Discovery of Potential M2 Channel Inhibitors Based on the Amantadine Scaffold via Virtual Screening and Pharmacophore Modeling

Abstract
:1. Introduction

2. Results and Discussion
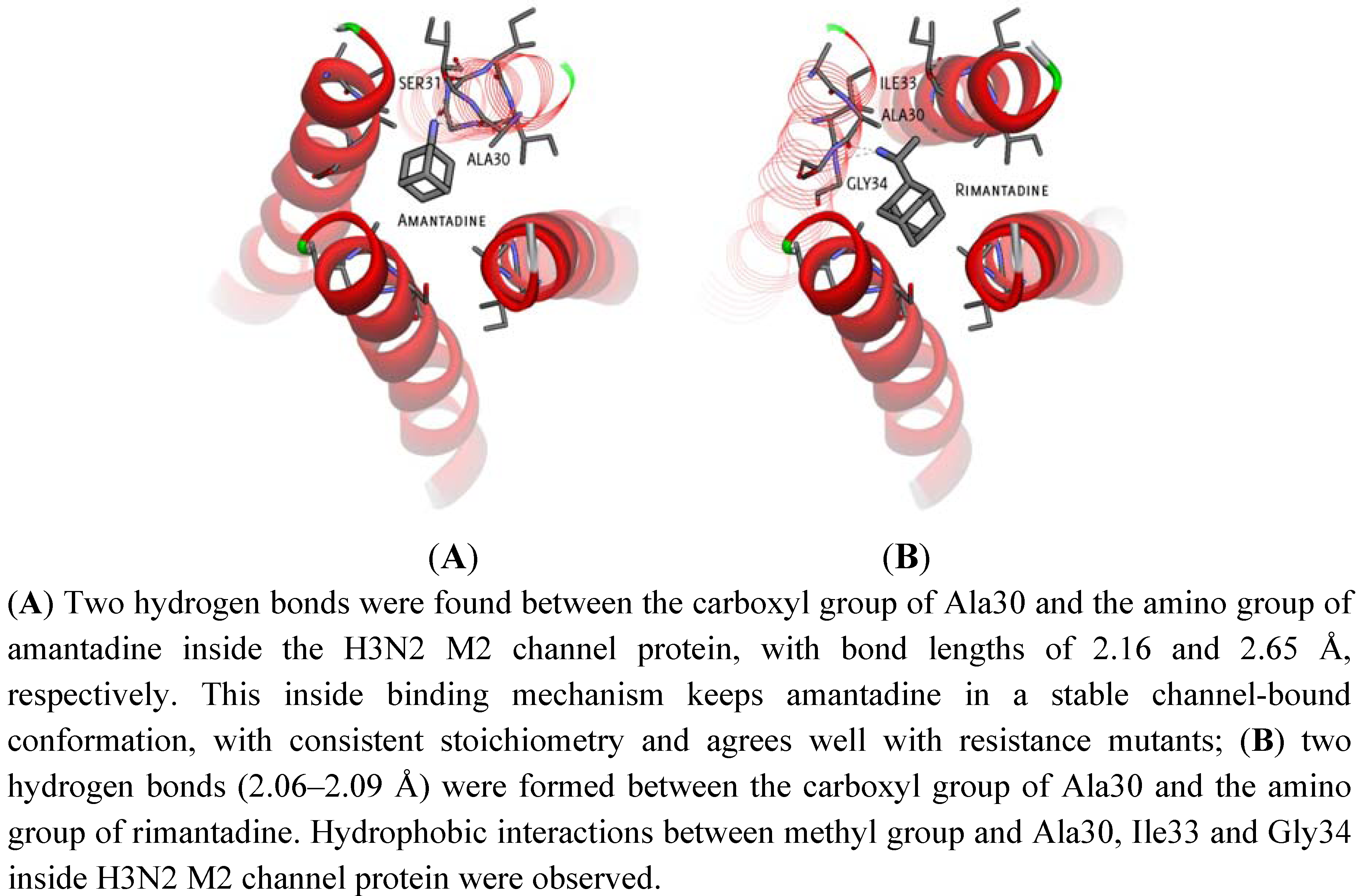
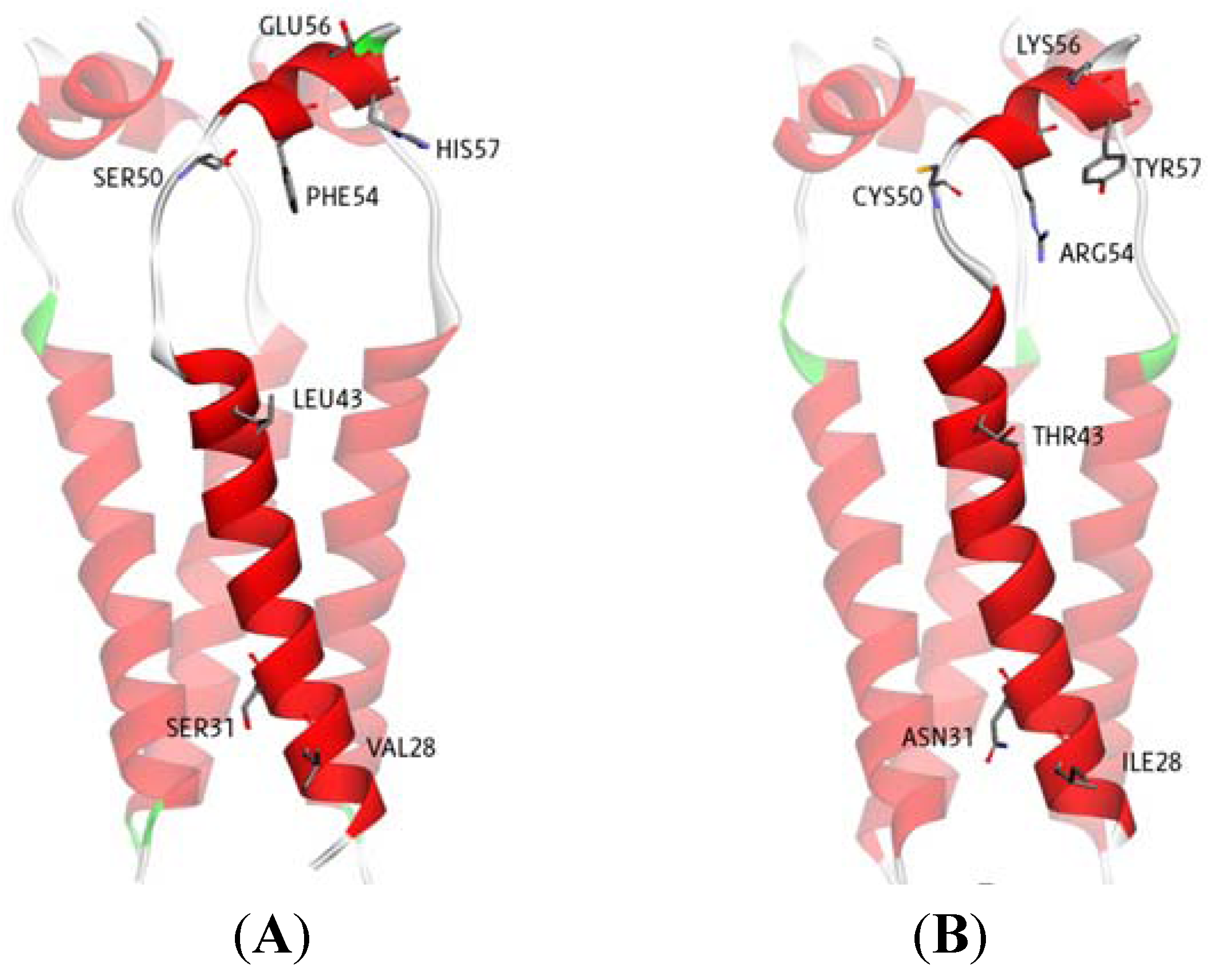
2.1. Binding Site Identification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Ligand | Binding energy (kcal/mol) | Possible binding sites with M2 channel proteins | ||
|---|---|---|---|---|---|
| H3N2 | 2009-H1N1 | H3N2 | 2009-H1N1 | ||
| Inside | Amantadine | −6.85 | −7.01 | Hydrogen bond with Ala30 | No Hydrogen bond |
| Inside | Rimantadine | −7.75 | −7.57 | Hydrogen bond with Ala30 | No Hydrogen bond |
| Hydrophobic interaction with Ile33 | Hydrophobic interaction with Ile33 | ||||
| Outside | Amantadine | −5.03 | −5.05 | Hydrogen bond with Asp44 | Hydrogen bond with Asp44 |
| Outside | Rimantadine | −5.53 | −4.92 | Hydrogen bond with Asp44 | Hydrogen bond with Asp44 |
| Hydrophobic interaction with Thr43 | |||||

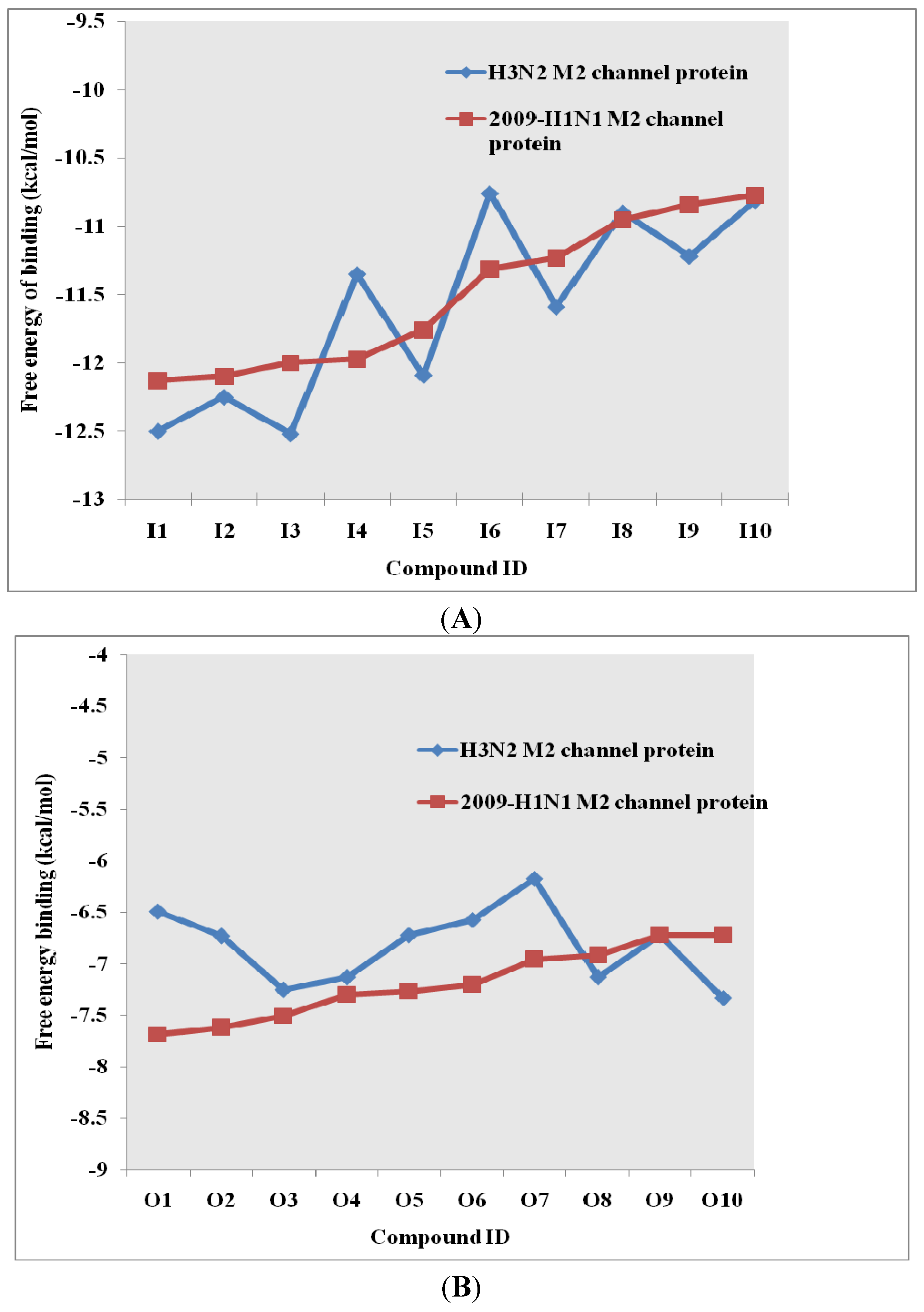
2.2. Docking Results and Ranking Top Hit Compounds Binding Inside and Outside M2 Channel Proteins
| Compound No. | 2D Chemical Structure | INSIDE Free energy of binding (kcal/mol) | OUTSIDE Free energy of binding (kcal/mol) | ||
|---|---|---|---|---|---|
| H3N2 | 2009-H1N1 | H3N2 | 2009-H1N1 | ||
| Group 1 (A1-A5) from [10] | |||||
| A1 | 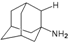 | −6.85 | −7.02 | −5.03 | −5.05 |
| A2 | 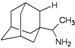 | −7.75 | −7.57 | −5.53 | −4.92 |
| A3 | 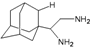 | −8.48 | −7.69 | −5.50 | −4.36 |
| A4 | 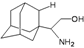 | −8.09 | −7.79 | −5.25 | −4.55 |
| A5 | 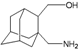 | −8.16 | −8.40 | −4.85 | −4.89 |
| Group 2 from [18] | |||||
| B1 | 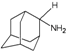 | −6.85 | −6.55 | −5.53 | −5.51 |
| B2 |  | −7.10 | −6.82 | −5.81 | −5.36 |
| B3 |  | −7.82 | −7.57 | −5.17 | −5.02 |
| B4 | 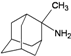 | −7.44 | −7.08 | −5.65 | −5.21 |
| B5 |  | −7.70 | −7.42 | −5.53 | −5.20 |
| B6 |  | −7.97 | −7.69 | −5.52 | −5.42 |
| B7 |  | −8.32 | −8.67 | −6.15 | −5.40 |
| B8 |  | −8.53 | −8.21 | −5.78 | −5.23 |
| B9 |  | −8.58 | −8.23 | −6.02 | −5.04 |
| B10 |  | −8.93 | −9.25 | −5.81 | −5.80 |
