Size-Dependent Electrocatalytic Activity of Gold Nanoparticles on HOPG and Highly Boron-Doped Diamond Surfaces

Abstract
:1. Introduction
2. Results
2.1. Substrate Characterization
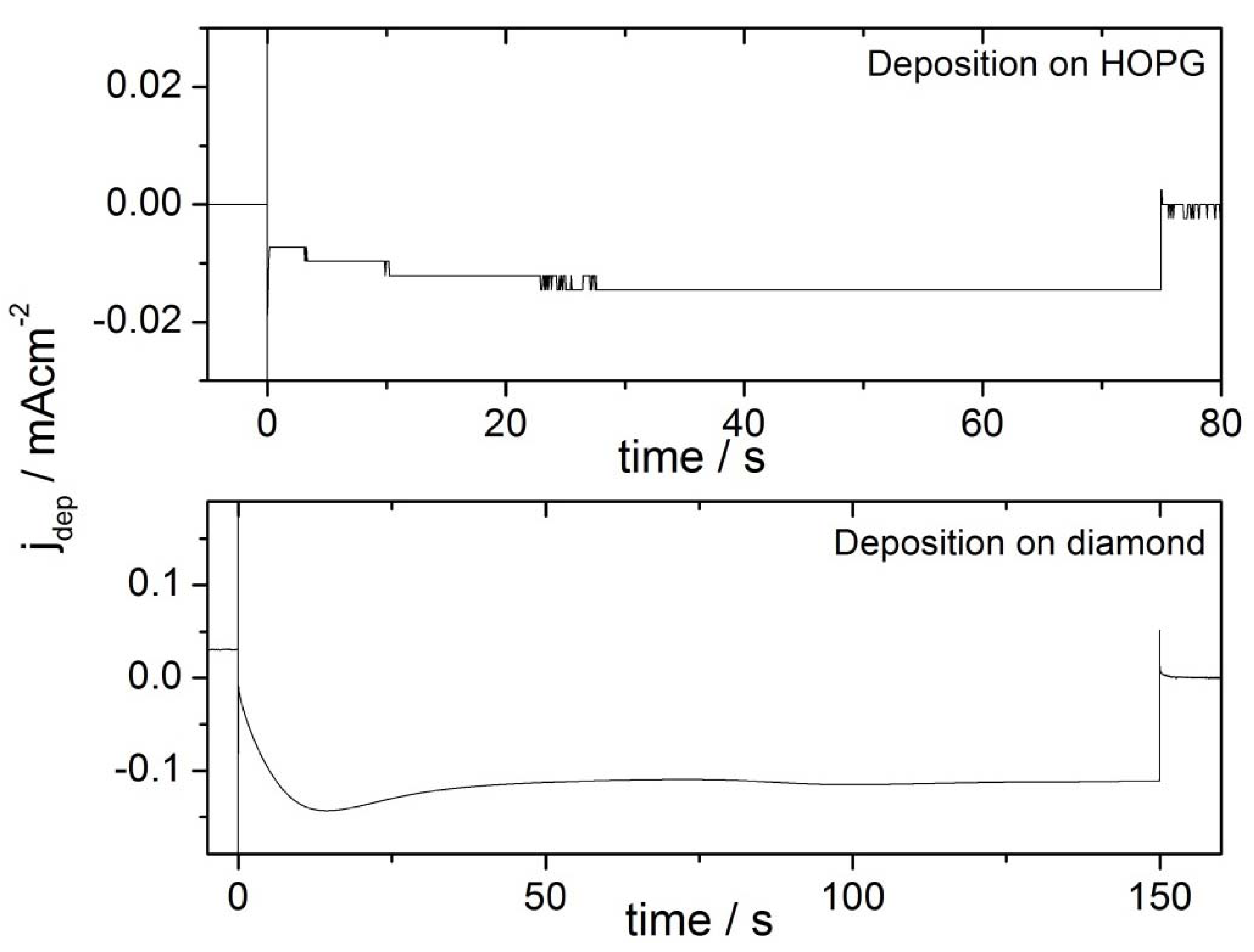
2.2. Deposition Transients

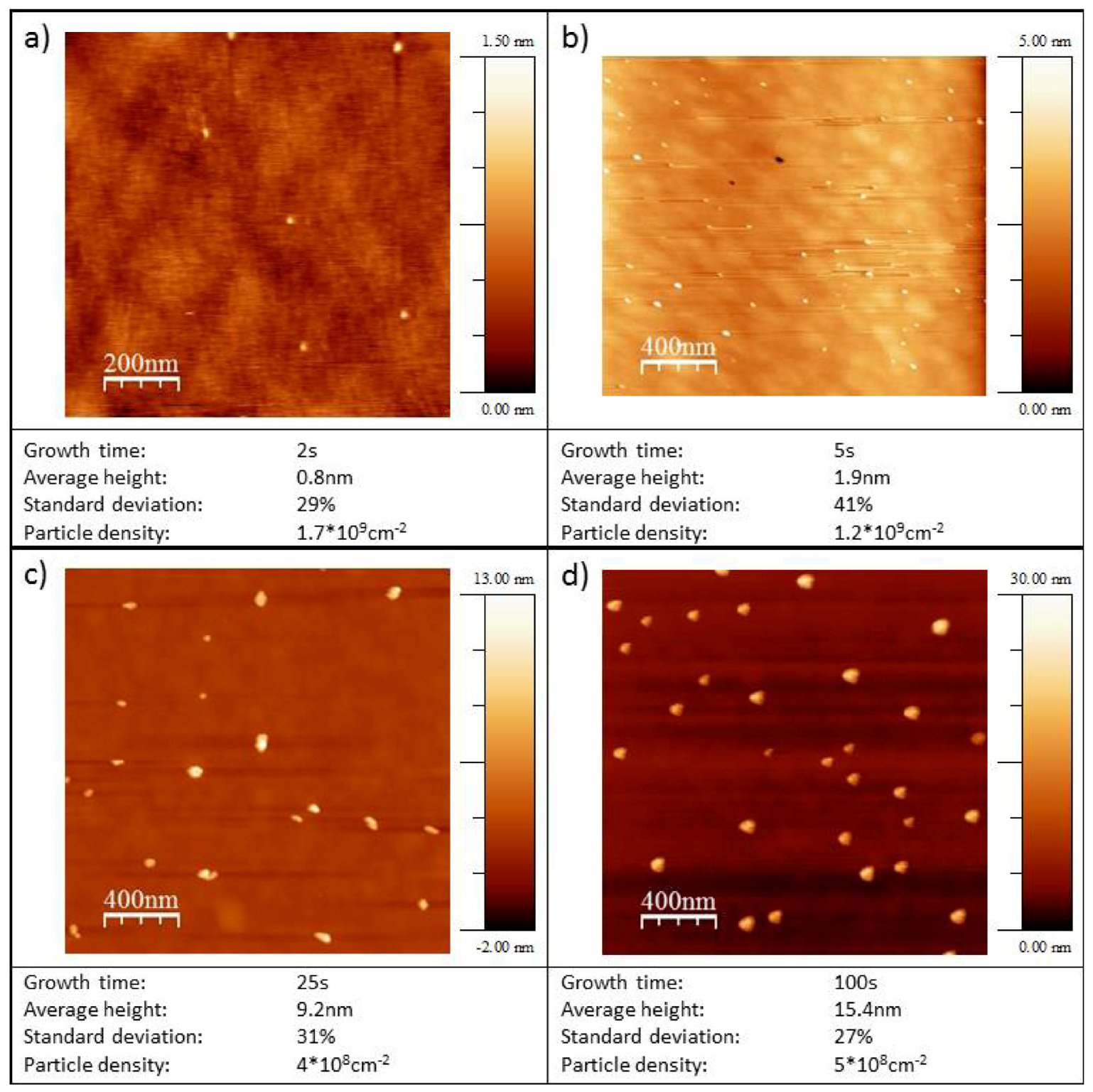
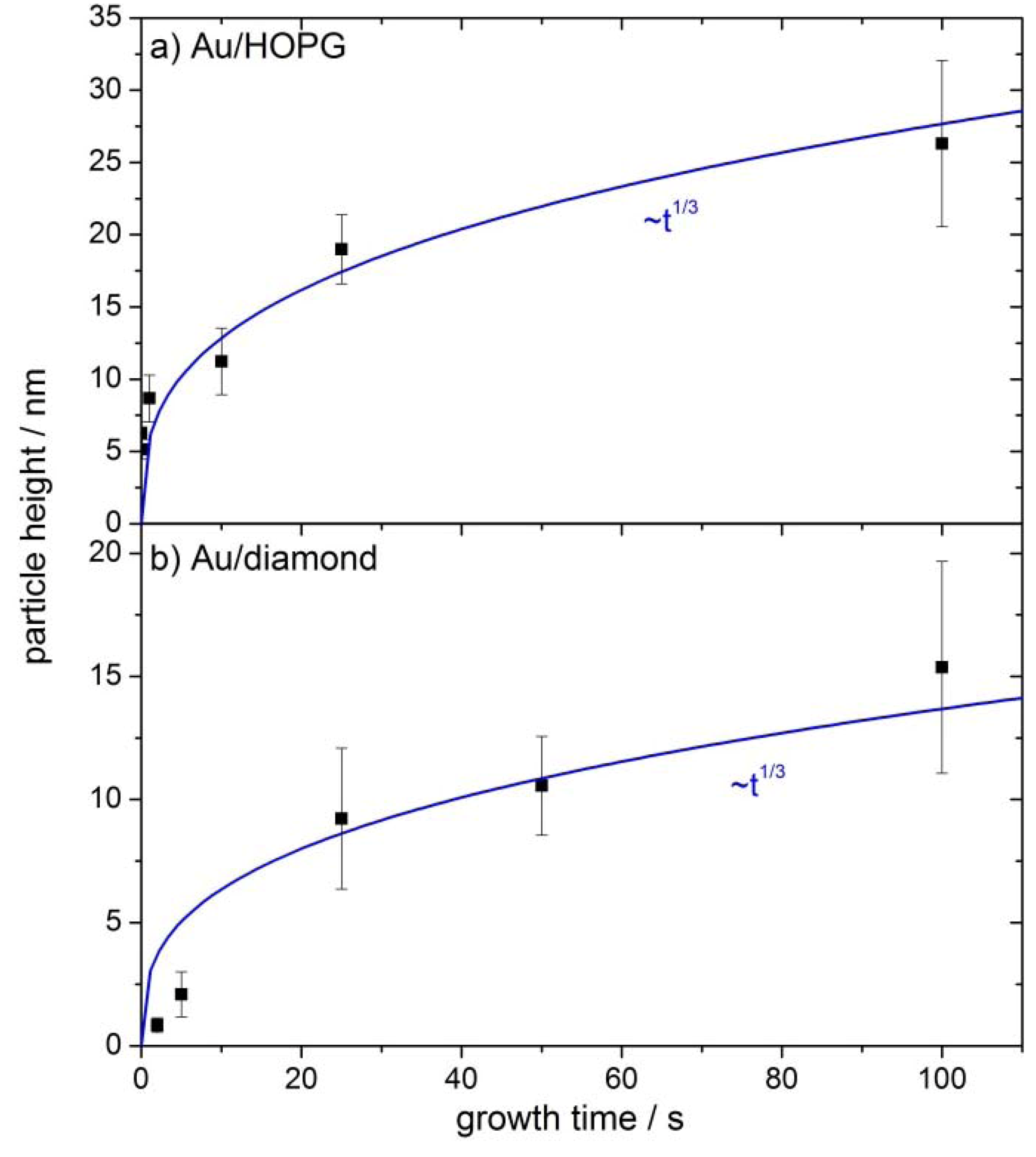
2.3. AFM Characterization of the Au/HOPG and Au/Diamond Surfaces




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Growth time tG / s | Particle density / 109 cm−2 | Average particle height / nm | Standard deviation particle height / % | |
|---|---|---|---|---|
| Au/HOPG | 0.01 | 1.2 | 6.3 | 24 |
| 0.1 | 1.6 | 5.2 | 13 | |
| 1 | 1.5 | 8.7 | 19 | |
| 10 | 3.1 | 11.2 | 21 | |
| 25 | 1.7 | 19 | 13 | |
| 100 | 2.3 | 26.3 | 22 | |
| Au/diamond | 2 | 1.40 | 0.8 | 35 |
| 5 | 0.9 | 2.1 | 44 | |
| 25 | 0.4 | 9.2 | 31 | |
| 50 | 0.2 | 10.6 | 19 | |
| 100 | 0.5 | 15.4 | 28 |
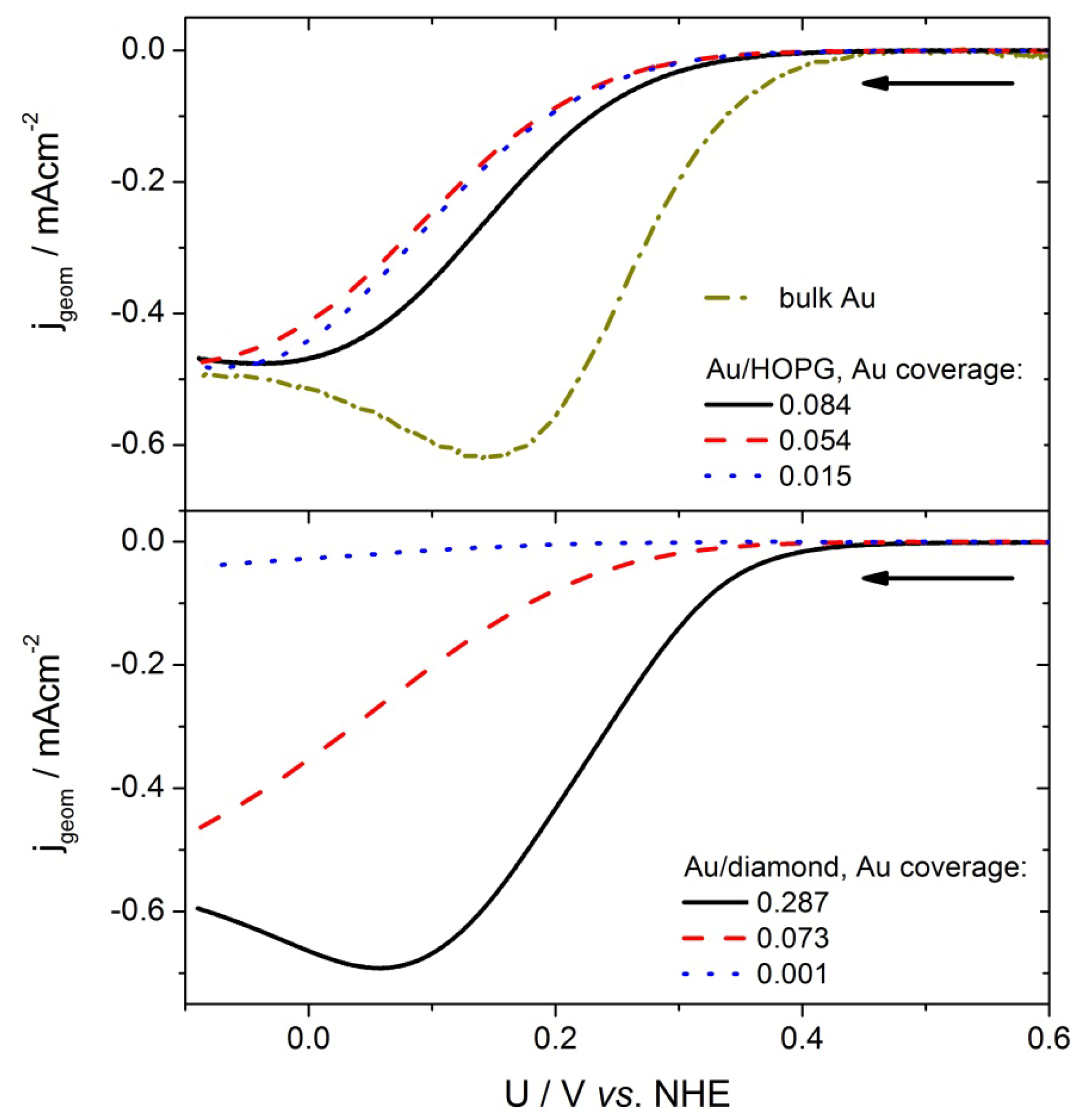
2.4. Catalytic Activity Measurements
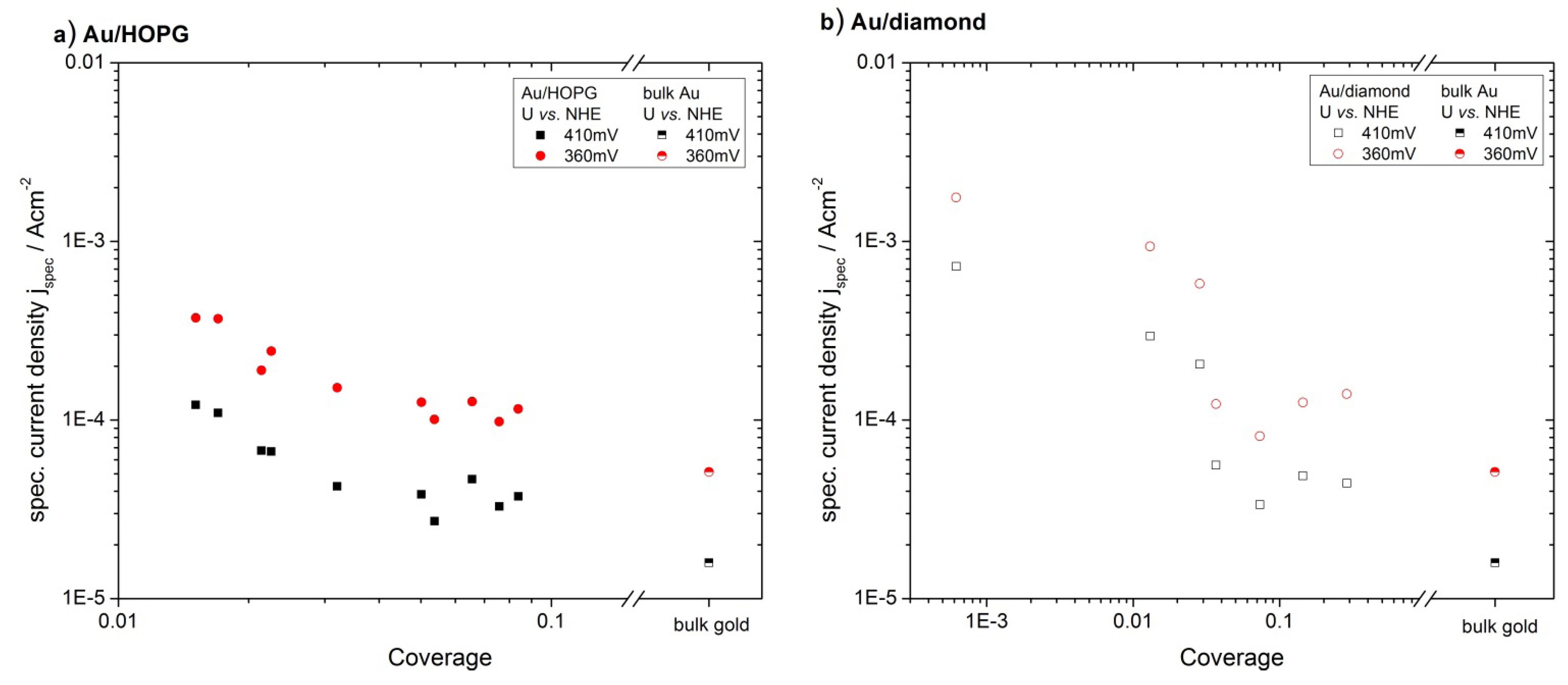
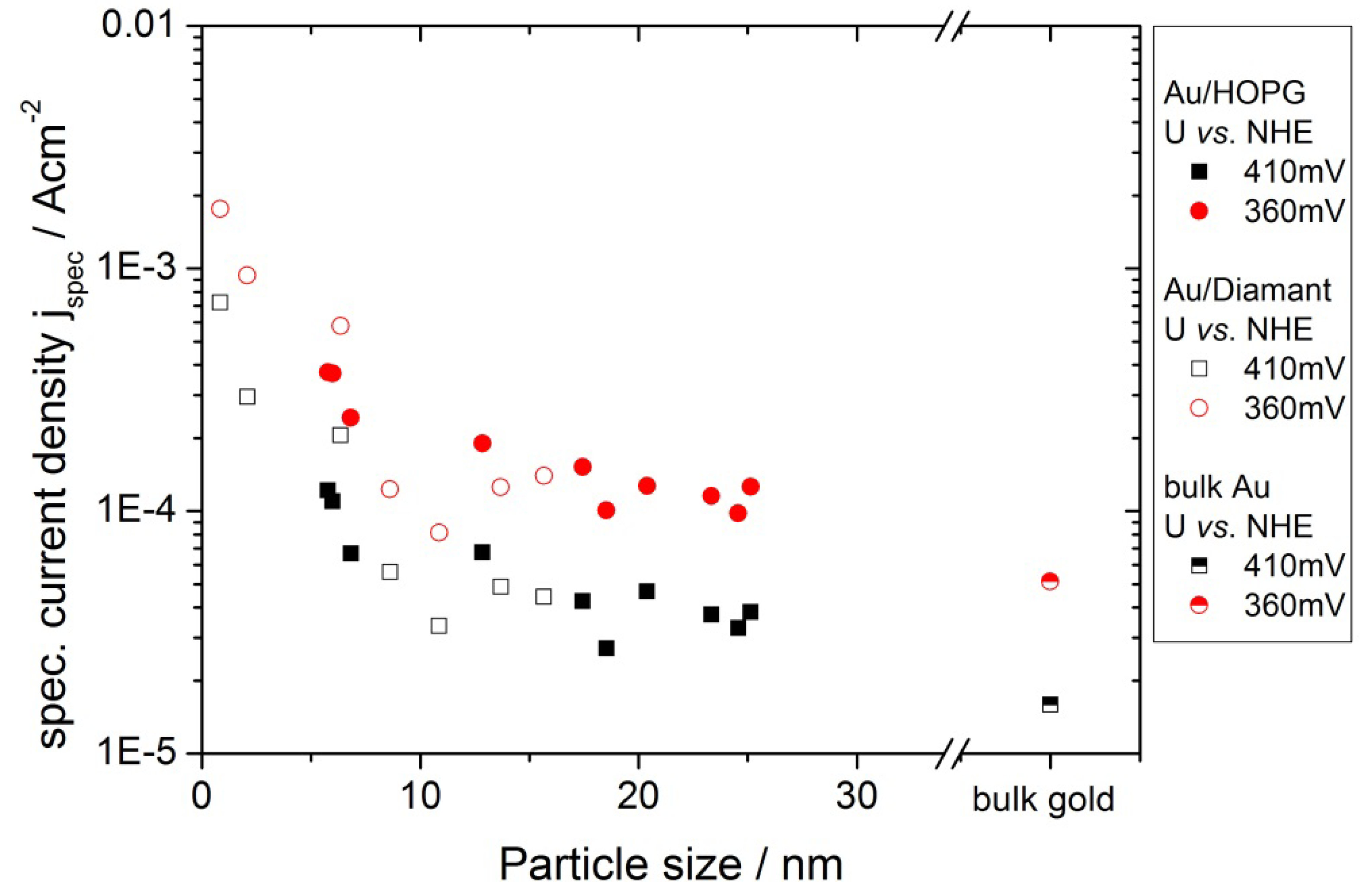
2.4.1 Oxygen Reduction Reaction


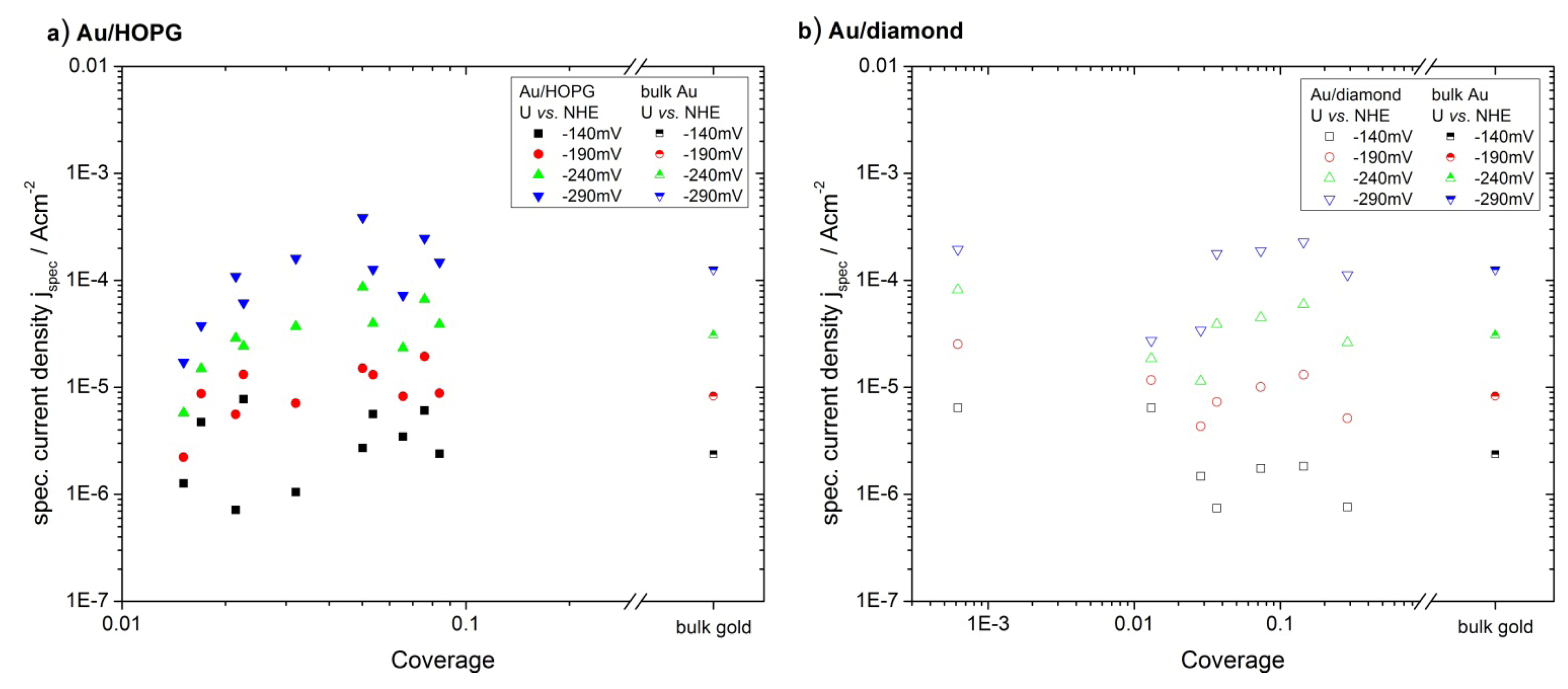
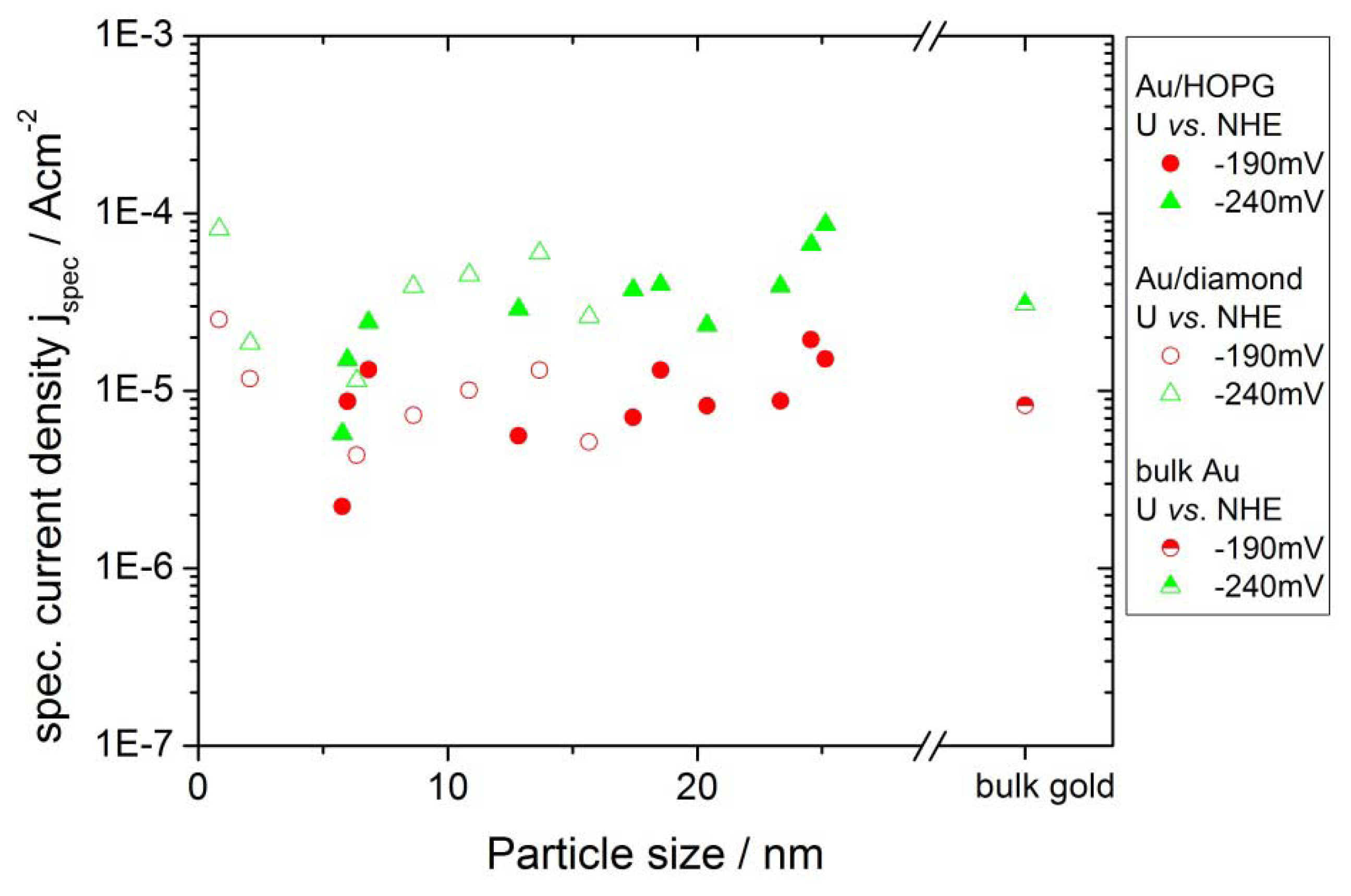
2.4.2. Hydrogen Evolution Reaction
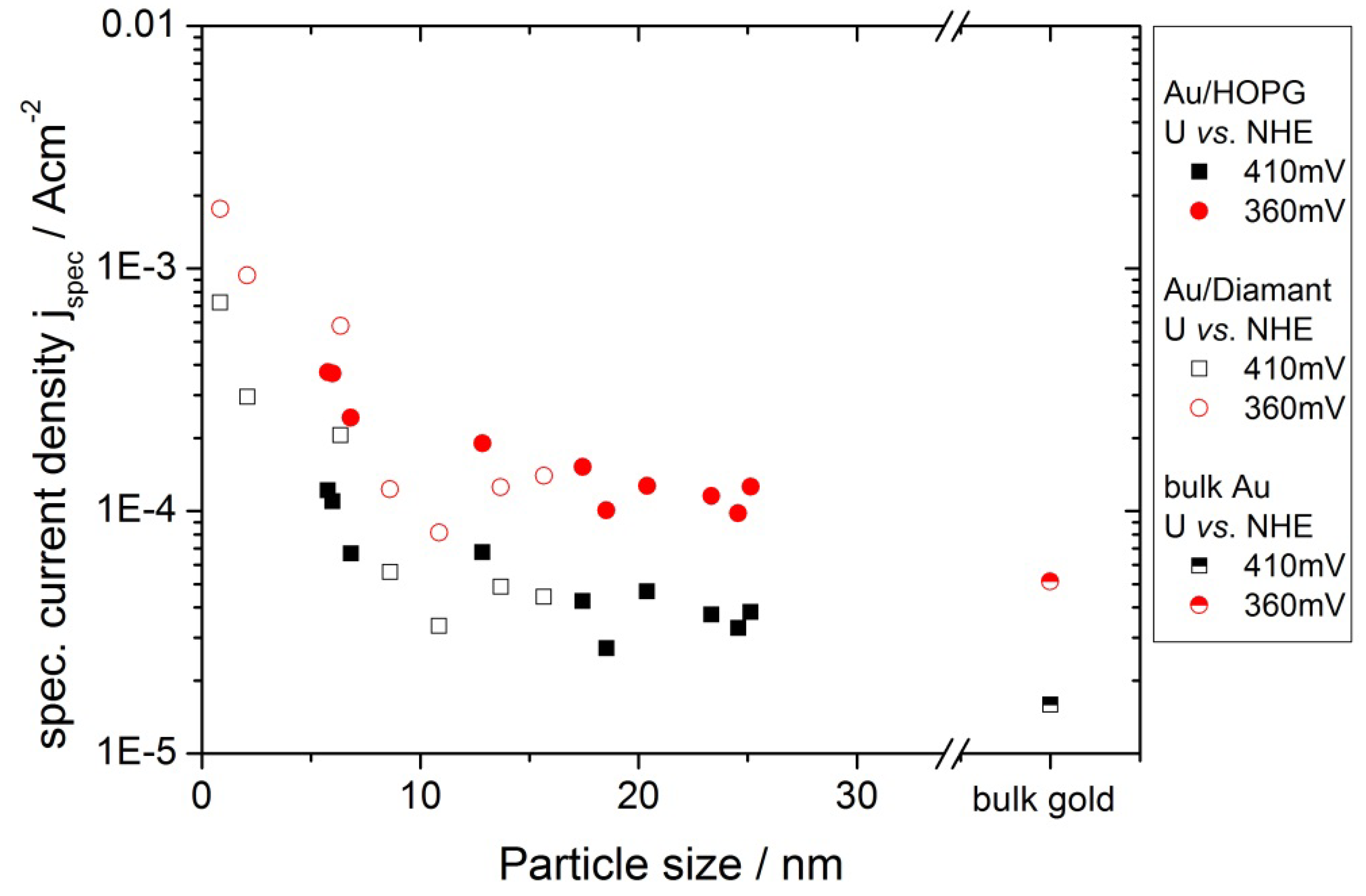


3. Discussion
3.1. Nanoparticle Deposition
3.2. Activity of Supported Au Nanoparticles for Oxygen Reduction

3.3. Activity of Supported Au Nanoparticles for Hydrogen Evolution
4. Experimental Section
4.1. General
