ CO2 + H2O | ||||
| Species | Position | Eads (eV) |
|---|---|---|
| H | bridge-b | −2.41 |
| hollow | −1.88 | |
| H + H | bridge-b + bridge-b | −4.75 |
| bridge-b + bridge-b inline | −4.72 | |
| O | bridge-b | −3.49 |
| hollow | −3.42 | |
| 3-fold hollow | −3.35 | |
| OH | bridge-b | −2.63 |
| O2 + H | hollow-a + bridge-b | −2.73 |
| hollow-b + bridge-b | −2.43 | |
| bridge-b + bridge-b | −2.38 | |
| hollow-a + hollow | −2.12 | |
| OOH | hollow-a | −1.09 |
| bridge-b | −0.96 | |
| hollow-b | −0.95 | |
| OH + O | bridge-b + bridge-b (H facing O) | −6.24 |
| bridge-b + bridge-b (O facing O) | −5.98 | |
| bridge-b + bridge-a | −5.46 | |
| CO + OOH | bridge-b + bridge-b | −1.65 |
| bridge-b + bridge-x | −1.64 | |
| hollow + top | −1.58 | |
| hollow + bridge-b | −1.09 | |
| OC-OOH | top | stabilized by −0.73 eV |
| w. r. t. | ||
| CO2 + OH | gas phase + bridge-b | most stable CO + OOH |
| −2.74 |


3.1.2. The role of a Reducible Metal Oxide for OOH Formation
3.1.3. Reaction between Molecular Oxygen and Hydrogen (O2 + H2 ➔ OH + OH)
3.1.4. Reaction between CO and OOH (CO + OOH ➔ CO2 + OH)
3.2. Hydrogen Peroxide Formation (HOOH)
3.3. Water Formation on Au
3.3.1. OH Formation (O + H ➔ OH)

3.3.2. OH + H ➔ H2O
3.3.3. Disproportionation of OHs (OH + OH ➔ H2O + O)
4. Discussion
5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon-monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 2, 405–408. [Google Scholar]
- Wigley, T.M.L.; Richels, R.; Edmonds, J.A. Economic and environmental choices in the stabilization of atmospheric CO2 concentrations. Nature 1996, 379, 240–243. [Google Scholar] [CrossRef]
- Velu, S.; Suzuki, K.; Kapoor, M.P.; Ohashi, F.; Osaki, T. Selective production of hydrogen for fuel cells via oxidative steam reforming of methanol over CuZnAl(Zr)-oxide catalysts. Appl. Catal. A-Gen. 2001, 213, 47–63. [Google Scholar] [CrossRef]
- Nieuwenhuys, B.E. The surface science approach toward understanding automotive exhaust conversion catalysis at the atomic level. Adv. Catal. 1999, 44, 259–328. [Google Scholar] [CrossRef]
- Grisel, R.; Weststrate, K.J.; Gluhoi, A.; Nieuwenhuys, B.E. Catalysis by gold nanoparticles. Gold Bull. 2002, 3, 39–45. [Google Scholar]
- Oetjen, H.F.; Schmidt, V.M.; Stimming, U.; Trila, F. Performance data of a proton exchange membrane fuel cell using H2/CO as fuel gas. J. Electrochem. Soc. 1996, 143, 3838–3842. [Google Scholar] [CrossRef]
- Grisel, R.J.H.; Nieuwenhuys, B.E. Selective oxidation of CO over supported Au catalysts. J. Catal. 2001, 199, 48–59. [Google Scholar]
- Korotkikh, O.; Farrauto, R. Selective catalytic oxidation of CO in H2: Fuel cell applications. Catal. Today 2000, 62, 249–254. [Google Scholar] [CrossRef]
- Kandoi, S.; Gokhale, A.A.; Grabow, L.C.; Dumesic, J.A.; Mavrikakis, M. Why Au and Cu are more selective than Pt for preferential oxidation of CO at low temperature. Catal. Lett. 2004, 93, 93–100. [Google Scholar] [CrossRef]
- Cameron, D.; Holliday, R.; Thompson, D. Gold's future role in fuel cell systems. J. Power Sources 2003, 118, 298–303. [Google Scholar] [CrossRef]
- Piccolo, L.; Daly, H.; Valcarcel, A.; Meunier, F.C. Promotional effect of H2 on CO oxidation over Au/TiO2 studied by operando infrared spectroscopy. Appl. Catal. B-Environ 2009, 86, 190–195. [Google Scholar] [CrossRef]
