3.1. General
NMR spectra were recorded on Bruker Avance DPX 300 or DPX 400 instruments. Chemical shifts are reported in ppm using TMS (d = 0.0) as the internal standard in CDCl
3 or relative to 2.50 ppm for
1H and 39.99 ppm for
13C in [
d6-DMSO] or 3.31 ppm for
1H and 49.15 ppm for
13C in CD
3OD. Structural assignments were based on
1H,
13C, DEPT135 and 2D-spetra, COSY, HSQC, HMBC, and NOESY. EI-Mass and ESI spectra were recorded on a Finnigan MAT 95XL spectrometer. IR spectra were obtained on a Thermo Nicolet FT-IR Nexus spectrometer using a Smart Endurance reflection cell. For ozonolysis was used an OZ-500 Ozone Generator produced by Fishcer Technology. Silica gel Kieselgel 60G (Merck) was used for Flash Chromatography. The solvents were purified by standard methods. All reactions were carried out in inert atmospheres (nitrogen). The synthesis of compounds
1a and
14a has been described elsewhere [
7].
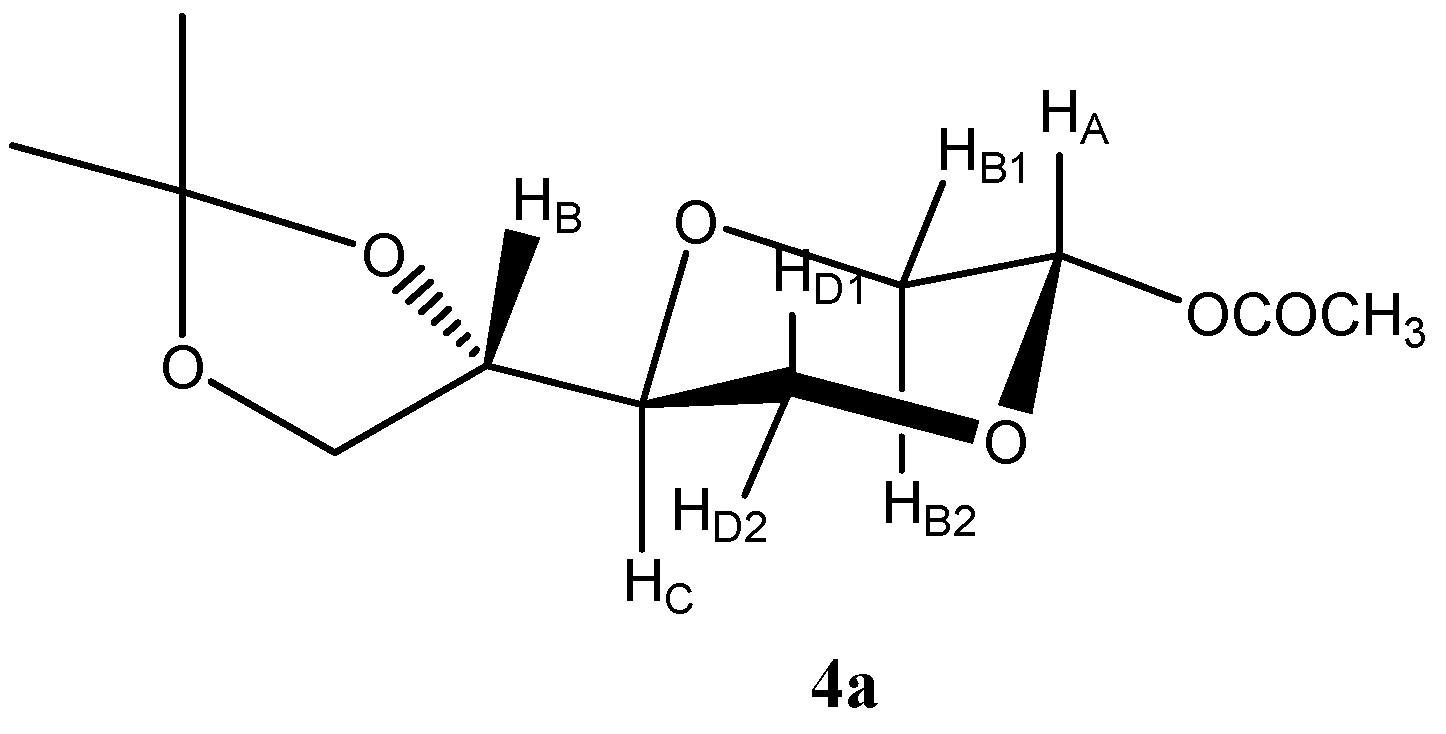
(2S,5S)-5-[(4S)-2,2-Dimethyl-1,3-dioxolan-4-yl]-2-acetyloxy-1,4-dioxane (
4a)
and (2R,5S)-5-[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]-2-acetyloxy-1,4-dioxane (
4b). To a solution of
3 (1.0 g, 4.9 mmol) in dry pyridine (15 mL) was added acetic anhydride (0.62 g, 6 mmol) at 0–5 °C and the reaction mixture was stirred for 6 hours. The solution was concentrated under reduced
in vacuo overnight, yielding the crude product in 87% yield as an oily solid material, which was used in the subsequent reaction step without further purification. The anomeric ratio
4a:
4b (
trans-
cis ratio) was determined to be 4:1 by NMR. The products exhibited the following spectroscopic properties: The
trans-product
4a (
Figure 1) was assigned the following signals:
1H-NMR (CDCl
3, 400 MHz): δ 1.36, 1.43 (s, 2 × 3H, (CH
3)
2C), 2.11 (s, 3H, CH
3COO), 3.48 (dd,
J = 8.0 Hz, 11.4 Hz, 1H, H
B1), 3.63 (m, 1H, H
C), 3.69 (dd,
J = 9.4 Hz, 11.4 Hz, 1H, H
D1), 3.81 (dd,
J = 6.8 Hz, 8.0 Hz, 1H, (CH
3)
2C-O-CH
2), 3.89 (dd,
J = 2.6 Hz, 11.4 Hz, 1H, H
D2), 3.93 (dd,
J = 2.8 Hz, 11.4 Hz, 1H, H
B2), 4.00 (dd,
J = 6.8 Hz, 8.4 Hz, 1H, (CH
3)
2C-O-CH
2), 4.15 (m, 1H, (CH
3)
2C-O-CH-), 5.74 (dd,
J = 2.8 Hz, 8.4 Hz, 1H, H
A) ppm.
13C-NMR (CDCl
3, 100 MHz): δ 20.9, 25.2, 26.3, 65.2, 65.6, 66.9, 74.2, 74.2, 89.4, 109.7, 169.0 ppm. The protons NMR spectrum of the corresponding
cis- compound,
4b, could not be fully assigned due to the peaks overlap with
4a, however, the carbon NMR spectrum of the
cis- compound was assigned the following signals:
13C-NMR (CDCl
3, 100 MHz): δ 21.1, 25.2, 26.2, 61.0, 64.9, 67.6, 74.9, 75.1, 88.4, 109.7, 169.8 ppm. The mixture exhibited the following mass spectrum: MS (EI)
m/z: 247 (M
++1), 231(M
+-CH
3), 187(M
+-OAc), 145 (C
6H
9O
4). Elem. Anal. calcd. for C
11H
18O
6: C 53.65, H 7.37; found C, 53.84, H 7.45.
Figure 1.
Structure of compound 4a.
Figure 1.
Structure of compound 4a.
