2. Results and Discussion
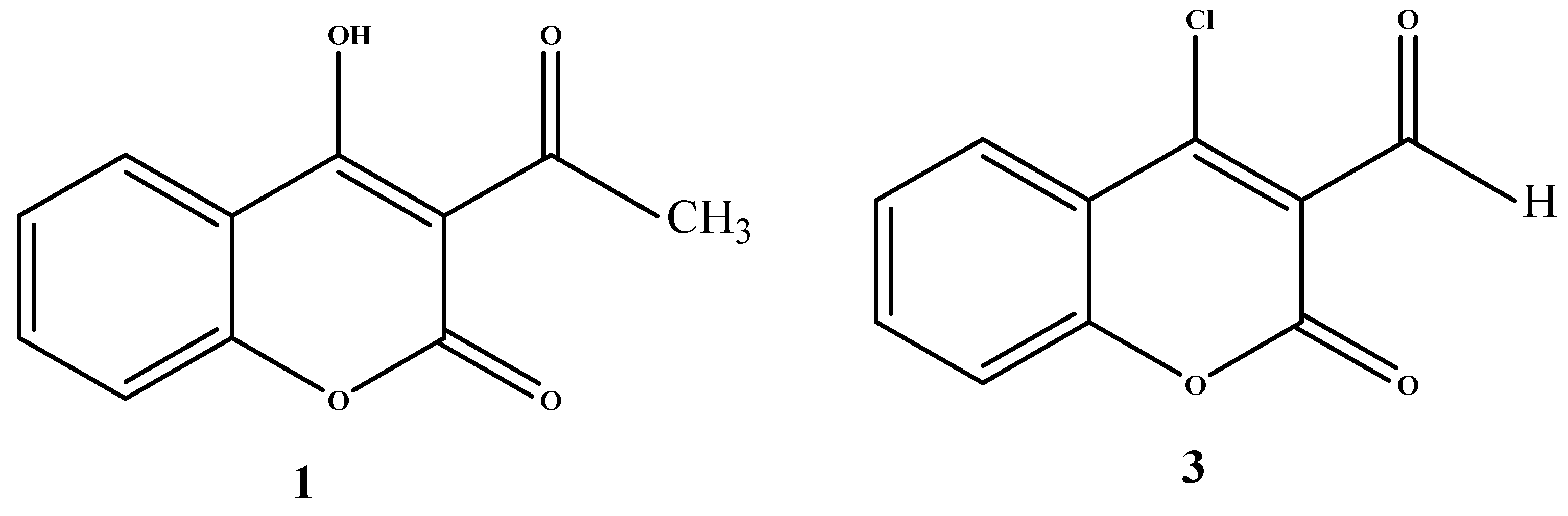
For acetylation of the 4-hydroxycoumarin to give 3-acetyl-4-hydroxycoumarin (
1), the method of Dholakia
et al. [
14] was employed, using glacial acetic acid as acetylating agent in the presence of POCl
3. The reaction was rapid, without involving any kind of competition from the intramolecular condensation of 4-hydroxycoumarin [
15]. This compound was characterized by IR,
1H- and
13C-NMR. The
1H-NMR shows the aromatic protons as a multiplet between 7.24 and 8.01 ppm. A singlet at 2.74 ppm was assigned to the methyl protons while the OH signal appeared at 17.72 ppm. This very high chemical shift might be explained by an intermolecular hydrogen bond.