| Group 3 from [21] | |||||
| 1a |  | −8.70 | −8.48 | −6.04 | −6.11 |
| 1b |  | −9.46 | −8.91 | −6.49 | −5.51 |
| 1c |  | −9.39 | −8.89 | −6.55 | −3.04 |
| 1d |  | −9.20 | −9.62 | −4.71 | −2.60 |
| 1e |  | −8.99 | −9.81 | −4.45 | −0.30 |
| 1f |  | −8.73 | −8.54 | −5.45 | −5.01 |
| 1g |  | −9.91 | −9.80 | −5.32 | −2.52 |
| 1h |  | −9.53 | −9.07 | −4.62 | −0.15 |
| 1i |  | −10.29 | −10.02 | −7.33 | −6.72 |
| 2a |  | −10.47 | −10.14 | −5.79 | −3.63 |
| 2b |  | −10.48 | −10.23 | −5.42 | −3.27 |
| 2c |  | −10.69 | −11.23 | −3.47 | +3.03 |
| 2d |  | −10.26 | −11.35 | −4.61 | −2.52 |
| 2e |  | −9.97 | −11.14 | −0.34 | −1.90 |
| 2f |  | −10.30 | −10.61 | −5.98 | −5.56 |
| 2g |  | −11.21 | −11.48 | −6.16 | −5.60 |
| 3a |  | −10.18 | −10.40 | −4.96 | −3.93 |
| 3b |  | −9.17 | −10.81 | +3.01 | +4.05 |
| III |  | −9.96 | −9.62 | −6.72 | −7.27 |
| IV |  | −10.20 | −10.10 | −6.57 | −7.20 |
| V |  | −9.71 | −9.44 | −5.88 | −5.17 |
| Group 4 from [22,23] | |||||
| M1 |  | −8.55 | −8.44 | −5.77 | −5.69 |
| M2 |  | −9.03 | −8.75 | −5.53 | −5.89 |
| M3 |  | −8.58 | −8.43 | −5.61 | −5.63 |
| M4 |  | −9.05 | −8.87 | −5.85 | −5.81 |
| M5 |  | −8.60 | −8.23 | −5.56 | −5.64 |
| M6 | 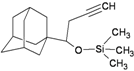 | −8.47 | −8.95 | −3.62 | −4.76 |
| M7 |  | −8.21 | −7.97 | −5.53 | −5.23 |
| M8 | 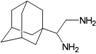 | −8.65 | −7.95 | −5.97 | −4.57 |
| M9 |  | −9.14 | −9.20 | −4.91 | −5.88 |
| M10 |  | −8.47 | −8.25 | −5.05 | −5.06 |
| TaM11 | 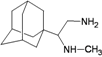 | −8.44 | −8.00 | −5.62 | −4.10 |
| M12 |  | −9.38 | −9.59 | −5.03 | −5.97 |
| M13 |  | −9.42 | −9.31 | −4.16 | −5.89 |
| M14 |  | −6.73 | −6.89 | −5.07 | −4.40 |
| M15 |  | −9.09 | −8.91 | −5.75 | −5.70 |
| M16 |  | −9.04 | −8.27 | −5.73 | −5.04 |
| M17 |  | −9.58 | −9.48 | −4.19 | −5.69 |
| M18 | 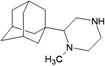 | −9.70 | −8.87 | −4.34 | −4.71 |
| M19 |  | −10.03 | −9.33 | −4.23 | −4.67 |
| M20 |  | −9.48 | −9.27 | −5.94 | −5.14 |
| M21 |  | −9.72 | −9.61 | −5.91 | −5.12 |
| M22 |  | −10.19 | −9.49 | −6.15 | −4.09 |
| M23 |  | −10.63 | −10.08 | −5.49 | −2.87 |
| M24 |  | −11.59 | −11.23 | −7.26 | −3.36 |
| Bananin |  | −9.67 | −9.39 | −3.83 | −4.40 |
| Iodobananin(IBN) |  | −9.60 | −10.16 | −4.96 | −5.36 |
| Eubananin(EUB) |  | −8.54 | −8.51 | +1.71 | +3.06 |