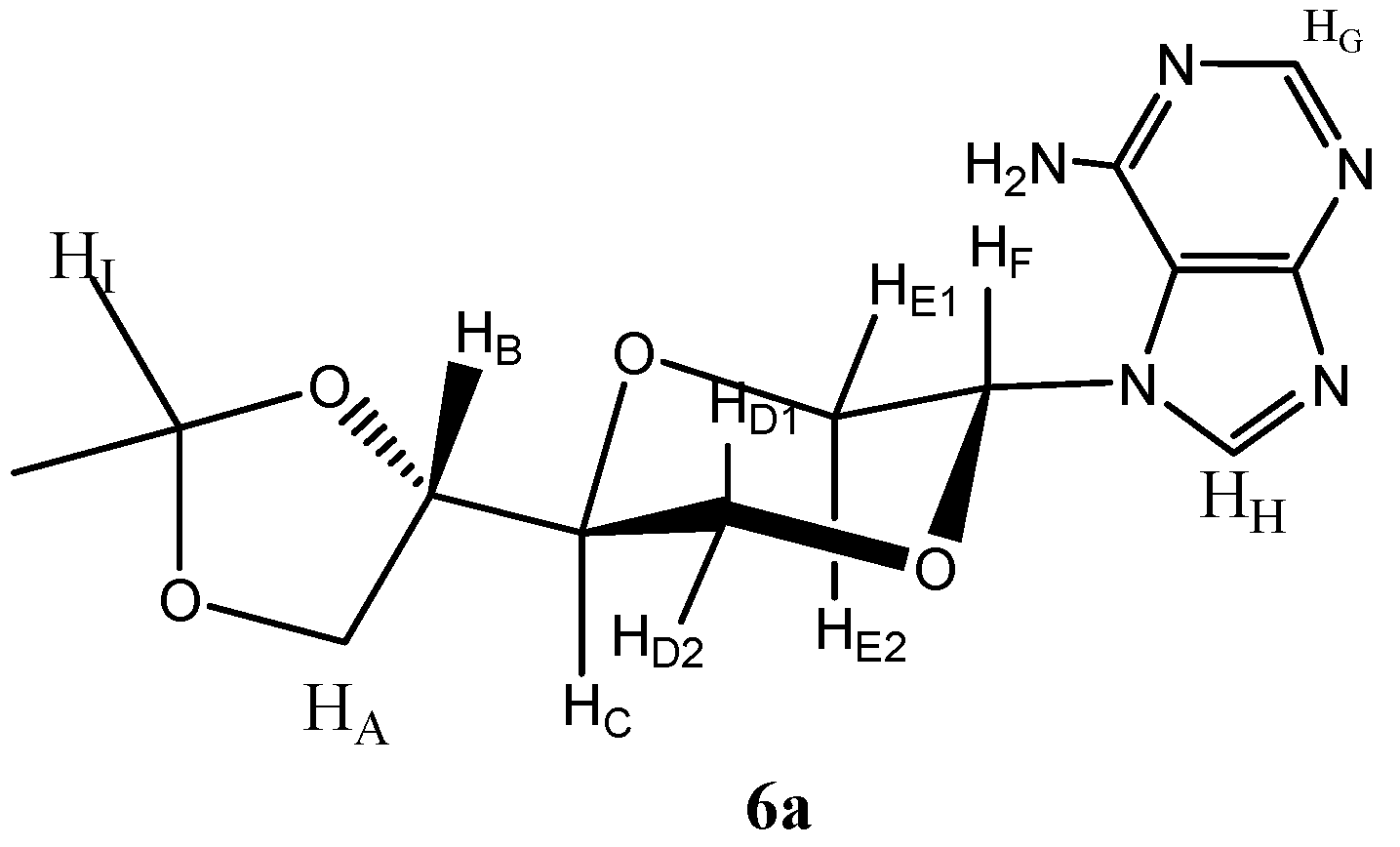
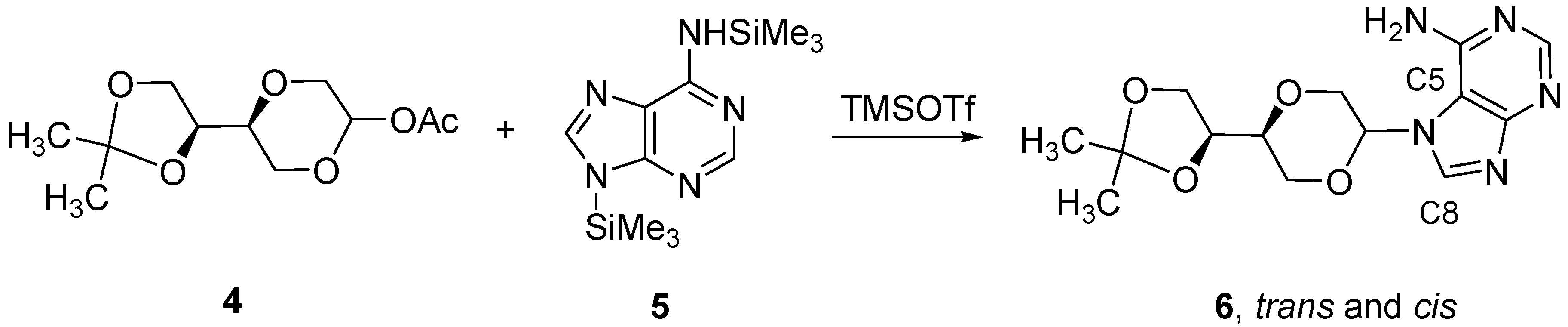
N-7-{(2R,5S)-5-[(4S)-(2,2-Dimethyl-1,3-dioxolan-4-yl)]-1,4-dioxan-2-yl}adenine (
6a)
and N-7-{(2S,5S)-5-[(4S)-(2,2-dimethyl-1,3-dioxolan-4-yl)]-1,4-dioxan-2-yl}adenine (
6b). The mixture of adenine (1.35 g, 10 mmol) and ammonium sulfate (124 mg, 0.9 mmol) in hexamethyldisilazane (HMDS, 35 mL) was refluxed overnight. The solvent was evaporated and the residue was dissolved in dry dichloroethane (20 mL). To this solution, compound
4 (0.85 g, 3.5 mmol) was added. The solution was cooled to 0 °C and TMSOTf (0.75 mL, 4 mmol) was added. The solution was stirred for 8 hours at room temperature. The chloroform (50 mL) was added and the solution was washed twice with saturated NaHCO
3 solution (15 mL). The aqueous phase was extracted twice with chloroform (50 mL), and the combined organic layers was dried over anhydrous MgSO
4 and filtered and concentrated under reduced pressure. The residue was purified by flash chromatography using a mixture of dichloromethane and methanol (19:1) as the eluent. A white solid (0.26 g, 23%) was obtained which was identified as a 3:2 mixture of the
trans- and
cis- products
6a and
6b. From the mixture of isomers was extracted the following spectroscopic properties for isomer
6a (
Figure 2):
1H-NMR (CDCl
3, 400 MHz): δ 1.47, 1.48 (s, 2 × 3H, H
I), 3.87–3.91 (m, 3H, H
B), 3.97–4.03 (m, 3H, H
A and H
E), 4.04 (dd,
J = 12 Hz, 6 Hz, 1H, H
D2), 4.07 (dd,
J = 8.4 Hz, 6.8 Hz, 1H, H
A), 4.21–4.27 (m, 2H, H
C and H
D), 5.72 (dd,
J = 8.8 Hz, 4 Hz, H
F), 5.99 (br.s, 2H, NH
2), 8.11 (s, 1H, H
G), 8.54 (s, 1H, H
H) ppm.