- Rossignol, C.; Arrii, S.; Morfin, F.; Piccolo, L.; Caps, V.; Rousset, J.L. Selective oxidation of CO over model gold-based catalysts in the presence of H2. J. Catal. 2005, 230, 476–483. [Google Scholar] [CrossRef]
- Schumacher, B.; Denkwitz, Y.; Plzak, V.; Kinne, M.; Behm, R.J. Kinetics, mechanism, and the influence of H2 on the CO oxidation reaction on a Au/TiO2 catalyst. J. Catal. 2004, 224, 449–462. [Google Scholar] [CrossRef]
- Hussain, A.; Curulla Ferre, D.; Gracia, J.; Nieuwenhuys, B.E.; Niemantsverdriet, J.W. DFT study of CO and NO adsorption on low index and stepped surfaces of gold. Surf. Sci. 2009, 603, 2734–2741. [Google Scholar] [CrossRef]
- Barrio, L.; Liu, P.; Rodriguez, J.A.; Campos-Martin, J.M.; Fierro, J.L.G. A density functional theory study of the dissociation of H2 on gold clusters: Importance of fluxionality and ensemble effects. J. Chem. Phys. 2006, 125, 164715–164720. [Google Scholar]
- Kresse, G.; Hafner, J. Abinitio molecular-dynamics for liquid-metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation-energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Structure Data of Elements and Intermetallic Phases, Landolt-Bornstein, New Series; Hellwege, K-H. (Ed.) Springer: Berlin, Germany, 1971; Volume IIIb.
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Pulay, P. Convergence acceleration of iterative sequences—The case of scf iteration. Chem. Phys. Lett. 1980, 73, 393–398. [Google Scholar] [CrossRef]
- Head, J.D. Computation of vibrational frequencies for adsorbates on surfaces. Int. J. Quantum Chem. 1997, 65, 827–838. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Varganov, S.A.; Olson, R.M.; Gordon, M.S.; Mills, G.; Metiu, H. A study of the reactions of molecular hydrogen with small gold clusters. J. Chem. Phys. 2004, 120, 5169–5175. [Google Scholar]
- Okumura, M.; Kitagawa, Y.; Haruta, M.; Yamaguchi, K. The interaction of neutral and charged Au clusters with O-2, CO and H2. Appl. Catal. A-Gen. 2005, 291, 37–44. [Google Scholar] [CrossRef]
- Corma, A.; Boronat, M.; Gonzalez, S.; Illas, F. On the activation of molecular hydrogen by gold: A theoretical approximation to the nature of potential active sites. Chem. Commun. 2007, 32, 3371–3373. [Google Scholar]
- Corma, A.; Serna, P. Chemoselective hydrogenation of nitro compounds with supported gold catalysts. Science 2006, 313, 332–334. [Google Scholar] [CrossRef]
- Stobinski, L.; Zommer, L.; Dus, R. Molecular hydrogen interactions with discontinuous and continuous thin gold films. Appl. Surf. Sci. 1999, 141, 319–325. [Google Scholar] [CrossRef]
- Gluhoi, A.C.; Vreeburg, H.S.; Bakker, J.W.; Nieuwenhuys, B.E. Activation of CO, O2 and H2 on gold-based catalysts. Appl. Catal. A-Gen. 2005, 291, 145–150. [Google Scholar] [CrossRef]
- Nakamara, I.; Mantoku, H.; Furukawa, T.; Fujitani, T. Active sites for hydrogen dissociation over TiOx/Au(111) surfaces. J. Phys. Chem. C 2011, 115, 16074–16080. [Google Scholar] [CrossRef]
- Barrio, L.; Liu, P.; Rodriguez, J.A.; Campos-Martin, J.M.; Fierro, J.L.G. Effects of hydrogen on the reactivity of O-2 toward gold nanoparticles and surfaces. J. Phys. Chem. C 2007, 111, 19001–19008. [Google Scholar]
- Quinet, E.; Piccolo, L.; Morfin, F.; Avenier, P.; Diehl, F.; Caps, V.; Rousset, J.L. On the mechanism of hydrogen-promoted gold-catalyzed CO oxidation. J. Catal. 2009, 268, 384–389. [Google Scholar] [CrossRef]