4.2. Preparation of the HOPG and Diamond Electrodes
4.3. Electrochemical Gold Deposition and Characterization of the Nanostructured Surfaces
| Nucleation pulse | Growth pulse | |||
|---|---|---|---|---|
| Potential vs. NHE | Pulse duration | Potential vs. NHE | Pulse duration | |
| HOPG | −0.04 V | 31 ms | 1.085 V | 1 s–100 s |
| Diamond | −0.14 V | 10 ms | 0.895 V | 2 s–150 s |
5. Conclusions
Acknowledgements
Conflict of Interest
References and Notes
- Daniel, M.C.; Astruc, D. Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef]
- Hutchings, G.J. Catalysis by gold. Catal. Today 2005, 100, 55–61. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon-monoxide at a temperature far below 0-degrees-C. Chem. Lett. 1987, 2, 405–408. [Google Scholar]
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef]
- Zhang, Y.; Suryanarayanan, V.; Nakazawa, I.; Yoshihara, S.; Shirakashi, T. Electrochemical behavior of Au nanoparticle deposited on as-grown and O-terminated diamond electrodes for oxygen reduction in alkaline solution. Electrochim. Acta 2004, 49, 5235–5240. [Google Scholar] [CrossRef]
- Sine, G.; Duo, I.; El Roustom, B.; Foti, G.; Comninellis, C. Deposition of clusters and nanoparticles onto boron-doped diamond electrodes for electrocatalysis. J. Appl. Electrochem. 2006, 36, 847–862. [Google Scholar] [CrossRef]
- El-Deab, M.S.; Ohsaka, T. An extraordinary electrocatalytic reduction of oxygen on gold nanoparticles-electrodeposited gold electrodes. Electrochem. Commun. 2002, 4, 288–292. [Google Scholar] [CrossRef]