13C-NMR (CDCl
3, 100 MHz): δ 25.2, 26.4, 66.1, 68.5, 60.4, 74.1, 82.0, 110.3, 111.1, 144.0, 151.3, 154.0, 161.6 ppm. For the mixture of
6a and
6b: IR (neat): 3418, 3290, 3149, 2979, 2905, 1625, 1595, 1062 cm
-1. HRMS (ESI): Calcd. for C
14H
19N
5O
4 [M+Na]
+ 322.1516, Found 322.1513.
Figure 2.
Structure of compound 6a.
Figure 2.
Structure of compound 6a.
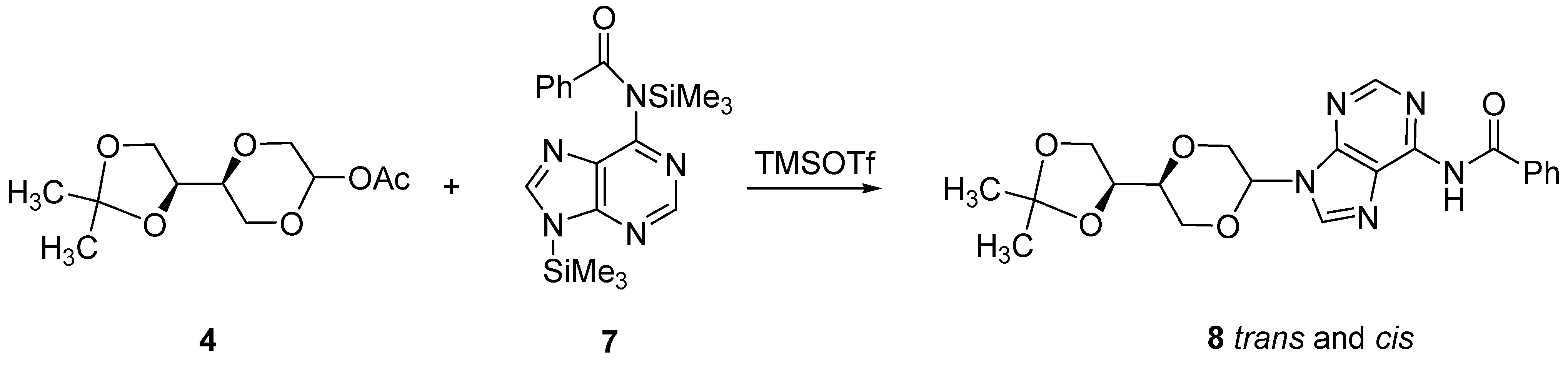
N-6-Benzoyl-N-9-{(2R,5S)-5-[(4S)-(2,2-dimethyl-1,3-dioxolan-4-yl)]-1,4-dioxan-2-yl} adenine (
8a)
and N-6-benzoyl-N-9-{(2S,5S)-5-[(4S)-(2,2-dimethyl-1,3-dioxolan-4-yl)]-1,4-dioxan-2-yl}-adenine (
8b). N6-benzoyladenine (2.065 g, 8.7 mmol) and ammonium sulfate (120 mg, 0.9 mmol) in HMDS (50 mL) was refluxed overnight. The solution was cooled and concentrated under reduced pressure. The residue was dissolved in dry acetonitrile (20 mL) and added a solution of
6 (0.77 g, 3.1 mmol) in dry acetonitrile (5 mL). The mixture was then cooled to 0 °C and added TMSOTf (0.6 mL, 3.3 mmol). The mixture was stirred for four hours. Then chloroform (50 mL) was added and the solution washed twice with saturated NaHCO
3 solution (20 mL). The organic phase was dried over anhydrous MgSO
4 filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography using a gradient eluent system, first diethyl ether, followed by a mixture of dichloromethane and methanol (19:1) to afford the product as a yellow solid (0.55 g, 42%) as a 3:1 mixture of
trans- and
cis products
8a and
8b. Pure
8a was obtained by repeated chromatography. Product
8a (
Figure 3) exhibited the following spectroscopic properties:
1H-NMR (CDCl
3, 400 MHz): δ 1.41, 1.52 (s, 2 × 3H, H
G), 3.48 (dd,
J = 11.4 Hz, 9.4 Hz, 1H, H
E2), 3.79–3.84 (m, 1H, H
C), 3.94–3.97 (m, 1H, H
A), 4.00 (dd,
J = 11.6 Hz, 10.8 Hz, 1H, H
D1), 4.09 (dd,
J = 8.4 Hz, 6.8 Hz, 1H, H
A), 4.17 (dd,
J = 11.6 Hz, 2.6 Hz, 1H, H
D2), 4.20–4.25 (m, 1H, H
B), 4.54 (dd,
J = 11.4 Hz, 2.6 Hz, H
E1), 6.64 (dd,
J = 9.2 Hz, 2.8 Hz, 1H, H
F), 7.43–7.49 (m, 2H, H
K), 7.50–7.54 (m, 1H, H
L), 8.23–8.28 (m, 2H, H
J) ppm.
13C-NMR (CDCl
3, 100 MHz): δ 25.4, 26.4, 62.8, 65.0, 66.6, 74.1, 74.4, 81.9, 110.3, 115.2, 128.4, 130.1, 132.4, 137.5, 142.1, 144.9, 149.3, 157.2, 175.6 ppm. For the mixture of
8a and
8b: IR (neat): 3214, 2985, 1635, 1597, 1480, 1116, 1067, 1048 cm
−1. HRMS (ESI): Calcd. for C
21H
23N
5O
5 [M+H]
+ 426.1778, Found 426.1776.
Figure 3.
Structure of compound 8a.
Figure 3.
Structure of compound 8a.