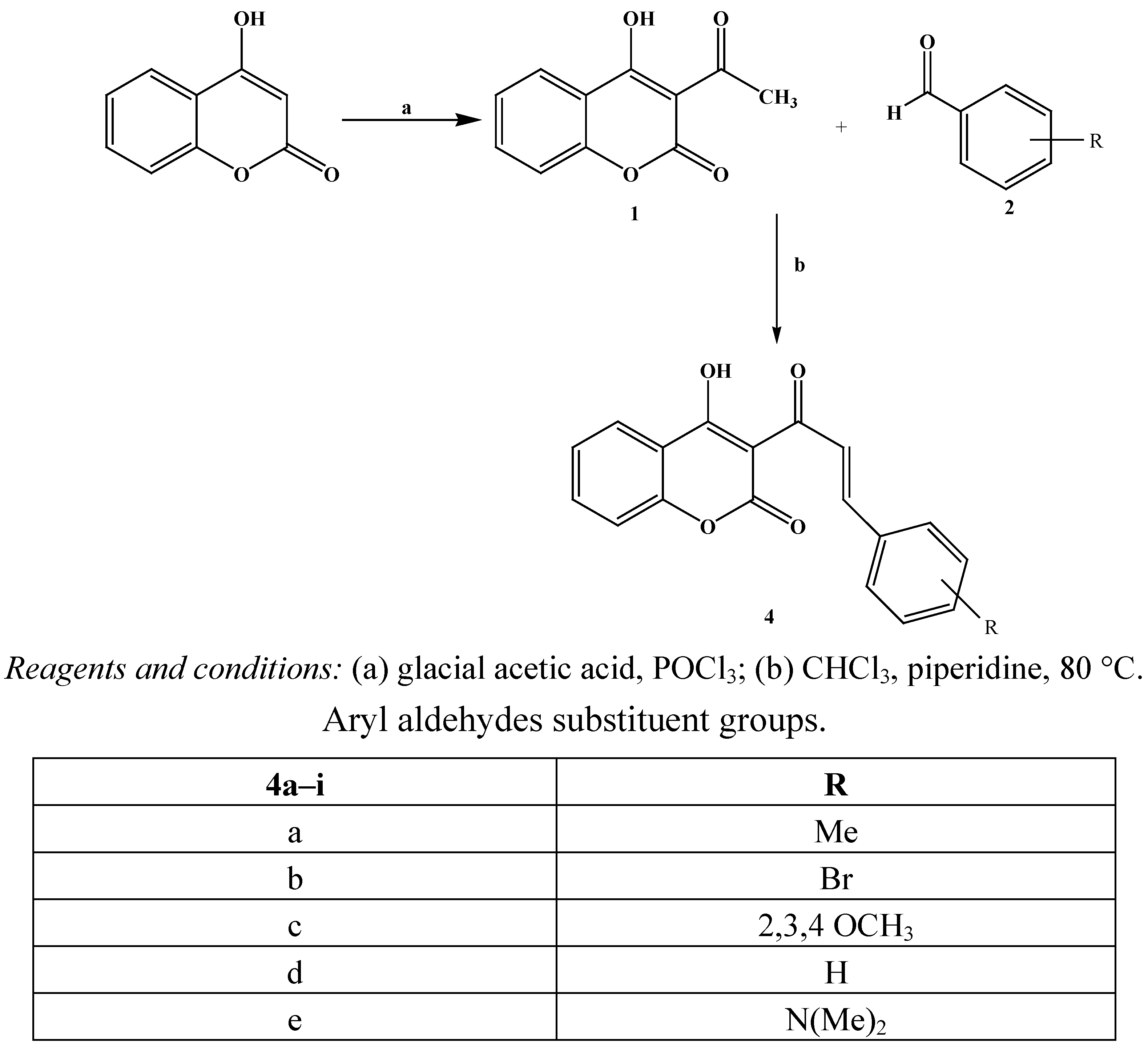
The synthesis of coumarinic chalcones was achieved in one step using a new pathway by refluxing 3-acetyl-4-hydroxy coumarin
1 with various aryl or heteroaryl aldehydes in the presence of piperidine in chloroform. The reaction studied and the products obtained are depicted in
Scheme 1.
Scheme 1.
Synthesis of coumarinic chalcones 4.
Scheme 1.
Synthesis of coumarinic chalcones 4.
In conventional chalcone synthesis methods, the time for completion of reactions at room temperature is very long, ranging from 24 to 36 h [
16,
17]. A small alteration in reaction conditions, using chloroform as solvent with a mild organic base, for example, piperidine, reduced the reaction time in most cases, to 1–1.5 h. Moreover, the isolation of product
4 was facilitated.