| Vanillinbananin(VBN) |  | −8.70 | −9.22 | −4.58 | −4.74 |
| Group 5 from [24] | |||||
| D1 |  | −8.48 | −8.26 | −5.38 | −5.06 |
| D2 |  | −7.83 | −7.94 | −5.58 | −5.20 |
| D3 |  | −7.38 | −6.91 | −4.27 | −3.86 |
| D4 |  | −7.46 | −7.29 | −5.69 | −5.53 |
| D5 | 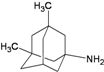 | −8.01 | −7.93 | −5.61 | −5.70 |
| D6 | 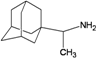 | −7.79 | −7.59 | −5.17 | −5.15 |
| D7 |  | −8.54 | −8.27 | −5.72 | −5.60 |
| D8 | 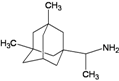 | −9.10 | −8.86 | −6.12 | −5.72 |
| D9 |  | −7.12 | −7.37 | −5.51 | −4.77 |
| D10 |  | −7.75 | −7.84 | −4.30 | −4.93 |
| D11 |  | −8.17 | −8.50 | −4.78 | −4.98 |
| D12 |  | −7.96 | −8.65 | −4.47 | −5.19 |
| D13 |  | −8.77 | −8.86 | −4.69 | −5.93 |
| D14 |  | −9.28 | −9.62 | −5.05 | −6.13 |
| D15 | 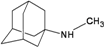 | −6.99 | −6.83 | −4.97 | −4.88 |
| D16 |  | −7.17 | −7.02 | −5.01 | −4.50 |
| Group 6 from [25,26] | |||||
| N1 |  | −9.38 | −9.74 | −5.51 | −5.51 |
| N2 |  | −8.57 | −8.26 | −5.65 | −5.17 |
| N3 |  | −12.09 | −11.76 | −6.72 | −6.72 |
| N4 | 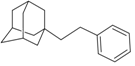 | −9.62 | −9.91 | −5.91 | −6.21 |
| N5 |  | −9.76 | −9.87 | −5.42 | −4.82 |
| N6 |  | −10.44 | −10.31 | −5.55 | −3.69 |
| N7 | 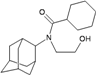 | −10.15 | −10.70 | −5.54 | −5.25 |
| N8 |  | −7.37 | −7.06 | −5.22 | −4.77 |
| N9 | 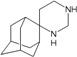 | −8.50 | −7.58 | −5.50 | −5.06 |
| N10 |  | −8.83 | −8.50 | −5.18 | −5.23 |
| N11 |  | −9.24 | −8.97 | −5.25 | −5.36 |
| N12 | 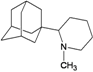 | −9.53 | −9.34 | −5.94 | −6.46 |
| N13 |  | −10.49 | −10.41 | −5.76 | −5.29 |
| N14 |  | −12.25 | −12.10 | −6.68 | −3.99 |
| N15 |  | −10.98 | −10.12 | −5.68 | −4.78 |
| N16 |  | −12.50 | −12.13 | −6.44 | −3.86 |
| N17 | 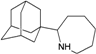 | −9.73 | −9.47 | −4.49 | −3.30 |
| N18 |  | −9.97 | −9.73 | −3.64 | −3.36 |
| N19 |  | −12.52 | −12.00 | −1.18 | +4.19 |
| N20 |  | −6.98 | −7.26 | −5.08 | −4.60 |
| N21 |  | −9.06 | −9.30 | −5.20 | −5.64 |
| Group 7 | |||||
| 1 | 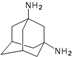 | −7.50 | −7.72 | −5.16 | −5.06 |
| 2 |  | −8.21 | −6.87 | −6.24 | −4.37 |
| 3 |  | −8.08 | −8.18 | −5.35 | −5.27 |
| 4 | 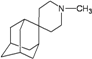 | −9.24 | −8.97 | −5.25 | −5.36 |