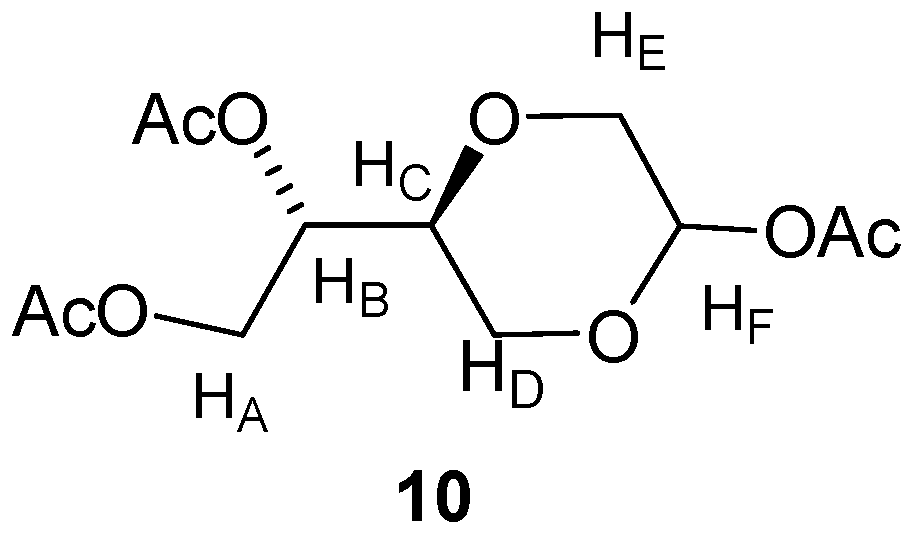
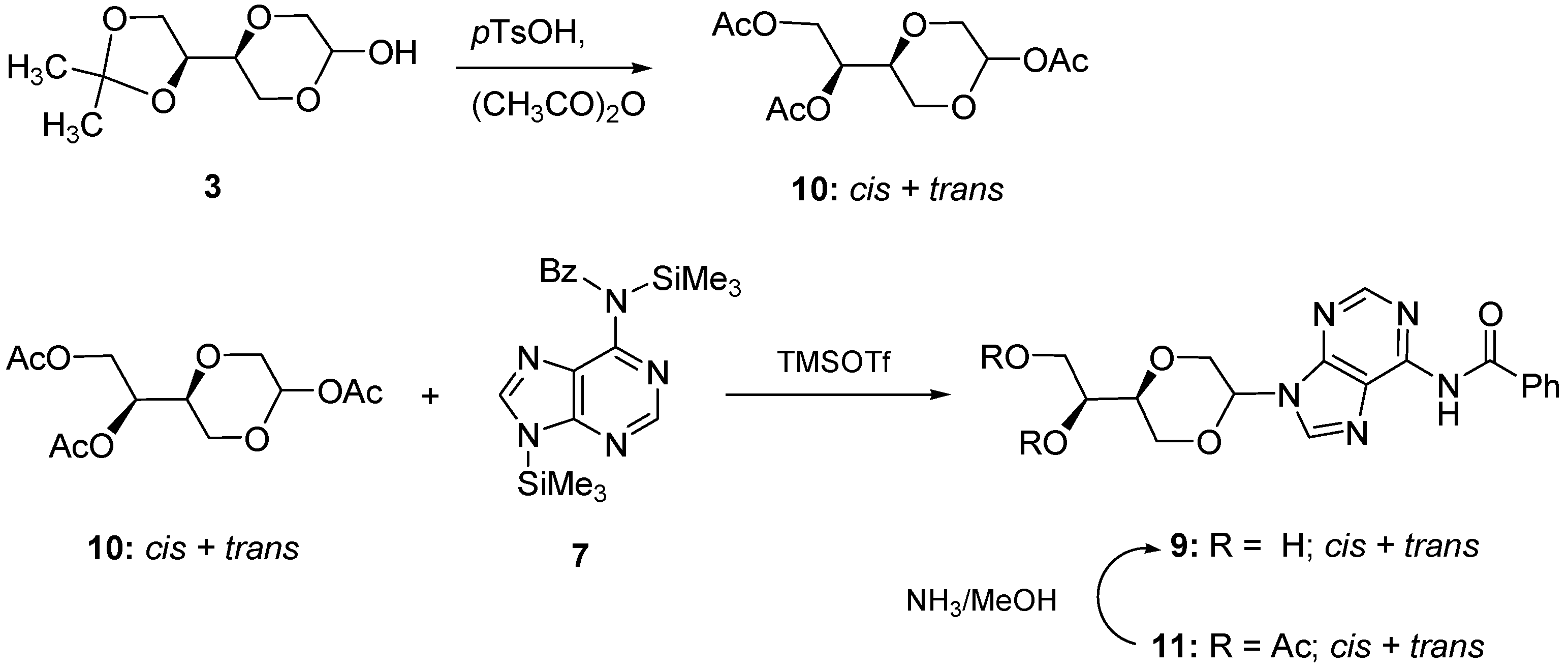
(2R,5S)-5-[(1S)-(1,2-Diacetyloxy)]-2-acetyloxy-1,4-dioxane (
10a)
and (2S,5S)-5-[(1S)-(1,2-diacetyl-oxy)]-2-acetyloxy-1,4-dioxane (
10b). Compound
3 (2.2 g, 10.8 mmol) and
p-TsOH (2.26 g, 11.9 mmol) was dissolved in acetic anhydride (50 mL). The mixture was stirred for 7 hours at room temperature and then poured into ice water. To the mixture was neutralized with sodium bicarbonate, and then extracted with ethyl acetate. The organic phase was dried over anhydrous MgSO
4 filtration and evaporated under reduced pressure. The residue was purified by flash chromatography using a mixture of diethyl ether and
n-hexane (2:1) as the eluent. The product appeared as yellow oil, (2.35 g, 75%). The diastereomers were not separated, however the NMR of the one diastereomer (
Figure 4) was assigned the NMR signals:
1H-NMR (CDCl
3, 400 MHz): δ 2.07, 2.14, 2.15 (s, 3 × 3H, CH
3), 3.59–3.61(m, 1H, H
D), 3.83 (dd,
J = 12.8 Hz, 2H, 1H, H
E), 3.89–3.98 (m, 3H, H
E, H
D and H
C), 4.16 (dd,
J = 12 Hz, 6.8 Hz, 1H, H
A), 4.36 (dd,
J = 11.8 Hz, 4.2 Hz, 1H, H
A), 5.15–5.18 (m, 1H, H
B), 5.87 (d, 2Hz, 1H, H
F) ppm.
13C-NMR (CDCl
3, 100 MHz): δ 20.7, 20.8, 21.1, 61.2, 62.1, 68.1, 69.8, 73.6, 88.2, 169.8, 170.2, 170.5 ppm. The NMR data for the other diastereomer were the following:
1H-NMR (CDCl
3, 400 MHz): δ 2.10, 2.11, 2.16 (s, 3 × 3H, CH
3), 3.78–3.82(m, 1H, H
C), 3.83 (dd, 12.8Hz, 2Hz, 1H, H
E), 3.93 (d, 12.8Hz, 1H, H
E), 4.13–4.26 (m, 5H, H
B, H
D and H
A), 5.87 (d, 2Hz, 1H, H
F) ppm.
13C-NMR (CDCl
3, 100 MHz): δ 20.7, 20.8, 21.4, 63.1, 63.4, 67.6, 68.8, 74.2, 89.0, 169.7, 170.5, 170.6 ppm. IR (neat): 2959, 1737, 1219, 1044, 1014 cm
−1. MS (m/z): 289.3, 243.3, 231.3, 217.3.
Figure 4.
Structure of compound 10.
Figure 4.
Structure of compound 10.
N-6-Benzoyl-N-9-{(2R,5S)-5-[(1S)-(1,2-diacetyloxy)]-1,4-dioxan-2-yl}adenine (11a) and N-6-benzoyl-N-9-{(2S,5S)-5-[(1S)-(1,2-diacetyloxy)]-1,4-dioxan-2-yl}adenine (11b). N-6-benzoyladenine (1.09 g, 4.6 mmol) and ammonium sulfate (62 mg, 0.47 mmol) in HMDS (50 mL) was refluxed overnight. The mixture containing the silylated N-6-benzoyladenine 7 was concentrated under reduced pressure, dissolved in dry dichloroethane (24 mL) and then added a solution of 10 (0.74 g, 2.6 mmol) in dry dichloromethane (6 mL). This mixture was added TMSOTf (0.94 mL, 5.2 mmol) at 0 °C and stirred overnight at room temperature. Chloroform (50 mL) was added to the mixture which was washed with saturated NaHCO3 solution (25 mL). The aqueous phase was extracted with chloroform (50 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered and concentrated under reduced pressure. The residue was purified by flash chromatography using a mixture of dichloromethane and methanol (9/1) as the eluent to afford the product (0.67 g, 56%) as a mixture of the trans and cis products, 11a and 11b. The product was used for the next reaction without further purification. The NMR was too complex to allow reasonable assignments of the signals. IR (neat): 2958, 1737, 1636, 1216.1045, 1023 cm−1. HRMS (ESI): Calcd. for C22H23N5O7 [M+H]+ 470.1677, Found 470.1663.