All the products were obtained as solids and their purity was checked by thin layer chromatography (eluant: hexane/ethyl acetate, 1/1. v/v). All the synthesized compounds have been characterized on the basis of their physical data and spectral analysis. The 1H-NMR spectra are consistent with the molecular structure 4. The 13C-NMR spectrum of 4c in DMSO showed two downfield signals at δ 162.42 (ketone CO) and δ 157.41 (lactone CO) as well as one upfield signal at δ 55.48 (OCH3). Measurement of the spectrum using the DEPT technique showed that the HC carbons of the coumarinic ring appear at δ 134.5, 131.3, 125.6 and 124.11. The IR spectra of compounds 4a–i showed bands resulting from the OH, (ketone) C=O and (lactone) (C=O) stretching in the region 3,305–3,320 cm−1, 1,670–1,710 cm−1 and 1,720–1,750 cm−1, respectively, in all the cases. All this evidence was supportive of the formation of compounds 4a–i.
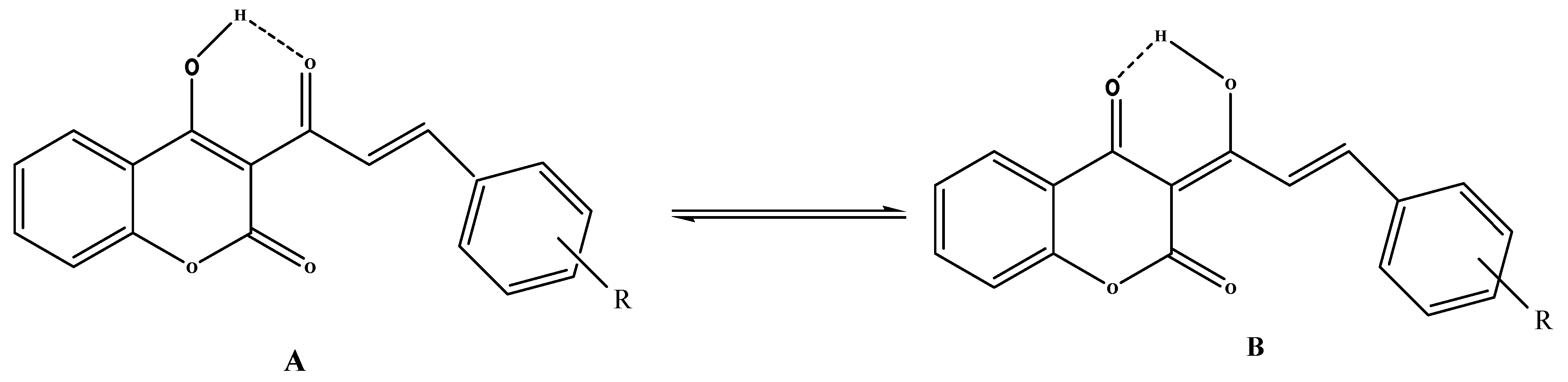
The coumarinic 4-arylbut-3-en-2-one structure was deduced from an analysis of HMBC spectra which indicate that both ethylenic protons correlate with the ketone carbon atom via
J2 and
J3 coupling constants, and C
3 respectively. Like 3-acetyl-4-hydroxycoumarin, compounds
4a–i can exist in several tautomeric forms. The most stable tautomers
A and
B are stabilized by intramolecular hydrogen bonding (
Scheme 2).
Scheme 2.
Tautomeric forms of compounds 4.
Scheme 2.
Tautomeric forms of compounds 4.
A range of activities have been claimed to arise from the fusion of some coumarins with pyrazole rings [
18,
19,
20]. Morever substituted pyrazoles have recently been used as analytical reagents in the complexation of transition metal ions [
21,
22]. We have attempted to exploit this behaviour of aldehyde groups in 4 chloro-3-formylcoumarin with the purpose of finding a satisfactory route by which to synthesize substituted chromen[4,3-c]pyrazol-4(1
H)-ones bearing hydrogen or alkyl groups on N-2.