| 5 |  | −8.41 | −7.75 | −5.21 | −4.69 |
| 6 |  | −8.46 | −8.25 | −5.20 | −4.93 |
| 7 | 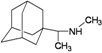 | −7.62 | −7.45 | −5.00 | −4.42 |
| 8 |  | −8.14 | −7.78 | −5.09 | −4.64 |
| 9 | 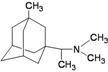 | −8.85 | −8.42 | −5.27 | −5.05 |
| 10 |  | −9.35 | −8.97 | −5.59 | −5.35 |
| 11 |  | −9.23 | −9.51 | −6.17 | −6.96 |
| 12 |  | −10.59 | −10.11 | −7.13 | −6.92 |
| 13 | 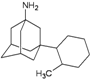 | −10.51 | −10.28 | −6.73 | −7.62 |
| 14 |  | −11.21 | −10.61 | −7.13 | −7.30 |
| 15 |  | −10.67 | −10.46 | −6.49 | −7.68 |
| 16 |  | −11.22 | −10.84 | −7.25 | −7.50 |
| 17 |  | −8.04 | −8.23 | −5.36 | −5.18 |
| 18 |  | −8.80 | −8.00 | −6.37 | −6.03 |
| 19 |  | −9.16 | −9.47 | −6.17 | −6.95 |
| 20 |  | −9.67 | −9.67 | −6.12 | −6.73 |
| 21 |  | −10.00 | −10.50 | −5.99 | −5.41 |
| 22 | 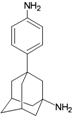 | −9.64 | −10.03 | −5.55 | −5.32 |
| 23 |  | −10.02 | −10.79 | −5.77 | −5.71 |
| 24 |  | −10.52 | −11.20 | −5.88 | −5.57 |
| 25 |  | −9.30 | −9.69 | −5.52 | −5.51 |
| 26 | 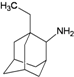 | −7.80 | −7.57 | −5.57 | −5.49 |
| 27 |  | −9.95 | −9.93 | −6.15 | −6.61 |
| 28 |  | −10.81 | −10.77 | −5.66 | −3.76 |
| 29 |  | −8.95 | −9.23 | −5.31 | −6.24 |
| 30 |  | −9.43 | −10.12 | −5.68 | −5.38 |
| 31 | 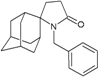 | −10.47 | −10.53 | −6.06 | −5.32 |
| 32 | 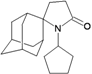 | −10.60 | −10.70 | −6.11 | −5.99 |
| 33 |  | −9.51 | −9.83 | −4.80 | −5.36 |
| 34 |  | −9.70 | −9.97 | −5.37 | −5.62 |
| 35 |  | −10.00 | −10.23 | −5.69 | −5.71 |
| 37 |  | −8.78 | −9.17 | −5.53 | −5.63 |
| 38 |  | −9.45 | −8.43 | −5.86 | −5.89 |
| 39 |  | −9.18 | −8.77 | −5.32 | −5.40 |
| 40 | 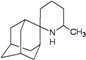 | −8.53 | −8.13 | −5.58 | −5.25 |
| 41 |  | −9.10 | −8.78 | −5.55 | −5.56 |
| 42 | 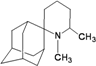 | −9.61 | −9.25 | −5.71 | −5.73 |
| 43 | 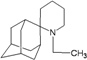 | −9.42 | −9.01 | −5.26 | −5.04 |
| 44 |  | −9.07 | −9.14 | −5.01 | −5.55 |
| 45 |  | −9.51 | −9.55 | −3.97 | −5.73 |
| 46 |  | −9.63 | −9.81 | −3.46 | −5.69 |
| 47 |  | −9.90 | −10.25 | −3.53 | −5.59 |
| 48 |  | −9.01 | −8.66 | −5.96 | −5.91 |
| 49 |  | −9.25 | −9.21 | −5.46 | −5.55 |