- Hussain, A.; Muller, A.J.; Nieuwenhuys, B.E.; Gracia, J.M.; Niemantsverdriet, J.W. Two gold surfaces and a cluster with remarkable reactivity for co oxidation, a density functional theory study. Top. Catal. 2011, 54, 415–423. [Google Scholar] [CrossRef]
- Hussain, A.; Gracia, J.; Nieuwenhuys, B.; Niemantsverdriet, J.W. Chemistry of O- and H-containing species on the (001) surface of anatase TiO2: A DFT study. ChemPhysChem 2010, 11, 2375–2382. [Google Scholar] [CrossRef]
- Grisel, R.J.H.; Weststrate, C.J.; Goossens, A.; Craje, M.W.J.; Van der Kraan, A.M.; Nieuwenhuys, B.E. Oxidation of CO over Au/MOx/Al2O3 multi-component catalysts in a hydrogen-rich environment. Catal. Today 2002, 72, 123–132. [Google Scholar]
- Ojifinni, R.A.; Froemming, N.S.; Gong, J.; Pan, M.; Kim, T.S.; White, J.M.; Henkelman, G.; Mullins, C.B. Water-enhanced low-temperature CO oxidation and isotope effects on atomic oxygen-covered Au(111). J. Am. Chem. Soc. 2008, 130, 6801–6812. [Google Scholar]
- Bollinger, M.A.; Vannice, M.A. A kinetic and DRIFTS study of low-temperature carbon monoxide oxidation over Au-TiO2 catalysts. Appl. Catal. B-Environ. 1996, 8, 417–443. [Google Scholar]
- Boccuzzi, F.; Chiorino, A.; Manzoli, M.; Lu, P.; Akita, T.; Ichikawa, S.; Haruta, M. Au/TiO2 nanosized samples: A catalytic, TEM, and FTIR study of the effect of calcination temperature on the CO oxidation. J. Catal. 2001, 202, 256–267. [Google Scholar] [CrossRef]
- Date, M.; Haruta, M. Moisture effect on CO oxidation over Au/TiO2 catalyst. J. Catal. 2001, 201, 221–224. [Google Scholar] [CrossRef]
- Date, M.; Okumura, M.; Tsubota, S.; Haruta, M. Vital role of moisture in the catalytic activity of supported gold nanoparticles. Angew. Chem. Int. Ed. 2004, 43, 2129–2132. [Google Scholar] [CrossRef]
- Daniells, S.T.; Makkee, M.; Moulijn, J.A. The effect of high-temperature pre-treatment and water on the low temperature CO oxidation with Au/Fe2O3 catalysts. Catal. Lett. 2005, 100, 39–47. [Google Scholar] [CrossRef]
- Shou, M.; Takekawa, H.; Ju, D.Y.; Hagiwara, T.; Lu, D.L.; Tanaka, K. Activation of a Au/TiO2 catalyst by loading a large amount of Fe-oxide: Oxidation of CO enhanced by H2 and H2O. Catal. Lett. 2006, 108, 119–124. [Google Scholar] [CrossRef]
- Date, M.; Imai, H.; Tsubota, S.; Haruta, M. In situ measurements under flow condition of the CO oxidation over supported gold nanoparticles. Catal. Today 2007, 122, 222–225. [Google Scholar] [CrossRef]
- Sample Availability: Computational models are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hussain, A.; Gracia, J.; Niemantsverdriet, J.W.; Nieuwenhuys, B.E. The Beneficial Effect of Hydrogen on CO Oxidation over Au Catalysts. A Computational Study. Molecules 2011, 16, 9582-9599. https://doi.org/10.3390/molecules16119582
Hussain A, Gracia J, Niemantsverdriet JW, Nieuwenhuys BE. The Beneficial Effect of Hydrogen on CO Oxidation over Au Catalysts. A Computational Study. Molecules. 2011; 16(11):9582-9599. https://doi.org/10.3390/molecules16119582
Chicago/Turabian StyleHussain, Akhtar, Jose Gracia, J. W. Niemantsverdriet, and B. E. Nieuwenhuys. 2011. "The Beneficial Effect of Hydrogen on CO Oxidation over Au Catalysts. A Computational Study" Molecules 16, no. 11: 9582-9599. https://doi.org/10.3390/molecules16119582