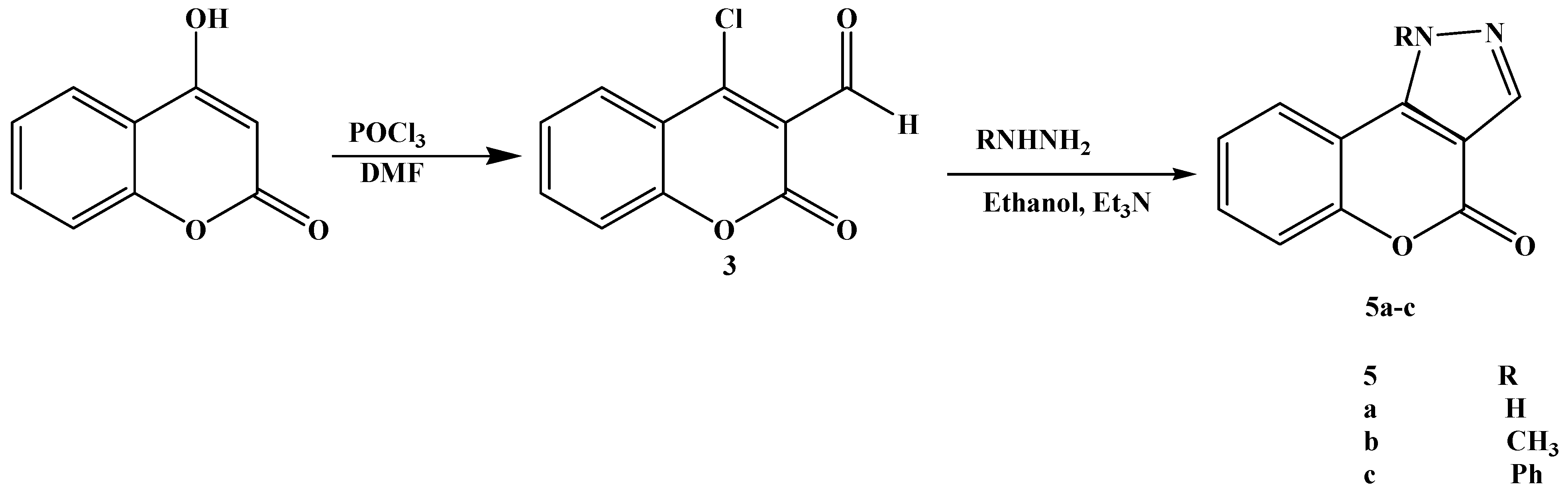
The compound
3 was prepared from 4-hydroxycoumarin under Vilsmeier conditions (POCl
3/DMF). The literature [
23,
24] claims the formation of mixture of carbaldehyde
3 and 4-chlorocoumarin without any indication of their ratio. Steinführer
et al. [
25] reported that the reaction mixture contained a mixture of 4-chloro-3-coumarin carbaldehyde 3(65%) with 4-chlorocoumarin as a side product (20%). After further optimization we could obtain up to 95% of the carbaldehyde
3. The reaction pathway is depicted in
Scheme 3.
Scheme 3.
Synthesis of chromen[4,3-c]pyrazol-4(1H)-ones 5.
Scheme 3.
Synthesis of chromen[4,3-c]pyrazol-4(1H)-ones 5.
TLC was used to monitor the progress of the reaction. The spectroscopic data MS, IR and
1H-NMR are in agreement with the structure of 4-chloro-3-formylcoumarin. The heating of 3-formyl-4-chloro-coumarin
3 and the appropriate hydrazine nitrogen compounds in ethanol at reflux for 2 h led to the quantitative formation of the substituted chromen[4,3-c]pyrazol-4-ones
5 in short times and in high yields (67 to 89%). The proposed structures of the new substituted chromen[4,3-c]pyrazol-4(1
H)-ones 5 were validated by their spectral data, which were consistent with available literature data for similar substitution patterns in coumarin derivatives [
26,
27].