| 50 | 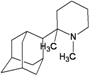 | −9.85 | −9.61 | −5.15 | −5.04 |
| 51 | 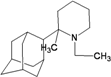 | −10.11 | −9.83 | −4.14 | −4.91 |
| 52 | 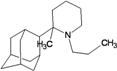 | −10.41 | −10.86 | −4.17 | −4.56 |
| 53 |  | −11.35 | −11.97 | −5.26 | −3.29 |
| 54 |  | −9.15 | −8.86 | −5.69 | −5.19 |
| 55 | 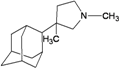 | −9.55 | −9.34 | −5.37 | −5.06 |
| 56 | 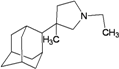 | −9.60 | −9.59 | −5.39 | −4.77 |
| 57 |  | −9.85 | −9.70 | −5.45 | −4.42 |
| 58 |  | −10.10 | −10.01 | −5.00 | −4.37 |
| 59 |  | −10.74 | −10.33 | −4.91 | −5.01 |
| 60 |  | −10.76 | −11.31 | −5.47 | −3.82 |
| 61 |  | −9.14 | −9.23 | −4.59 | −5.69 |
| 62 | 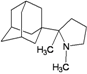 | −9.50 | −9.15 | −4.04 | −5.33 |
| 63 |  | −9.88 | −9.55 | −3.69 | −4.42 |
| 64 | 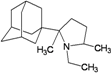 | −10.15 | −10.49 | −3.49 | −3.72 |
| 65 | 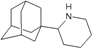 | −9.03 | −8.74 | −5.55 | −5.90 |
| 66 | 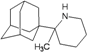 | −9.46 | −9.27 | −4.12 | −5.28 |
| 67 |  | −10.06 | −9.72 | −4.47 | −3.54 |
| 68 | 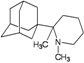 | −9.92 | −9.78 | −4.13 | −5.56 |
| 69 | 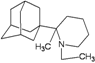 | −10.31 | −10.19 | −3.75 | −4.61 |
| 70 | 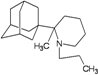 | −10.62 | −10.38 | −3.57 | −4.70 |
| 71 |  | −8.30 | −7.81 | −5.66 | −5.55 |
| 72 |  | −8.60 | −8.11 | −4.77 | −4.77 |
| 73 |  | −8.84 | −8.76 | −4.35 | −4.26 |
| 74 |  | −9.25 | −9.04 | −3.86 | −3.89 |
| 75 | 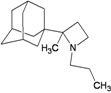 | −9.62 | −9.25 | −4.39 | −3.78 |
| 76 |  | −9.80 | −9.45 | −4.07 | −3.80 |
| 77 |  | −10.10 | −9.82 | −3.95 | −1.91 |
| 78 | 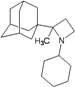 | −10.90 | −10.95 | −4.60 | −4.48 |
| 79 |  | −7.44 | −7.08 | −5.08 | −4.83 |
| 80 | 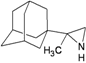 | −7.87 | −7.66 | −4.93 | −4.56 |
| 81 | 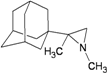 | −7.98 | −7.92 | −4.38 | −4.91 |
| 82 | 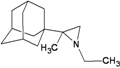 | −8.29 | −8.11 | −4.34 | −4.59 |
| 83 | 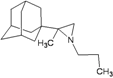 | −8.76 | −8.61 | −4.56 | −4.41 |
| 84 | 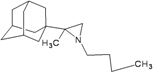 | −9.01 | −8.82 | −4.65 | −4.30 |
| 85 |  | −9.35 | −8.77 | −4.66 | −4.21 |
| 86 | 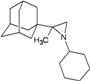 | −10.19 | −10.15 | −5.37 | −5.29 |