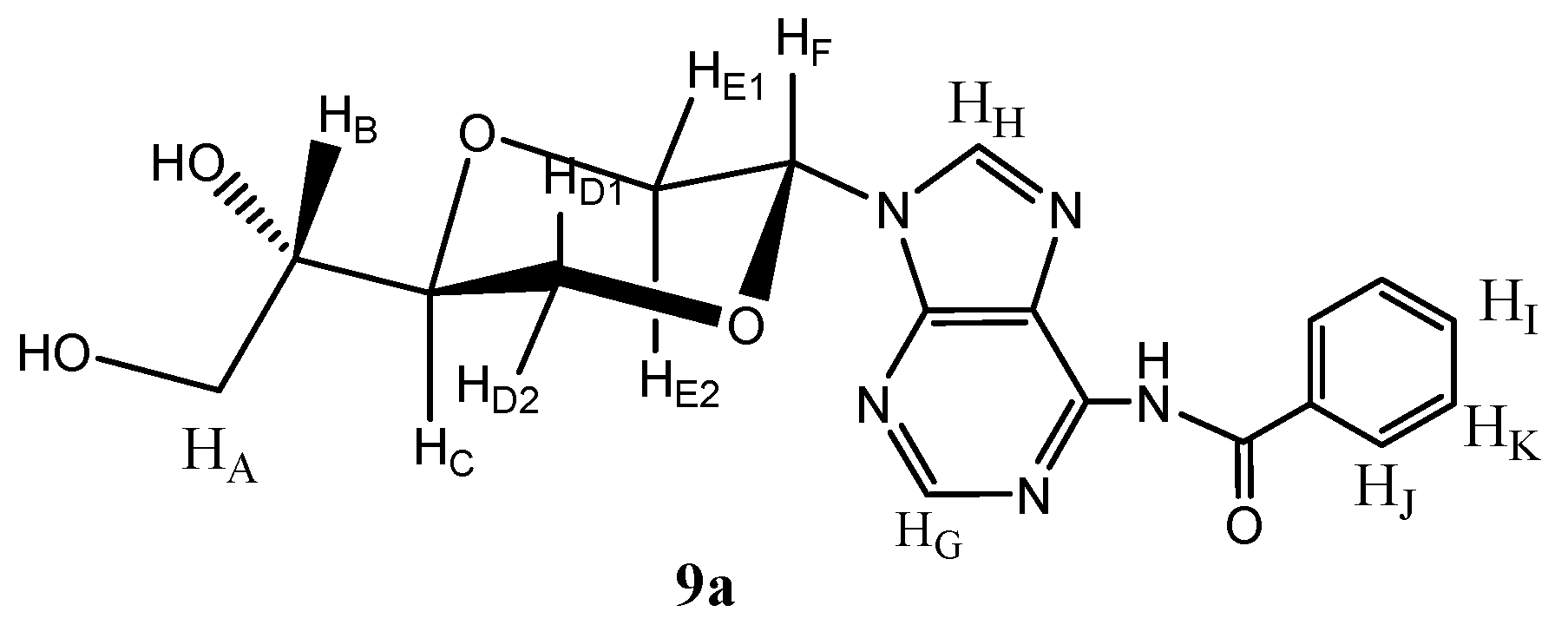
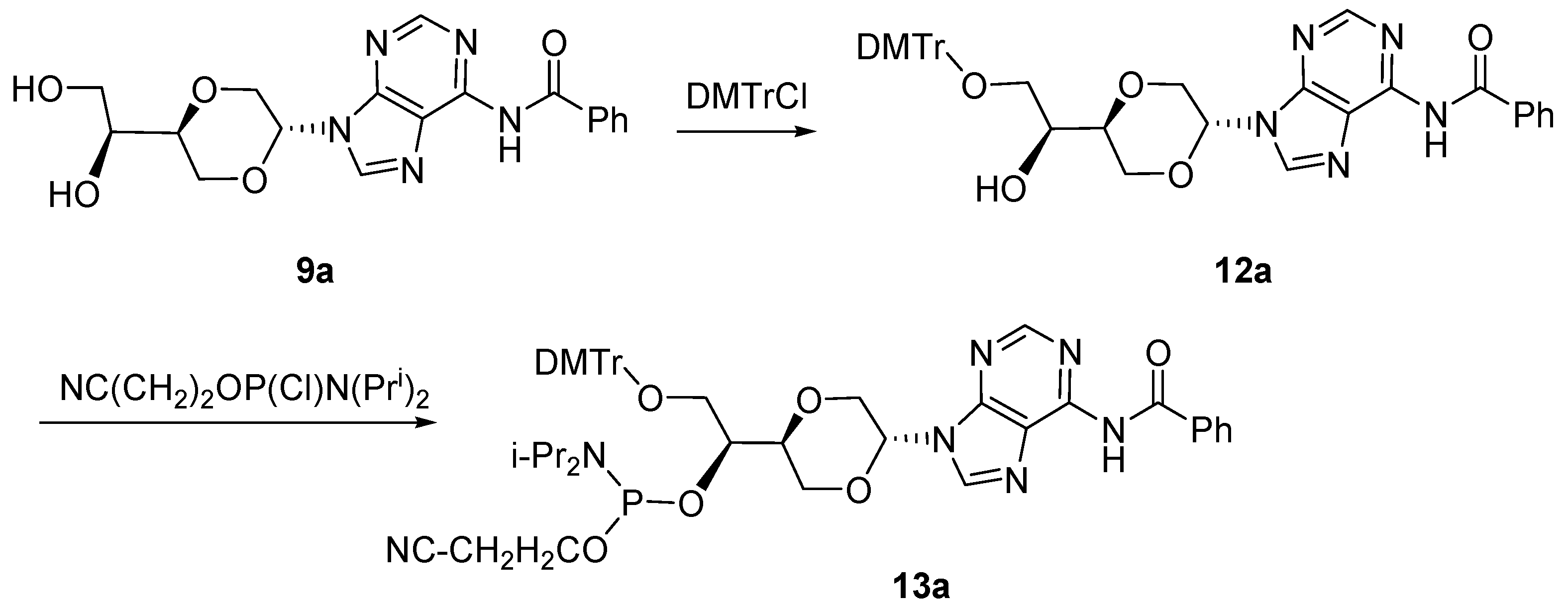
N-6-Benzoyl-N-9-{(2R,5S)-5-[(1S)-(1,2-dihydroxyl)]-1,4-dioxan-2-yl}adenine (
9a)
and N-6-benzoyl-N-9-{(2S,5S)-5-[(1S)-(1,2-dihydroxyl)]-1,4-dioxan-2-yl}adenine (
9b). Compound
11 (1.27 g, 30 mL) was dissolved in 7N ammonia in methanol (30 mL) and stirred for 7 hours at room temperature. The solution was concentrated and purified by flash chromatography using a mixture of dichloromethane and methanol (19/1) as the eluent. The pure
trans- product
9a was isolated (140 mg, 13.5%,
Figure 5). Product
9a exhibited the following spectroscopic properties:
1H-NMR (CD
3OD, 400 MHz): δ 3.59 (dd,
J = 11.2 Hz, 9.4 Hz, 1H, H
E2), 3.60–3.70 (m, 3H, H
A and H
B), 3.89 (dt,
J = 10.6 Hz, 2.8 Hz, 1H, H
C), 4.13 (dd,
J = 11.8, 10.6 Hz, 1H, H
D1), 4.24 (dd,
J = 11.8, 2.8 Hz, 1H, H
D2), 4.37 (dd,
J = 11.2 Hz, 2.6 Hz, 1H, H
E1), 6.55 (dd,
J = 9.4, 2.6 Hz, 1H, H
F), 7.41–7.45 (m, 1H, H
I), 7.49–7.53 (m, 2H, H
K), 8.21–8.24 (m, 1H, H
C), 8.24 (s, 1H, H
G), 8.67 (s, 1H, H
H) ppm.