The
1H-NMR spectra of compound
5a, for example, showed the expected signals: An exchangeable NH proton and the typical doublet for the coumarin 9-H in the δ 7.96–8.20 ppm range, validated by the
13C spectral pattern of the coumarin ring. All the chromen[4,3-c]pyrazol-4(1
H)-ones
5a–c displayed IR stretching at 1,711–1,742 cm
−1 associated with lactone carbonyl groups, also confirmed by the signals appearing between δ 155 and 162 ppm in the
13C-NMR spectra. The derivatives
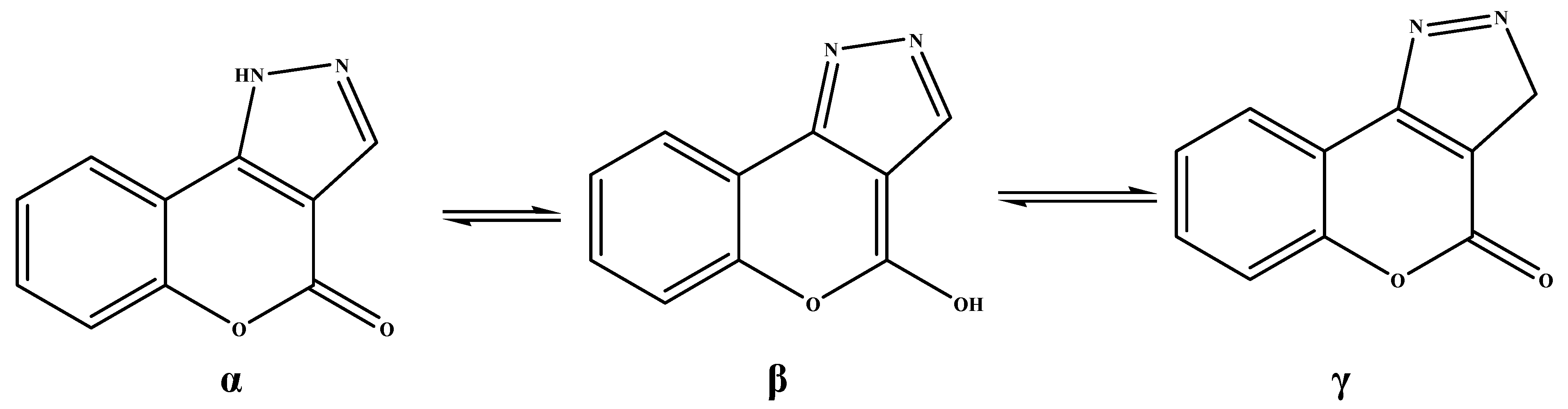
5a can exist in
α, β and
γ tautomeric forms, as depicted in
Scheme 4.
Ring tautomerism is common in heteroaromatic compounds and has been widely studied, but literature relating to the chromen[4,3-c]pyrazol-4(1
H)-ones nucleus gave poor attention to this tautomerism problem [
28,
29]. The authors of the cited references assign two opposite tautomeric structures to the same compound and the
1H-NMR spectroscopic data for this product are not very detailed.
Scheme 4.
Tautomeric forms of chromen[4,3-c]pyrazol-4(1H)-ones 5.
Scheme 4.
Tautomeric forms of chromen[4,3-c]pyrazol-4(1H)-ones 5.
In our case the β tautomer can be ruled out because all the IR spectra of the chromen[4,3-c]pyrazol-4(1H)-ones 5a–c, recorded in chloroform solution, showed lactone bands at 1,704–1,712 cm−1, confirmed by typical carbonyl patterns in the 13C-NMR spectra. 1H- and 13C-NMR analyses show that compounds 5 exist as the tautomeric form containing a proton on the N atom.
3. Antioxidant Activities
There is an increasing interest in antioxidants, particularly in those intended to prevent the presumed deleterious effects of free radicals in the human body, and to prevent the deterioration of fats and other constituents of foodstuffs. In both cases, there is a preference for antioxidants from natural rather than from synthetic sources. There has therefore been a parallel increase in the use of methods for estimating the efficiency of such substances as antioxidants.
One such method that is currently popular is based upon the use of the stable free radical diphenylpicrylhydrazyl (DPPH). The purpose of our study was to examine the use of the parameter “EC50” (equivalent concentration to give 50% effect) which is currently used in the interpretation of experimental data from the method.
When a solution of DPPH is mixed with that of a substance that can donate a hydrogen atom, this gives rise to the reduced form with the loss of this violet colour (although there would be expected to be a residual pale yellow colour from the picrylgroup still present). Representing the DPPH radical by Z
• and the donor molecule by AH, the primary reaction is:
where ZH is reduced form and A
• is free radical produced in this first step. This latter radical will then undergo further reactions which control the overall stoichiometry, that is, the number of molecules of DPPH reduced (decolorized) by one molecule of the reductant.