| 87 |  | −7.91 | −7.15 | −6.62 | −5.94 |
| 88 |  | −6.94 | −6.63 | −5.34 | −4.58 |
| 89 |  | −7.23 | −6.82 | −5.37 | −4.59 |
| 90 |  | −9.23 | −9.04 | −6.38 | −5.84 |
| 91 |  | −7.16 | −7.02 | −5.26 | −5.16 |
| 92 | 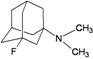 | −7.40 | −7.65 | −5.69 | −5.22 |
| 93 | 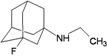 | −7.41 | −7.01 | −5.45 | −4.74 |
| 94 |  | −7.67 | −7.39 | −5.57 | −4.91 |
| 95 |  | −8.86 | −9.15 | −5.65 | −5.72 |
| 96 | 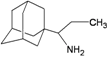 | −8.00 | −7.62 | −5.23 | −4.88 |
| 97 |  | −8.66 | −8.92 | −5.16 | −5.90 |
| 98 |  | −8.90 | −9.01 | −5.06 | −6.16 |
| 99 |  | −9.37 | −9.51 | −5.20 | −6.42 |
| 100 |  | −9.74 | −9.81 | −5.39 | −6.50 |
| Inside Ranking Comp. | Top 10 binding compounds (inside M2 channel protein) | Free energy of binding (kcal/mol) | Outside Ranking Comp. | Top 10 binding compounds(outside M2 channel protein) | Free energy of binding (kcal/mol) | |||
|---|---|---|---|---|---|---|---|---|
| H3N2 | 2009-H1N1 | H3N2 | 2009-H1N1 | |||||
| I1 |  | −12.50 | −12.13 | O1 |  | −6.49 | −7.68 | |
| I2 |  | −12.25 | −12.10 | O2 |  | −6.73 | −7.62 | |
| I3 |  | −12.52 | −12.00 | O3 |  | −7.25 | −7.50 | |
| I4 |  | −11.35 | −11.97 | O4 |  | −7.13 | −7.30 | |
| I5 |  | −12.09 | −11.76 | O5 |  | −6.72 | −7.27 | |
| I6 |  | −10.76 | −11.31 | O6 |  | −6.57 | −7.20 | |
| I7 |  | −11.59 | −11.23 | O7 | 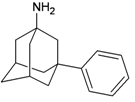 | −6.17 | −6.96 | |
| I8 |  | −10.90 | −10.95 | O8 |  | −7.13 | −6.92 | |
| I9 |  | −11.22 | −10.84 | O9 |  | −6.72 | −6.72 | |
| I10 | 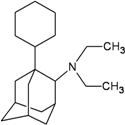 | −10.81 | −10.77 | O10 |  | −7.33 | −6.72 | |

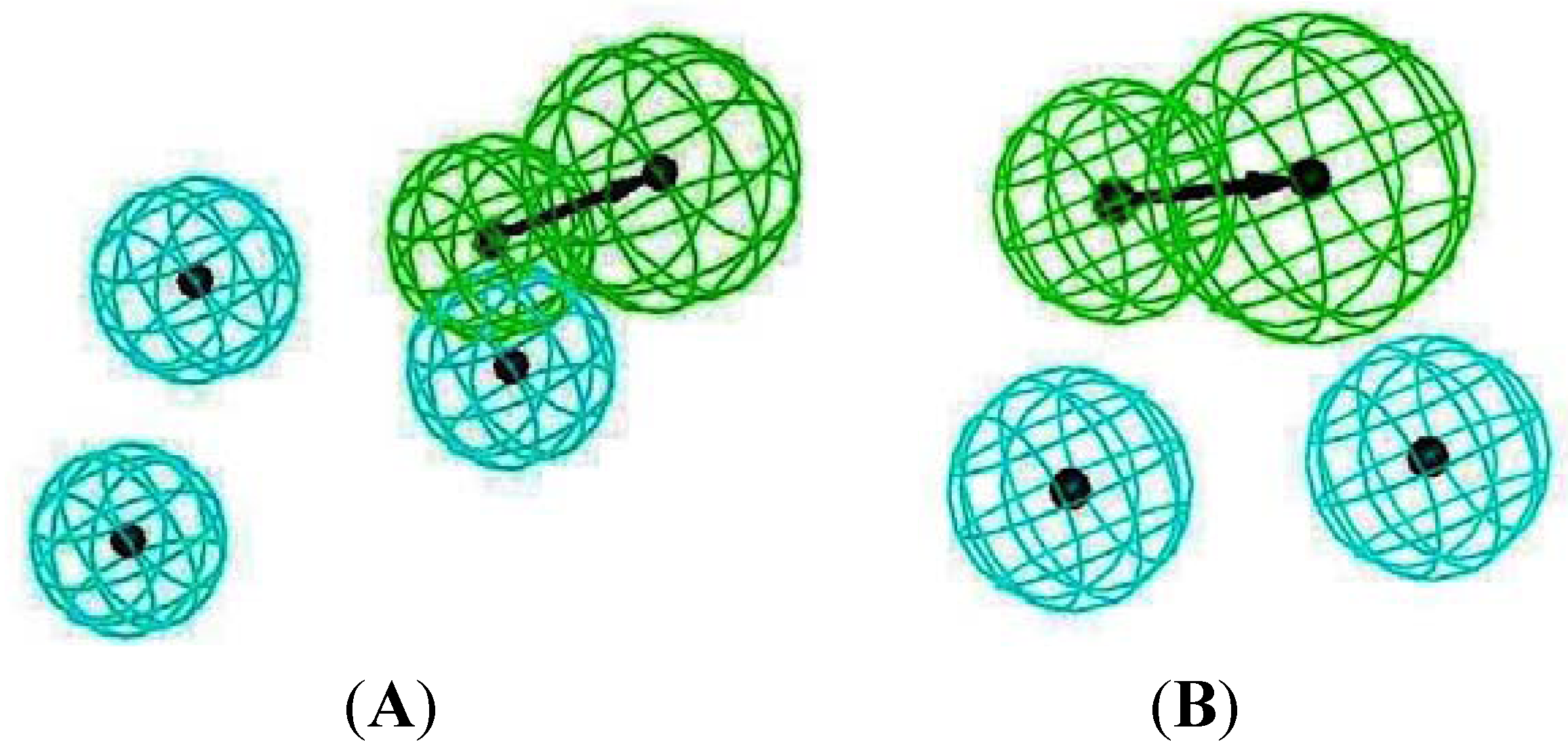
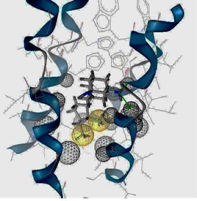
2.3. Pharmacophore Analysis for Top hits M2 Channel-Inhibitor Interactions
| Cartoon Representation of M2 protein complex | Protein-Ligand Interactions | Hypothesis |
|---|---|---|
| I5 inside M2 channel | ||
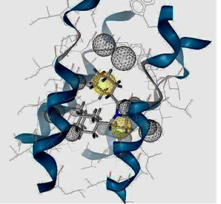 | 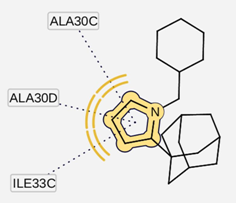 |  |
| I9 inside M2 channel | ||
 | 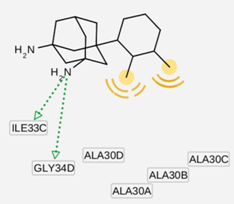 |  |
| I5 outside M2 channel | ||
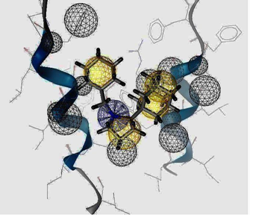 | 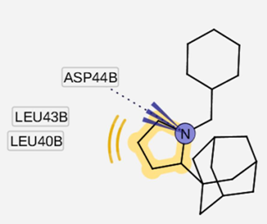 | 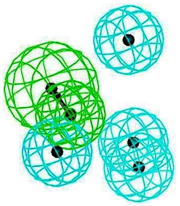 |