- Alexeyeva, N.; Kozlova, J.; Sammelselg, V.; Ritslaid, P.; Mandar, H.; Tammeveski, K. Electrochemical and surface characterisation of gold nanoparticle decorated multi-walled carbon nanotubes. Appl. Surf. Sci. 2010, 256, 3040–3046. [Google Scholar]
- Zhang, Y.R.; Asahina, S.; Yoshihara, S.; Shirakashi, T. Oxygen reduction on Au nanoparticle deposited boron-doped diamond films. Electrochim. Acta 2003, 48, 741–747. [Google Scholar] [CrossRef]
- El-Deab, M.S.; Sotomura, T.; Ohsaka, T. Morphological selection of gold nanoparticles electrodeposited on various substrates. J. Electrochem. Soc. 2005, 152, C730–C737. [Google Scholar] [CrossRef]
- Hernandez, J.; Solla-Gullon, J.; Herrero, E. Gold nanoparticles synthesized in a water-in-oil microemulsion: electrochemical characterization and effect of the surface structure on the oxygen reduction reaction. J. Electroanal. Chem. 2004, 574, 185–196. [Google Scholar] [CrossRef]
- Diao, P.; Zhang, D.F.; Guo, M.; Zhang, Q. Electrocatalytic oxidation of CO on supported gold nanoparticles and submicroparticles: Support and size effects in electrochemical systems. J. Catal. 2007, 250, 247–253. [Google Scholar]
- Rossmeisl, J.; Logadottir, A.; Norskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Chen, W.; Chen, S. Oxygen electroreduction catalyzed by gold nanoclusters: Strong core size effects. Angew. Chem. Int. Ed. 2009, 48, 4386–4389. [Google Scholar] [CrossRef]