The reaction (1) is therefore intended to provide the link with the reactions taking place in an oxidizing system, such as the autoxidation of a lipid or other unsatured substance; the DPPH molecule Z
• is thus intended to represent the free radicals formed in the system whose activity is to be suppressed by the substance AH
• Representing the DPPH radical by Z
• and the coumarinic chalcones by ROH, the initial reaction is then Z
• + ROH= ZH+ RO
• [
1], the free radical RO
• Evidently then reacts with another molecule of the same kind that was produced by a parallel reaction to (1) RO
• + RO
• = RO-OR [
2]. This therefore leads to the observed reduction of two molecules of DPPH by two molecules of coumarinic chalcones, that is, a 1:1 stoichiometry. One parameter that has been introduced recently for the interpretation of the results from the DPPH method, is the efficient concentration or EC
50 value (otherwise called the IC
50 value), defined as the concentration of substrate that causes 50% loss of the DPPH activity (colour), corresponding to the endpoint of the titration. It should be noted that in all cases, any residual (yellow) colour from the reduced form or any non-specific absorbance from the sample has to be taken into account in defining the “endpoint” of the titration, or the 50% point, this EC
50 parameter also has the drawback that the higher the antioxidant activity, the lower is the value of EC
50. This is a disadvantage particularly when results are presented graphically as a bar chart even if the same data are available in numerical forms. The reported derivatives
4a–i were tested for their antioxidant. The corresponding IC
50 values are summarized in
Table 1.
Table 1.
The EC50 values exhibited by coumarinic derivatives 4.
Table 1.
The EC50 values exhibited by coumarinic derivatives 4.
| Compounds 4a–i | EC50 (μM) |
|---|
| 4a | 2.07 |
| 4b | 2.25 |
| 4c | 2.29 |
| 4d | 2.30 |
| 4e | 2.35 |
| 1 | 2.35 |
| 3 | 2.45 |
| Trolox | 2.30 |
Compounds 4c and 4f showed potent antioxidant activity, while coumarinic chalcone 4f was inactive, even at a concentration of 3.21 mM. Compound 4e proved to be the most active compound in this study. Comparing the activity of compounds 4a–c, 4e and 4g it was concluded that there was no substantial difference when we change the nature of the group R.
4. Experimental
4.1. General
Flash chromatography was carried out on 0.04–0.063 mm (Merck) silica gel, thin layer chromatography was carried out on aluminium backed silica plates by Merck and plates were revealed using a UV 254 light. 1H-NMR (300 MHz) and 13C-NMR (75 MHz) spectra were recorded on a Varian VXR 300 instrument at 293 °K in CDCl3 or DMSO d-6. Spectra were internally referenced to TMS. Peaks are reported in ppm downfield of TMS. Multiplicities are reported as singlet (s), doublet (d), triplet (t), quartet (q), some combinations of these were made by DEPT editing of the spectra. The IR-spectra were recorded on a Philips Analytical PU 9800 spectrometer. The melting points of compounds were determined in open glass capillaries in a paraffin bath and are uncorrected. The MS-spectra were recorded on a AEI MS 902 S electron ionization spectrometer (EI = 70 eV). The absorption spectra were measured on a Beckmann K25 spectrophotometer.
4.2. Materials
1,1-Diphenyl-2-picrylhydrazyl (DPPH) was obtained from Sigma. All other chemicals were of analytical grade purity. The 4-hydroxycoumarin, aromatic aldehydes and nitrogen binucleophiles were purchased from Fluka. DMF were purified, dried and distilled over CaH2 prior to use.
4-Chloro-3-coumarincarbaldehyde (3). To a stirred mixture of 4-hydroxycoumarin (9.72 g, 0.06 mol) in anhydrous DMF (46.2 mL, 0.6 mol) were added dropwise POCl3 (27.6 g, 0.18 mol) at –10° to –5 °C. The reaction mixture was then stirred for 1 h at room temperature and heated and stirred for 2 h at 60 °C. After the reaction was completed, the mixture was poured onto crushed ice (200 g) under vigorous stirring. After storing the mixture overnight at 0 °C the pale yellow solid was collected by filtration and washed successively with aquous Na2CO3 (5%) and water, and then was air–dried. 4-chlorcoumarin (2.50 g, 20%) was separated by Soxhlet extraction as the second product. Recrystallization from acetone gave 8.99 g (72%) of 2 as a pale yellow powder with mp 115–120 °C; 1H-NMR δ 10.39 (1H, s, CH=O), 8.19–7.40 (4H, m, Ar-H); 13C-NMR δ 186.81 (11-C), 158.44 (2-C), 153.28 (4-C), 153.27 (9-C),135.68 (7-C), 127.65 (5-C), 125.56 (6-C), 118.39 (10-C), 118.22 (3-C), 117.20 (8-C); IR ν 2920, 2874, 1720, 1702, 1603, 1587, 1541 cm–1; MS, m/z: 208 (MH+, 11), 182 (31), 180 (100), 154(31), 152 (91), 124 (20), 101 (11).