| I9 outside M2 channel | ||
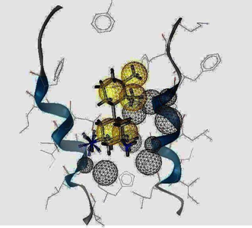 | 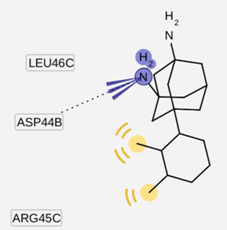 | 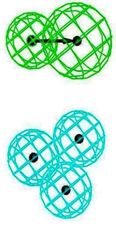 |
2.4. Common Pharmacophore Features of the Top Binding Compounds
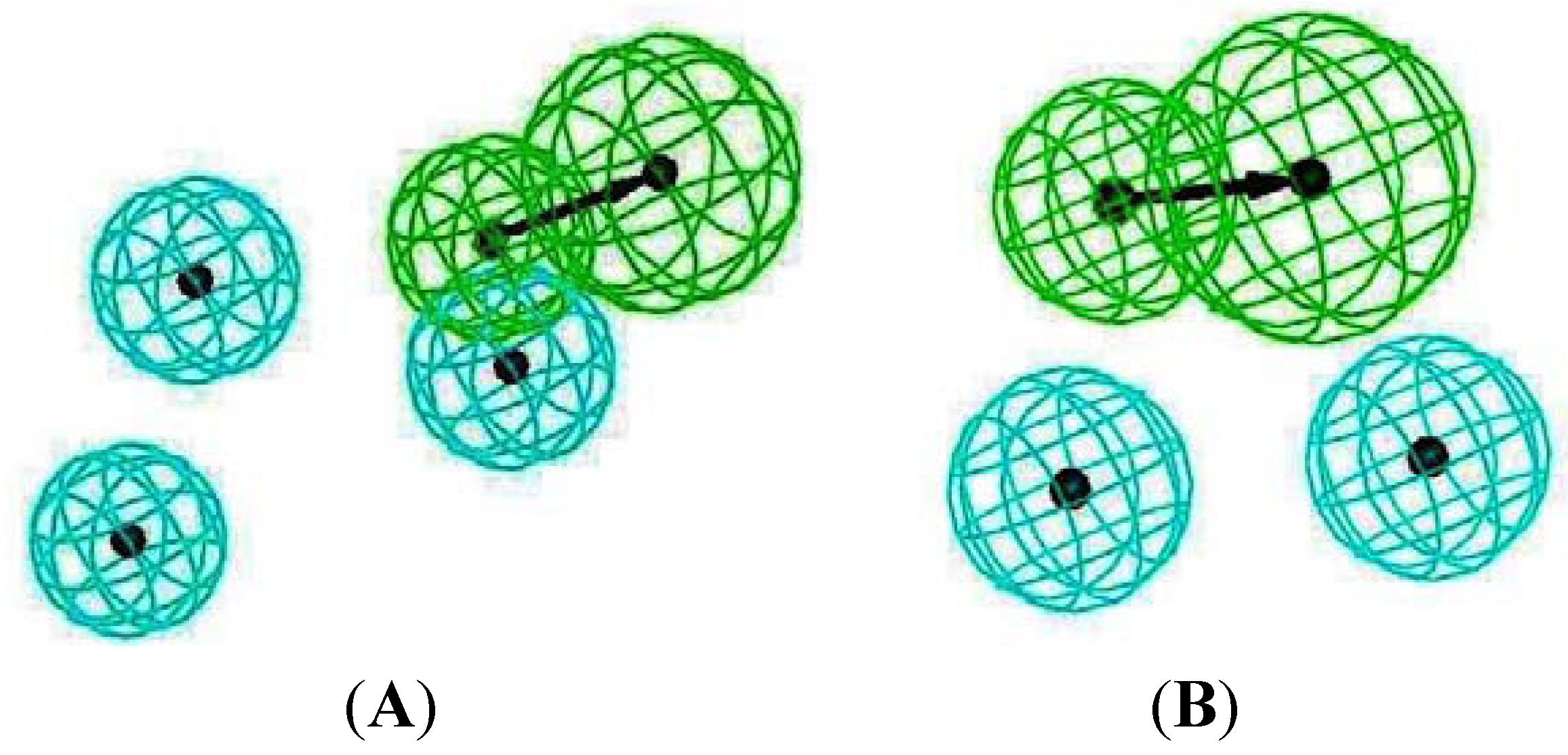
3. Experimental
3.1. Proteins and Inhibitor Preparation

3.2. Virtual Screening with Autodock 3.05
3.3. Pharmacophore Modeling
3.3.1. Generation of Structure-Based Pharmacophore Models Using LigandScout 3.01
3.3.2. Ligand-Based Pharmacophore Modeling Using Discovery Studio 2.5
4. Conclusions
Acknowledgments
References and Notes
- Sugrue, R.J.; Hay, A.J. Structural characteristics of the M2 protein of influenza A viruses: Evidence that it forms a tetrameric channel. Virology 1991, 180, 617–624. [Google Scholar] [CrossRef]
- Betakova, T. M2 protein-a proton channel of influenza A virus. Curr. Pharm. Des. 2007, 13, 3231–3235. [Google Scholar] [CrossRef]
- Holsinger, L.J.; Nichani, D.; Pinto, L.H.; Lamb, R.A. Influenza a virus M2 ion channel protein: A structure-function analysis. J. Viol. 1994, 68, 1551–1563. [Google Scholar]
- Jefferson, T.; Deeks, J.J.; Demicheli, V.; Rivetti, D.; Rudin, M. Amantadine and rimantadine for preventing and treating influenza A in adults. Cochrane Database Syst. Rev. 2004, 3, CD001169. [Google Scholar]
- Pielak, R.M.; Schnell, J.R.; Chou, J.J. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA 2009, 106, 7379–7384. [Google Scholar] [CrossRef]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. The 2009 H1N1 pandemic influenza virus: What next? MBio 2010, 1. [Google Scholar] [CrossRef]
- Qin, G.; Yu, K.; Shi, T.; Luo, C.; Li, G.; Zhu, W.; Jiang, H. How does influenza virus a escape from amantadine? J. Phys. Chem. B 2010, 114, 8487–8493. [Google Scholar]
- Laohpongspaisan, C.; Rungrotmongkol, T.; Intharathep, P.; Malaisree, M.; Decha, P.; Aruksakunwong, O.; Sompornpisut, P.; Hannongbua, S. Why amantadine loses its function in influenza m2 mutants: MD simulations. J. Chem. Inf. Model. 2009, 49, 847–852. [Google Scholar]
- Gayday, A.V.; Levandovskiy, I.A.; Byler, K.G.; Shubina, T.E. Mechanism of Influenza a M2 Ion-Channel Inhibition: A Docking and QSAR Study. In ICCS 2008 Proceedings of the 8th international conference on Computational Science, Kraków, Poland, 23-25 June 2008; pp. 360–368, Part II.