- Bron, M. Carbon black supported gold nanoparticles for oxygen electroreduction in acidic electrolyte solution. J. Electroanal. Chem. 2008, 624, 64–68. [Google Scholar] [CrossRef]
- Guerin, S.; Hayden, B.E.; Pletcher, D.; Rendall, M.E.; Suchsland, J.-P. A combinatorial approach to the study of particle size effects on supported electrocatalysts: Oxygen reduction on gold. J. Comb. Chem. 2006, 8, 679–686. [Google Scholar] [CrossRef]
- Inasaki, T.; Kobayashi, S. Particle size effects of gold on the kinetics of the oxygen reduction at chemically prepared Au/C catalysts. Electrochim. Acta 2009, 54, 4893–4897. [Google Scholar] [CrossRef]
- Limat, M.; El Roustom, B.; Jotterand, H.; Foti, G.; Comninellis, C. Electrochemical and morphological characterization of gold nanoparticles deposited on boron-doped diamond electrode. Electrochim. Acta 2009, 54, 2410–2416. [Google Scholar] [CrossRef]
- Enea, O.; Riedo, B.; Dietler, G. AFM study of Pt clusters electrochemically deposited onto boron-doped diamond films. Nano Lett. 2002, 2, 241–244. [Google Scholar] [CrossRef]
- El-Deab, M.S.; Okajima, T.; Ohsaka, T. Electrochemical reduction of oxygen on gold nanoparticle-electrodeposited glassy carbon electrodes. J. Electrochem. Soc. 2003, 150, A851–A857. [Google Scholar] [CrossRef]
- Zoval, J.V.; Lee, J.; Gorer, S.; Penner, R.M. Electrochemical preparation of platinum nanocrystallites with size selectivity on basal plane oriented graphite surfaces. J. Phys. Chem. B 1998, 102, 1166–1175. [Google Scholar]