13C-NMR (CD
3OD, 100 MHz): δ 63.8, 70.4, 70.8, 72.1, 76.3, 82.6, 115.5, 129.3, 131.0, 133.4, 138.2, 144.2, 145.2, 149.7, 156.9, 176.6ppm. IR (neat): 3286, 2876, 1635, 1424, 1285, 1115, 1063, 1021cm
-1. HRMS (ESI): Calcd. for C
18H
19N
5O
5 [M+H]
+ 470.1677, Found 470.1668.
Figure 5.
Structure of compound 9a.
Figure 5.
Structure of compound 9a.
N-6-Benzoyl-N-9-{(2S,5S)-5-[(1S)-hydroxyl-2-O-(4,4-dimethoxytrityl)-ethyl-1-yl]-1,4-dioxan-2-yl}-adenine (
12). Compound
9 (mixture of
9a and
9b, 139 mg, 0.36 mmol) and dimethoxyltrityl chloride (277 mg, 0.79 mmol) were dissolved in dry pyridine (10 mL). The solution was stirred at room temperature for 3 hours. The solution was concentrated
in vacuo and residue was purified by flash chromatography using first diethyl ether and then ethyl acetate as the eluent to provide the product (
12a and
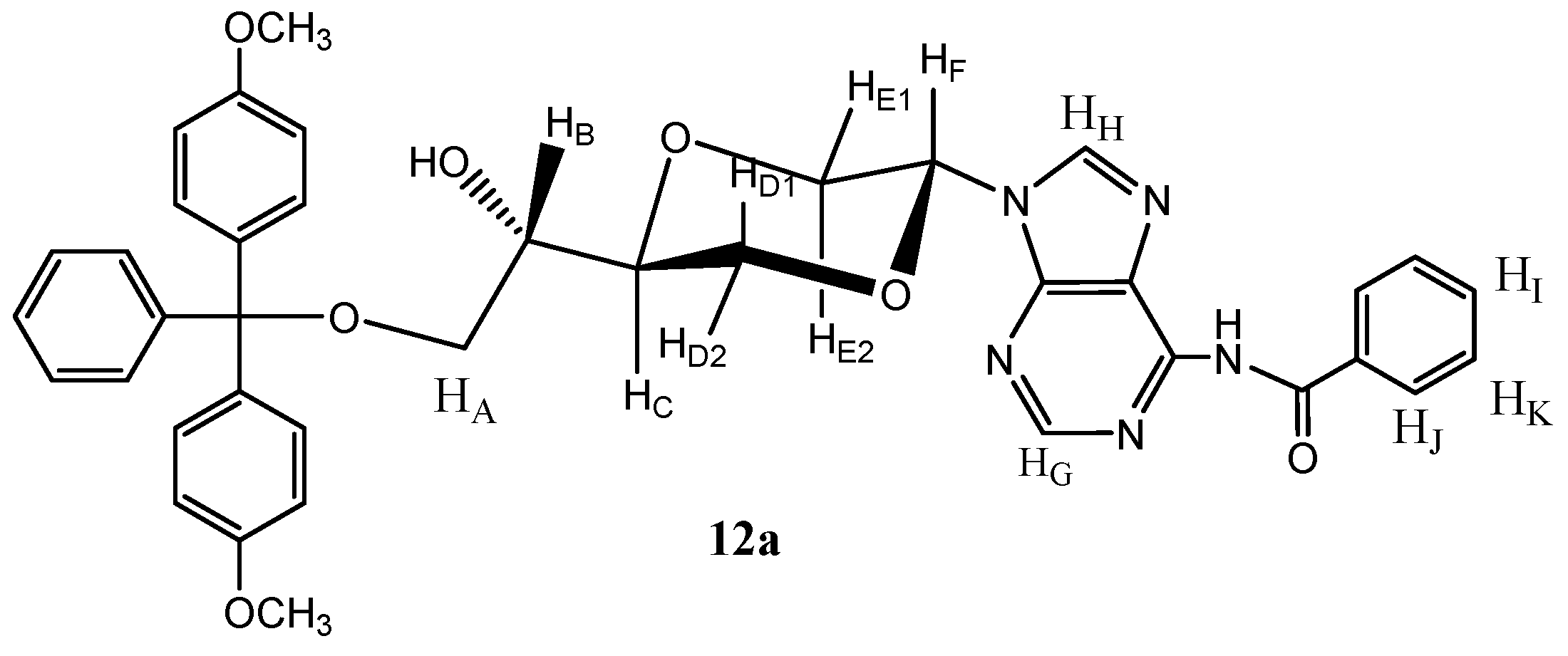
12b) as a white solid (100 mg, 40%). Product
12a (
Figure 6) was obtained upon repeated chromatography and exhibited the following spectroscopic properties:
1H-NMR (CDCl
3, 400 MHz): δ 2.82 (d,
J = 4.8 Hz, 1H, OH), 3.25 (dd,
J = 9.6 Hz, 5.4 Hz, 1H, H
A), 3.34 (dd,
J = 9.6 Hz, 5.6 Hz, H
A), 3.43 (dd,
J = 11.2 Hz, 9.2 Hz, H
E2), 3.75–3.82 (m, 1H, H
B), 3.77 (s, 2 × 3H, OCH
3), 3.86–3.90 (m, 1H, H
C), 4.00 (dd,
J = 12 Hz, 10.2 Hz, H
D1), 4.13 (dd,
J = 12 Hz, 2.8 Hz, H
D2), 4.43 (dd,
J = 11.2 Hz, 2.8 Hz, H
E), 6.60 (dd,
J = 9.2 Hz, 2.8 Hz, H
F), 6.82–6.86 (m, 5H, aromatic protons), 7.20–7.24 (m, 1H, aromatic proton), 7.26–7.37 (m, 5H, aromatic protons), 7.40–7.53 (m, 5H, aromatic protons), 8.13 (s, 1H, H
G), 8.15–8.25 (m, 2H, aromatic protons), 8.64 (s, 1H, H
H) ppm.
13C-NMR (CDCl
3, 100 MHz): δ 55.2, 63.8, 69.3, 69.4, 70.0, 75.5, 81.0, 86.5, 113.2, 126.9, 129.8, 130.0, 132.3, 135.7, 135.8, 137.1, 142.1, 143.0, 144.6, 148.4, 157.1, 158.6, 175.4 ppm. HRMS (ESI): Calcd. for C
39H
37N
5O
7 [M+Na]
+ 710.2591, Found 710.2588.
Figure 6.
Structure of compound 12a.
Figure 6.
Structure of compound 12a.