- Du, Q.S.; Huang, R.B.; Wang, S.Q.; Chou, K.C. Designing inhibitors of M2 proton channel against H1N1 swine influenza virus. PLoS One 2010, 5, e9388. [Google Scholar]
- Kukol, A.; Adams, P.D.; Rice, L.M.; Brunger, A.T.; Arkin, T.I. Experimentally based orientational refinement of membrane protein models: A structure for the Influenza A M2 H+ channel. J. Mol. Biol. 1999, 286, 951–962. [Google Scholar] [CrossRef]
- Darapaneni, V.; Prabhaker, V.K.; Kukol, A. Large-scale analysis of influenza A virus sequences reveals potential drug target sites of non-structural proteins. J. Gen. Virol. 2009, 90, 2124–2133. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kim, S.; Kovacs, F.; Cross, T.A. Structure of the transmembrane region of the M2 protein H(+) channel. Protein Sci. 2001, 10, 2241–2250. [Google Scholar]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [Google Scholar]
- Du, Q.-S.; Wang, S.-Q.; Huang, R.-B.; Chou, K.-C. Computational 3D structures of drug-targeting proteins in the 2009-H1N1 influenza A virus. Chem. Phys. Lett. 2009, 485, 191–195. [Google Scholar]
- Beauchemin, C.A.; McSharry, J.J.; Drusano, G.L.; Nguyen, J.T.; Went, G.T.; Ribeiro, R.M.; Perelson, A.S. Modeling amantadine treatment of influenza A virus in vitro. J. Theor. Biol. 2008, 254, 439–451. [Google Scholar] [CrossRef]
- Wang, J.; Schnell, J.R.; Chou, J.J. Amantadine partition and localization in phospholipid membrane: A solution NMR study. Biochem. Biophys. Res. Commun. 2004, 324, 212–217. [Google Scholar] [CrossRef]
- Eleftheratos, S.; Spearpoint, P.; Ortore, G.; Kolocouris, A.; Martinelli, A.; Martin, S.; Hay, A. Interaction of aminoadamantane derivatives with the influenza A virus M2 channel-docking using a pore blocking model. Bioorg. Med. Chem. Lett. 2010, 20, 4182–4187. [Google Scholar] [CrossRef]
- Klebe, G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discov. Today 2006, 11, 580–594. [Google Scholar] [CrossRef]
- Du, Q.S.; Huang, R.B.; Wang, C.H.; Li, X.M.; Chou, K.C. Energetic analysis of the two controversial drug binding sites of the M2 proton channel in influenza A virus. J. Theor. Biol. 2009, 259, 159–164. [Google Scholar] [CrossRef]
- Papanastasiou, I.; Tsotinis, A.; Kolocouris, N.; Prathalingam, S.R.; Kelly, J.M. Design, synthesis, and trypanocidal activity of new aminoadamantane derivatives. J. Med. Chem. 2008, 51, 1496–1500. [Google Scholar] [CrossRef]
- Tanner, J.A.; Zheng, B.J.; Zhou, J.; Watt, R.M.; Jiang, J.Q.; Wong, K.L.; Lin, Y.P.; Lu, L.Y.; He, M.L.; Kung, H.F.; et al. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chem. Biol. 2005, 12, 303–311. [Google Scholar] [CrossRef]
- De Clercq, E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [Google Scholar] [CrossRef]
- Tataridis, D.; Fytas, G.; Kolocouris, A.; Fytas, C.; Kolocouris, N.; Foscolos, G.B.; Padalko, E.; Neyts, J.; De Clercq, E. Influence of an additional 2-amino substituent of the 1-aminoethyl pharmacophore group on the potency of rimantadine against influenza virus A. Bioorg. Med. Chem. Lett. 2007, 17, 692–696. [Google Scholar]
- Stamatiou, G.; Foscolos, G.B.; Fytas, G.; Kolocouris, A.; Kolocouris, N.; Pannecouque, C.; Witvrouw, M.; Padalko, E.; Neyts, J.; de Clercq, E. Heterocyclic rimantadine analogues with antiviral activity. Bioorg. Med. Chem. 2003, 11, 5485–5492. [Google Scholar] [CrossRef]
- Balannik, V.; Wang, J.; Ohigashi, Y.; Jing, X.; Magavern, E.; Lamb, R.A.; Degrado, W.F.; Pinto, L.H. Design and pharmacological characterization of inhibitors of amantadine-resistant mutants of the M2 ion channel of influenza A virus. Biochemistry 2009, 48, 11872–11882. [Google Scholar] [CrossRef]
- Chang, T.-T.; Sun, M.-F.; Chen, H.-Y.; Tsai, F.-J.; Lin, J.-G.; Chen, C.Y.-C. Key features for designing M2 proton channel anti swine Flu inhibitors. J. Taiwan Inst. Chem. Eng. 2011, 42, 701–708. [Google Scholar] [CrossRef]
- Ieen Frisch, A.E.; Dennington, R., II.; Keith, T.A.; Millam, J.; Millan, J. GaussView Reference Version 4; Gaussian, Incorporated: Wallingford, CT, USA, 2007; ISBN:978-0-9727287-5-2. [Google Scholar]
- Discovery Studio® Visualizer 3.0, Accelrys Inc.: San Diego, CA, USA, 2009.
- Nguyen, H.T.; Le, L.; Truong, T.N. Top-hits for H1N1pdm identified by virtual screening using ensemble-based docking. PLoS Curr. 2009, 1, RN1030. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar]
- Wolber, G.; Langer, T. Ligandscout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Discovery Studio Version 2.5 (DS 2.5) User Manual, Accelrys Inc.: San Diego, CA, USA, 2009.
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Potential virtual lead identification in the discovery of renin inhibitors: application of ligand and structure-based pharmacophore modeling approaches. Eur. J. Med. Chem. 2011, 46, 2469–2476. [Google Scholar] [CrossRef]
- Lin, S.-K. Pharmacophore perception, development and use in drug design. Molecules 2000, 5, 987–989. [Google Scholar] [CrossRef]
- Taha, M.O.; Al-Bakri, A.G.; Zalloum, W.A. Discovery of potent inhibitors of pseudomonal quorum sensing via pharmacophore modeling and in silico screening. Bioorg. Med. Chem. Lett. 2006, 16, 5902–5906. [Google Scholar] [CrossRef]
- Sample Availability: Samples of 200 compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tran, L.; Choi, S.B.; Al-Najjar, B.O.; Yusuf, M.; Wahab, H.A.; Le, L. Discovery of Potential M2 Channel Inhibitors Based on the Amantadine Scaffold via Virtual Screening and Pharmacophore Modeling. Molecules 2011, 16, 10227-10255. https://doi.org/10.3390/molecules161210227
Tran L, Choi SB, Al-Najjar BO, Yusuf M, Wahab HA, Le L. Discovery of Potential M2 Channel Inhibitors Based on the Amantadine Scaffold via Virtual Screening and Pharmacophore Modeling. Molecules. 2011; 16(12):10227-10255. https://doi.org/10.3390/molecules161210227
Chicago/Turabian StyleTran, Linh, Sy Bing Choi, Belal O. Al-Najjar, Muhammad Yusuf, Habibah A. Wahab, and Ly Le. 2011. "Discovery of Potential M2 Channel Inhibitors Based on the Amantadine Scaffold via Virtual Screening and Pharmacophore Modeling" Molecules 16, no. 12: 10227-10255. https://doi.org/10.3390/molecules161210227