- Penner, R.M. Mesoscopic metal particles and wires by electrodeposition. J. Phys. Chem. B 2002, 106, 3339–3353. [Google Scholar] [CrossRef]
- Liu, H.; Penner, R.M. Size-selective electrodeposition of mesoscale metal particles in the uncoupled limit. J. Phys.Chem. B 2000, 104, 9131–9139. [Google Scholar] [CrossRef]
- Ueda, M.; Dietz, H.; Anders, A.; Kneppe, H.; Meixner, A.; Plieth, W. Double-pulse technique as an electrochemical tool for controlling the preparation of metallic nanoparticles. Electrochim. Acta 2002, 48, 377–386. [Google Scholar] [CrossRef]
- Sandmann, G.; Dietz, H.; Plieth, W. Preparation of silver nanoparticles on ITO surfaces by a double-pulse method. J. Electroanal. Chem. 2000, 491, 78–86. [Google Scholar] [CrossRef]
- Brülle, T.; Denisenko, A.; Sternschulte, H.; Stimming, U. Catalytic activity of platinum nanoparticles on highly boron-doped and 100-oriented epitaxial diamond towards HER and HOR. Phys. Chem. Chem. Phys. 2011, 13, 12883–12891. [Google Scholar]
- Wolfschmidt, H.; Bussar, R.; Stimming, U. Charge transfer reactions at nanostructured Au(111) surfaces: Influence of the substrate material on electrocatalytic activity. J. Phys. Condens. Matter 2008, 20, 374127. [Google Scholar] [CrossRef]
- Wolfschmidt, H.; Weingarth, D.; Stimming, U. Enhanced reactivity for hydrogen reactions at Pt nanoislands on Au(111). Chemphyschem 2010, 11, 1533–1541. [Google Scholar] [CrossRef]