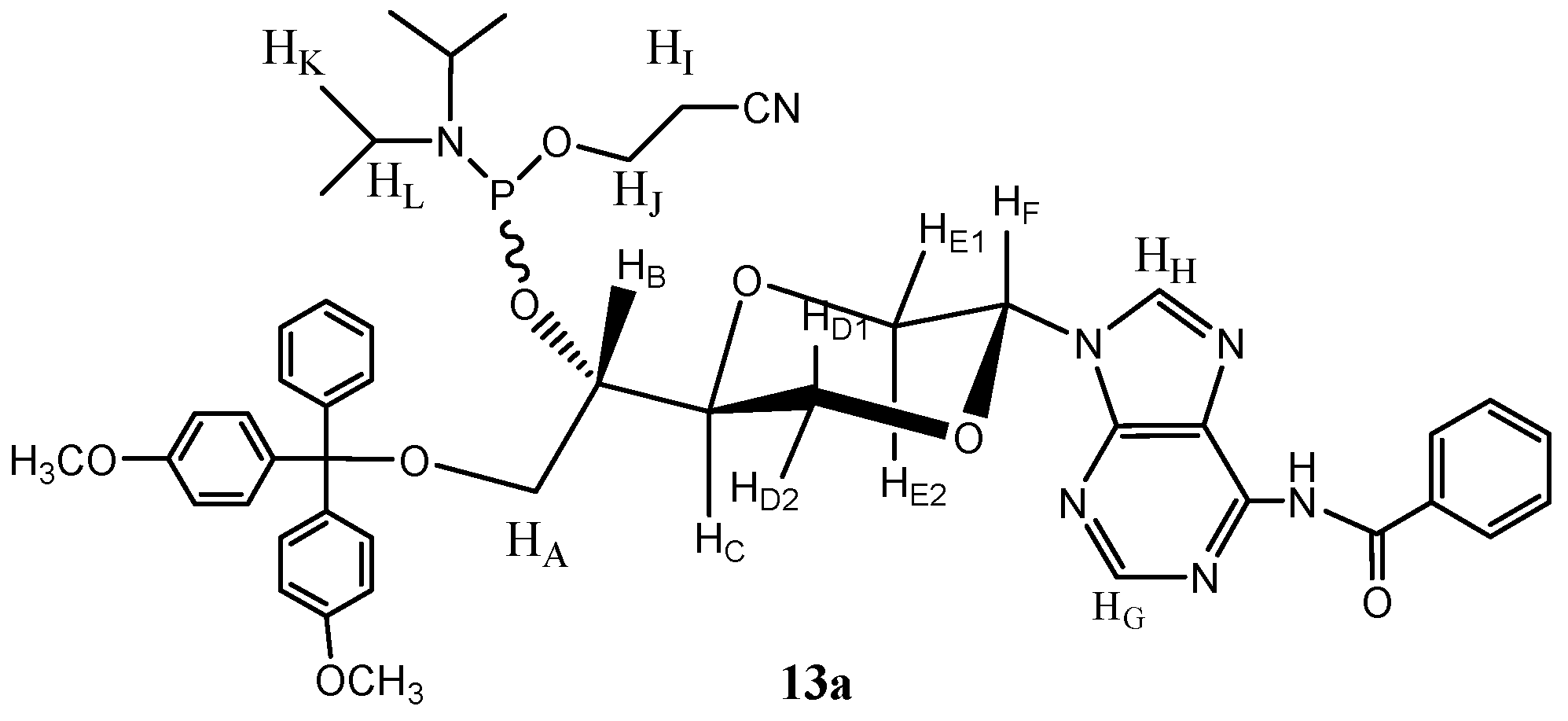
N-6-Benzoyl-N-9-{(2S,5S)-5-{(1S)-O-[2-cyanoethoxy(diisopropylamino)phosphino]-2-O-(4,4′-di-methoxytrityl)-ethyl-1-yl}-1,4-dioxan-2-yl}adenine (
13a). Compound
12 (mixture of
12a and
12b) (99 mg, 0.14 mmol) was dissolved in dry dicholormethane (6 mL) was then added
N,N-diisopropyl-ethylamine (50 µL, 0.29 mmol). Then
N,N-diisopropyl-2-cyanoethyl phosphoramidic chloride (105 µL, 0.47 mmol) was added. The resulting solution was stirred for 6 hours at room temperature. The solution was then concentrated and purified by flash chromatography using a mixture of ethyl acetate and n-hexane (4:1) as the eluent to give the
trans isomer
13a (65 mg, 51%) as a mixture of diastereomers due to the stereogenic phosphorus. Major diastereomer
13a (
Figure 7) had the following spectroscopic properties:
1H-NMR (CDCl
3, 400 MHz): δ 1.14 (d,
J = 7.2 Hz, 2 × 3H, H
K), 1.19 (d,
J = 7.2 Hz, 2 × 3H, H
K), 2.64 (dt,
J = 6.4, 2.4 Hz, 2H, H
I), 3.27 (dd,
J = 9.4, 5.2 Hz, 1H, H
A), 3.36 (dd,
J = 9.4, 6 Hz, 1H, H
A), 3.45 (dd,
J = 11.2, 9.4 Hz, 1H, H
E2), 3.56–3.70 (m, 2 × 1H, H
L), 3.771, 3.768 (s, 2 × 3H, OCH
3), 3.72–3.82 (m, 1H, H
J), 3.87–3.95 (m, 1H, H
J), 4.00–4.09 (m, 3 × 1H, H
B, H
D, H
C), 4.24 (d,
J = 10 Hz, 1H, H
D), 4.40 (dd,
J = 11.2, 2.6 Hz, 1H, H
E1), 6.61 (dd,
J = 9.4, 2.6 Hz, 1H, H
F), 6.81–6.85 (m, 4H, aromatic H), 7.19–7.22 (m, 1H, aromatic H), 7.27–7.30 (m, 2H, aromatic H), 7.34–7.36 (m, 4H, aromatic H), 7.41–7.48 (m, 4H, aromatic H), 7.52–7.56 (m, 4H, aromatic H), 8.14 (s, 1H, H
G), 8.22–8.25 (m, 2H, aromatic H), 8.69 (s, 1H, H
H), 12.59 (s, 1H, NH) ppm.
13C-NMR (CDCl
3, 100 MHz): δ 20.4, 20.5, 24.56, 24.64, 24.67, 24.74, 43.2, 43.4, 55.2, 58.0, 58.2, 60.4 ,69.55, 69.62, 72.1, 75.3, 75.4, 81.3, 86.4, 113.1, 114.4, 117.7, 126.8, 127.8, 128.2, 128.3, 129.9, 130.05, 130.09, 132.3, 135.95, 136.01, 137.2, 141.8, 143.3, 144.8, 148.4, 157.2, 158.5, 175.5 ppm.
31P-NMR (CDCl
3): 151.0, 152.0 (small) ppm. IR (neat): 3239, 3056, 2965, 2929, 2362, 2338, 1637, 1507, 1251, 1083, 788, 754, 719 cm
−1. HRMS (ESI): Calcd. for C
48H
54N
7O
8P [M+Na]
+ 910.3669, Found 910.3639.
Figure 7.
Structure of compound 13a.
Figure 7.
Structure of compound 13a.