4.3. General Procedure for the Preparation of 3-[3-Substituted phenyl prop-2-enoyl) chromen-2-ones 4a–g
3-Acetyl-4-hydroxyxcoumarin (0.031 mol) and the appropriate substituted aromatic aldehyde (0.03 mol) were dissolved in chloroform (30 mL). A catalytic amount of piperidine (0.02 mol) was added and the reaction mixture was refluxed for 1.5 h. The chloroform was distilled out and the residue was washed with methanol.
4.4. Characterization of Newly Synthesized Coumarinic Chalcones
3-((2E)-3-(p-Tolyl)prop-2-enoyl)-2(H)-chromen-2-one (4a). Solid (yield 75%), mp = 138 °C, IR: ν 3168 (-OH), 1622 (>C=O), 1577 (C=C), 1028(s) (sym) (C-O-C); 1H-NMR: δ (ppm) 2.84 (s, 3H, CH3); 7.28 (s, 1H), 7.60 (s, 1H), 7.2–8.2 (m, 8H, Ar-H), 17.82 (s, 1H, OH); 13C-NMR (ppm): 30.0 (OCH3); 178.4 (CO); 154.6 (C4); 135.6 (C2); 101.3–135.9 (Carom). C19H14O4 calc. C 74.50, H 4.57, O 20.91, found C 74.40, H 4.60, O 20.90.
3-[(2E)-3-(4-Bromophenyl)prop-2-enoyl]-2(H)-chromen-2-one (4b). Solid (yield 85%), mp = 182 °C, IR: ν 3168 (-OH), 1622 (>C=O), 1577 (C=C), 1028(s) (sym) (C-O-C); 1H-NMR: δ (ppm) 6.26–7.84 (m, 8H, Ar-H+Héthyl), 19.58 (s, 1H, OH); 13C-NMR (ppm): 191.2 (CO); 172.8 (C4); 172.1 (C2); 102.2–168.8 (Carom+Cethyl). C18H11O4Br calc. C 58.22, H 2.96, O 17.25, found C 58.30, H 2.90, O 17.30.
3-((2E)-3-(2,3,4-Trimethoxy-phenyl)prop-2-enoyl)-2(H)-chromen-2-one (4c). Solid (yield 83%), mp = 193 °C, IR: ν 3368 (-OH), 1716(s) (>C=O), 1577 (C=C), 1018(s) (sym) (C-O-C); 1H-NMR: δ (ppm) 7.30 (s, 1H), 7.51 (s, 1H) 6.90–7.96 (m, 7H, Ar-H), 19.50 (s, 1H, OH); 13C-NMR (ppm): 191.6 (CO); 59.7 (O-CH3); 59.8 (O-CH3); 60.0 (O-CH3) 103.47–153.3 (Carom); 164.8 (C4); 163.2 (C2); 102.8 (C3). C21H18O7 calc. C 65.96, H 4.71, O 29.31; found C 65.90, H 4.80, O 29.30.
3-[(2E)-3-(Naphtyl)prop-2-enoyl]-2(H)-chromen-2-one (4d). Solid (yield 80%), mp = 185 °C, IR: ν 3365 (-OH), 1718(s) (>C=O), 1578 (C=C), 1019(s) (sym) (C-O-C); 1H-NMR: δ (ppm) 7.30 (s, 1H), 7.51 (s, 1H), 6.75–7.92 (m, 11H, Ar-H), 19.40 (s,1H, OH); 13C-NMR (ppm): 192.6(CO); 102.47–154.2 (Carom); 164.6(C4); 164.2 (C2); 101.8 (C3). C22H14O4 calc. C 77.20, H 4.10, O 18.71; found C 77.10, H 4.10, O 18.70.