- Brülle, T.; Stimming, U. Platinum nanostructured HOPG - Preparation, characterization and reactivity. J. Electroanal. Chem. 2009, 636, 10–17. [Google Scholar] [CrossRef]
- Kibler, L.A. Hydrogen electrocatalysis. ChemPhysChem 2006, 7, 985–991. [Google Scholar] [CrossRef]
- Wolfschmidt, H.; Paschos, O.; Stimming, U. Fuel cell science: Theory, fundamentals, and biocatalysis. In Wiley Series on Electrocatalysis and Electrochemistry; Wieckowski, A., Nørskov, J.K., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2010; p. 1, Chapter 1. [Google Scholar]
- Maltez-da Costa, M.; de la Escosura-Muniz, A.; Merkoci, A. Electrochemical quantification of gold nanoparticles based on their catalytic properties toward hydrogen formation: Application in magnetoimmunoassays. Electrochem. Commun. 2010, 12, 1501–1504. [Google Scholar] [CrossRef]
- Liu, H.; Favier, F.; Ng, K.; Zach, M.P.; Penner, R.M. Size-selective electrodeposition of meso-scale metal particles: A general method. Electrochim. Acta 2001, 47, 671–677. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Stamenkovic, V.; Arenz, M.; Markovic, N.M.; Ross, P.N. Oxygen electrocatalysis in alkaline electrolyte: Pt(hkl), Au(hkl) and the effect of Pd-modification. Electrochim. Acta 2002, 47, 3765–3776. [Google Scholar] [CrossRef]
- Adzic, R.R.; Strbac, S.; Anastasijevic, N. Electrocatalysis of oxygen on single-crystal gold electrodes. Mater. Chem. Phys. 1989, 22, 349–375. [Google Scholar] [CrossRef]