3-((2E)-3-(4-Dimethylamino-phenyl)prop-2-enoyl)-2(H)-chromen-2-one (4e). Solid (yield 85%), mp = 192 °C, IR: ν 3125 (-OH), 1712(s) (>C=O), 1550 (C=C), 1028 (sym) (C-O-C); 1H-NMR: δ (ppm) 2.9 (s, 6H, CH3), 6.54–8.1 (m, 10H, Ar-H+Héthyl), 18.62 (s, 1H, OH); 13C-NMR (ppm): 191.0 (CO); 40.1 (CH3-N); 102.8–142.9 (Carom); 179.2 (C4); 162.2 (C2); 102.8 (C3). C20H17O4N calc. C 71.64, H 5.1, O 19.10, N 4.17; found C 71.60, H 5.10, O 19.30, N 4.20.
4.5. Synthesis of Aryl(alkyl)chromeno[4,3-c]pyrazol-4(1H)-one 5
The aldehyde 3 (0.41 g, 2 mmol) dissolved on boiling in ethanol (10 mL) and was then cooled to 15–20 °C. A solution prepared from aryl or alkylhydrazine hydrochloride (0.29 g, 2 mmol) and triethylamine (4 mmol) in 60%) ethanol (10 mL) was slowly added dropwise so that the temperature of the reaction mixture did not exceed 25 °C. After addition of the reagent was complete a yellowish precipitate formed rapidly. The precipitate was filtered off and recrystallized from ethanol and then twice more from DMF to give fine colourless needles
1H-Chromeno[4,3-c]pyrazol-4-one (5a). Solid (Yiled 67%), mp = 183 °C; IR: ν 3368 (-NH), 1716(s) (lactone >C=O), 1577 (C=C), 1018(s) (sym) (C-O-C); 1H-NMR: δ (ppm) 7.40 (s, 1H, Héthy), 7.13–7.46 (m, 4H, Ar-H), 13.2 (s, 1H, NH); 13C-NMR (ppm): 158.2 (C2); 102.1 (C3); 149.2 (C4); 146.7 (C=N); 122.1–151.2 (Carom). C10H5O2N2 calc. C 64.86, H 2.70, O 17.30, N 15.13; found C 64.80, H 2.70, O 17.20, N 15.2.
1-Methyl-1H-chromeno[4,3-c]pyrazol-4-one (5b). Solid(Yiled 75%), mp = 184 °C; IR: ν 1720(s) (lactone >C=O), 1578 (C=C), 1020(s) (sym) (C-O-C); 1H-NMR: δ (ppm). 3.5 (s, 3H, CH3), 6.4 (s, 1H, Héthy); 7.13–7.45 (m, Ar-H), 13C-NMR (ppm): 157.2 (C2); 112.2 (C3); 144.6 (C4); 140.6 (Céthyl); 122.1–151.2 (Carom). C11H7O2N2 calc. C 60.53, H 6.27, O 33.20; found C 60.50, H 6.30, O 33.20.
1-Phenyl-1H-chromeno[4,3-c]pyrazol-4-one (5c). Solid (Yiled 89%), mp = 185 °C; IR: ν 1718(s) (lactone >C=O), 1579 (C=C), 1022(s) (sym) (C-O-C); 1H-NMR: δ (ppm): 7.13–7.45 (m, Ar-H), 7.2 (Héthy) 13C-NMR (ppm): 158.2 (C2); 112.4 (C3); 144.2 (C4); 138.6 (Céthyl); 122.1–151.2 (Carom). C16H10O2N2 calc. C 58.89, H 3.06, O 9.81, N 8.58; found, C 58.90, H 3.10, O 9.80, N 8.60.
4.6. The Experimental Procedure for the Antioxidant Evaluation
The DPPH radical scavenging capacity of the obtained coumarinic derivatives was measured from the bleaching of purple colored ethanol solution of DPPH. The method described by Hatano et al. [32] was used. Half a millilitre of each sample concentration was mixed with an equal volume of DPPH ethanolic solution. After incubation for 30 minutes in darkness at 25 °C, absorbance was read at 520 nm wavelength. A mixture of DPPH solution (0.5 mL) and ethanol (0.5 mL) was taken as a blank (absorbance is equal to zero). Inhibitory concentration (IC50) values denoting the concentration (microgram of natural substance per milliliter of ethanol) required to scavenge 50% of DPPH radicals were calculated. All measurements were performed in triplicate. Results were expressed in inhibition percentage versus sample concentrations (mg/mL) at 30 minutes.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}