- Gottesfeld, S.; Raistrick, I.D.; Srinivasan, S. Oxygen reduction kinetics on a platinum RDE coated with a recast nafion film. J. Electrochem. Soc. 1987, 134, 1455–1462. [Google Scholar] [CrossRef]
- Zwetanova, A.; Juttner, K. The electrocatalytical influence of underpotential lead and thallium adsorbates on the cathodic reduction of oxygen on (111), (100) and (110) silver single-crystal surfaces. J. Electroanal. Chem. 1981, 119, 149–164. [Google Scholar] [CrossRef]
- Shao, M.H.; Adzic, R.R. Spectroscopic identification of the reaction intermediates in oxygen reduction on gold in alkaline solutions. J. Phys. Chem. B 2005, 109, 16563–16566. [Google Scholar]
- Horcas, I.; Fernandez, R.; Gomez-Rodriguez, J.M.; Colchero, J.; Gomez-Herrero, J.; Baro, A.M. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705. [Google Scholar] [CrossRef]
- Pietzka, C.; Denisenko, A.; Romanyuk, A.; Schafer, P.J.; Kibler, L.A.; Scharpf, J.; Kohn, E. Electronic surface barrier properties of boron-doped diamond oxidized by plasma treatment. Diam. Rel. Mater. 2010, 19, 213–216. [Google Scholar] [CrossRef]
- Denisenko, A.; Pietzka, C.; Romanyuk, A.; El-Hajj, H.; Kohn, E. The electronic surface barrier of boron-doped diamond by anodic oxidation. J. Appl. Phys. 2008, 103, 014904. [Google Scholar] [CrossRef]
- Denisenko, A.; Romanyuk, A.; Pietzka, C.; Scharpf, J.; Kohn, E. Surface structure and surface barrier characteristics of boron-doped diamond in electrolytes after CF4 plasma treatment in RF-barrel reactor. Diam. Rel. Mater. 2010, 19, 423–427. [Google Scholar] [CrossRef]
- Michri, A.A.; Pshchenichnikov, A.G.; Burshtein, R.K. Determining the actual surface area of smooth gold electrodes. Sov. Electrochem. 1972, 8, 351. [Google Scholar]
- Kondo, T.; Morita, J.; Hanaoka, K.; Takakusagi, S.; Tamura, K.; Takahasi, M.; Mizuki, J.I.; Uosaki, K. Structure of Au(111) and Au(100) single-crystal electrode surfaces at various potentials in sulfuric acid solution determined by in situ surface X-ray scattering. J. Phys. Chem. C 2007, 111, 13197–13204. [Google Scholar]
- Sample Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brülle, T.; Ju, W.; Niedermayr, P.; Denisenko, A.; Paschos, O.; Schneider, O.; Stimming, U. Size-Dependent Electrocatalytic Activity of Gold Nanoparticles on HOPG and Highly Boron-Doped Diamond Surfaces. Molecules 2011, 16, 10059-10077. https://doi.org/10.3390/molecules161210059
Brülle T, Ju W, Niedermayr P, Denisenko A, Paschos O, Schneider O, Stimming U. Size-Dependent Electrocatalytic Activity of Gold Nanoparticles on HOPG and Highly Boron-Doped Diamond Surfaces. Molecules. 2011; 16(12):10059-10077. https://doi.org/10.3390/molecules161210059
Chicago/Turabian StyleBrülle, Tine, Wenbo Ju, Philipp Niedermayr, Andrej Denisenko, Odysseas Paschos, Oliver Schneider, and Ulrich Stimming. 2011. "Size-Dependent Electrocatalytic Activity of Gold Nanoparticles on HOPG and Highly Boron-Doped Diamond Surfaces" Molecules 16, no. 12: 10059-10077. https://doi.org/10.3390/molecules161210059