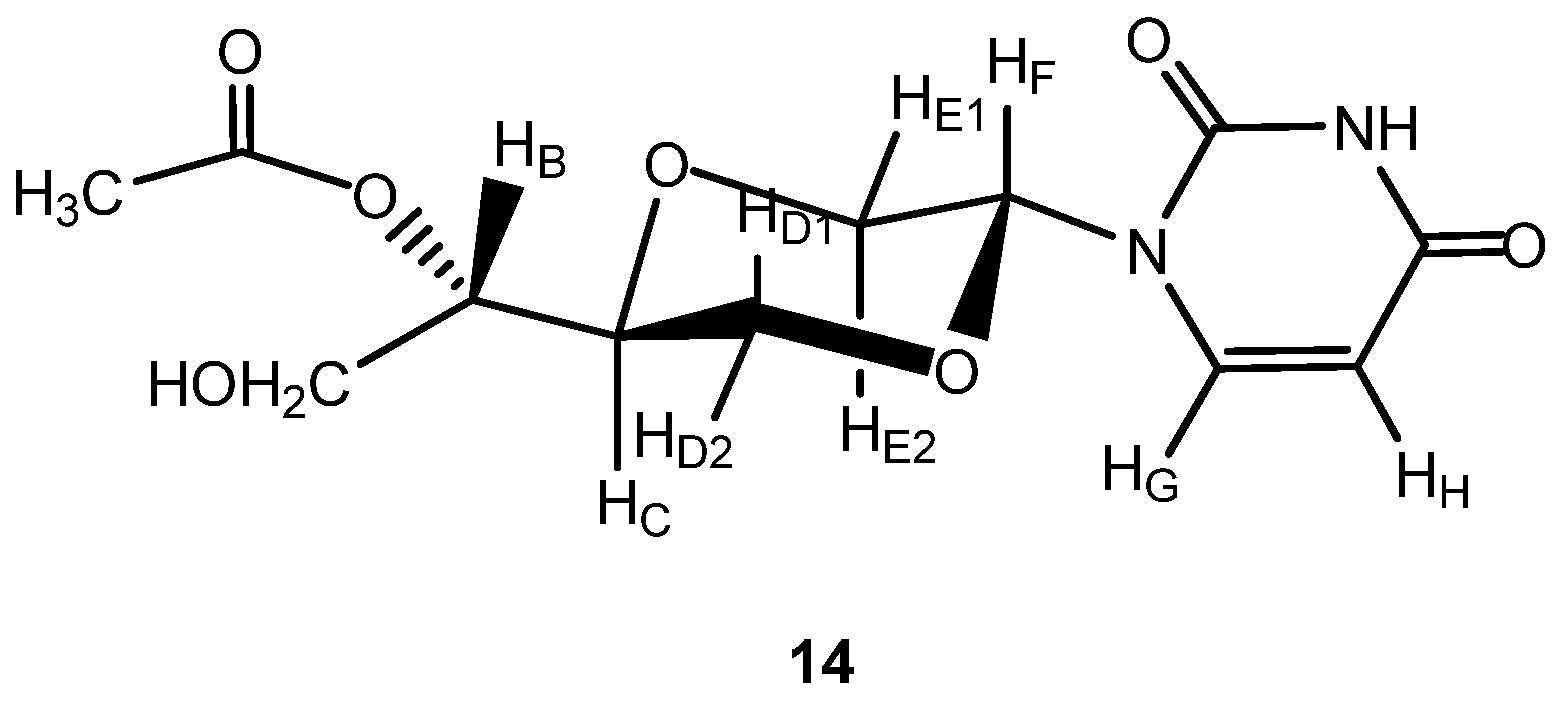
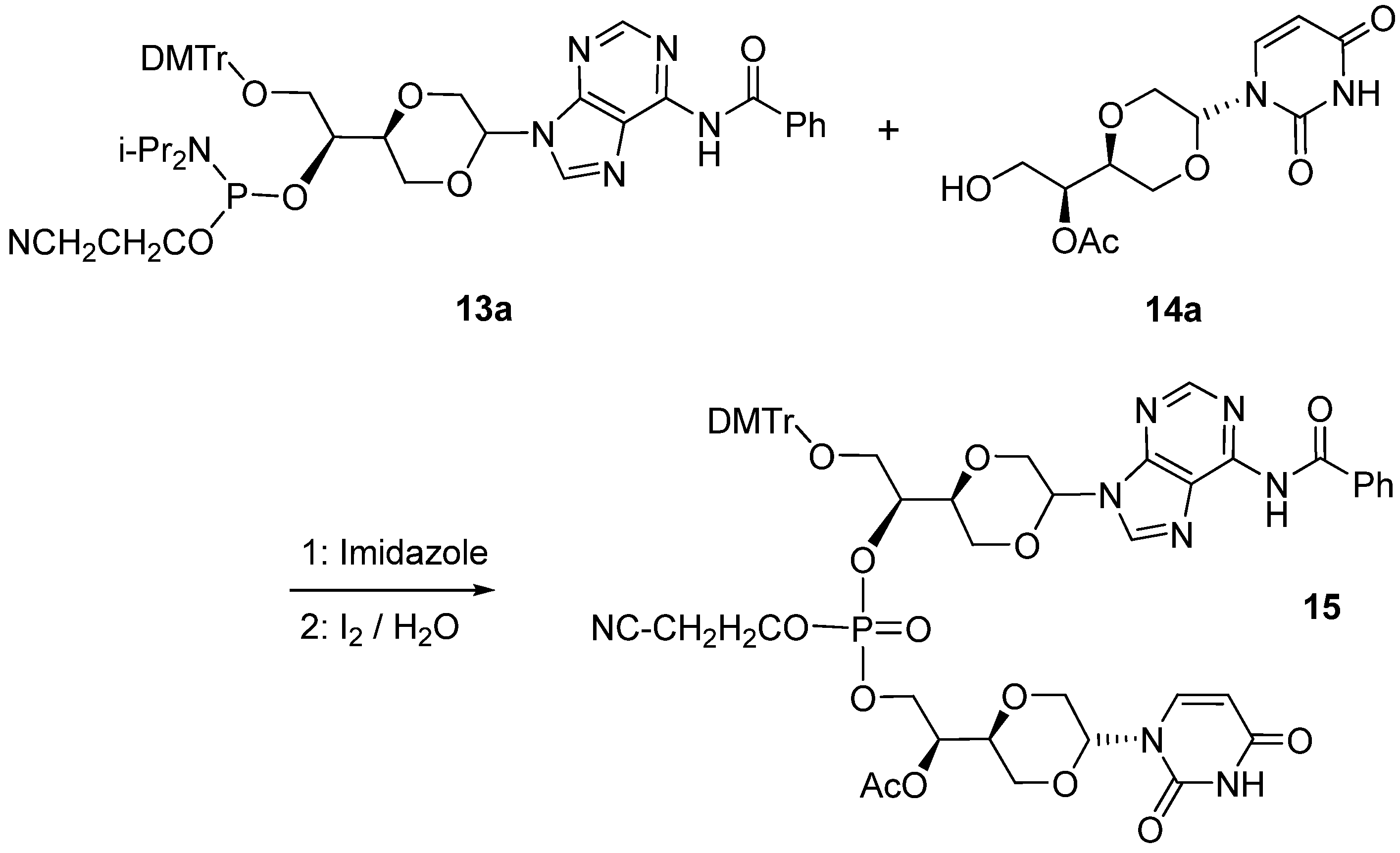
1-[(2R,5S)-5-[(1S)-Acetyloxy-2-hydroxylethyl-1-yl]-1,4-dioxan-2-yl]uracil (
14a). The synthesis of this compound has been reported earlier [
7]. Compound
14a (
Figure 8) exhibited the following spectroscopic properties:
1H-NMR (CD
3OD, 400 MHz): δ 2.07 (s, 3H, CH
3), 3.58 (dd,
J = 11.4 Hz, 9.8Hz, 1H, H
E2), 3.70–3.74 (m, 1H, H
B), 3.77–3.82 (m, 1H, H
C), 3.92–3.98 (m, 2H, H
E and H
D1), 4.06 (dd,
J = 11.6, 2.8 Hz, 1H, H
D2), 4.13 (dd,
J = 11.2, 6.4 Hz, 1H, H
A), 4.16 (dd,
J = 11.2 Hz, 5.4Hz, 1H, H
A), 5.69 (dd,
J = 10 Hz, 2.8 Hz, 1H, H
F), 5.70 (d,
J = 8 Hz, 1H, H
H), 7.71 (d,
J = 8 Hz, 1H, H
G) ppm.
13C-NMR (CD
3OD, 100 MHz): δ 20.9, 66.4, 69.2, 69.4, 69.8, 76.0, 80.0, 103.1, 142.3, 151.9, 166.0, 172.8 ppm. IR (neat): 3477, 3190, 3110, 3074, 2996, 2879, 1697, 1268, 1105 cm
−1. MS (EI): 230.3, 197(M
+-(CH
3COCHCH
2OH)), 189(M
+-uracil). HRMS (ESI): Calcd. for C
12H
16N
2O
7 [M+Na]
+ 323.0856, Found 323.0862.
Figure 8.
Structure of compound 14.
Figure 8.
Structure of compound 14.
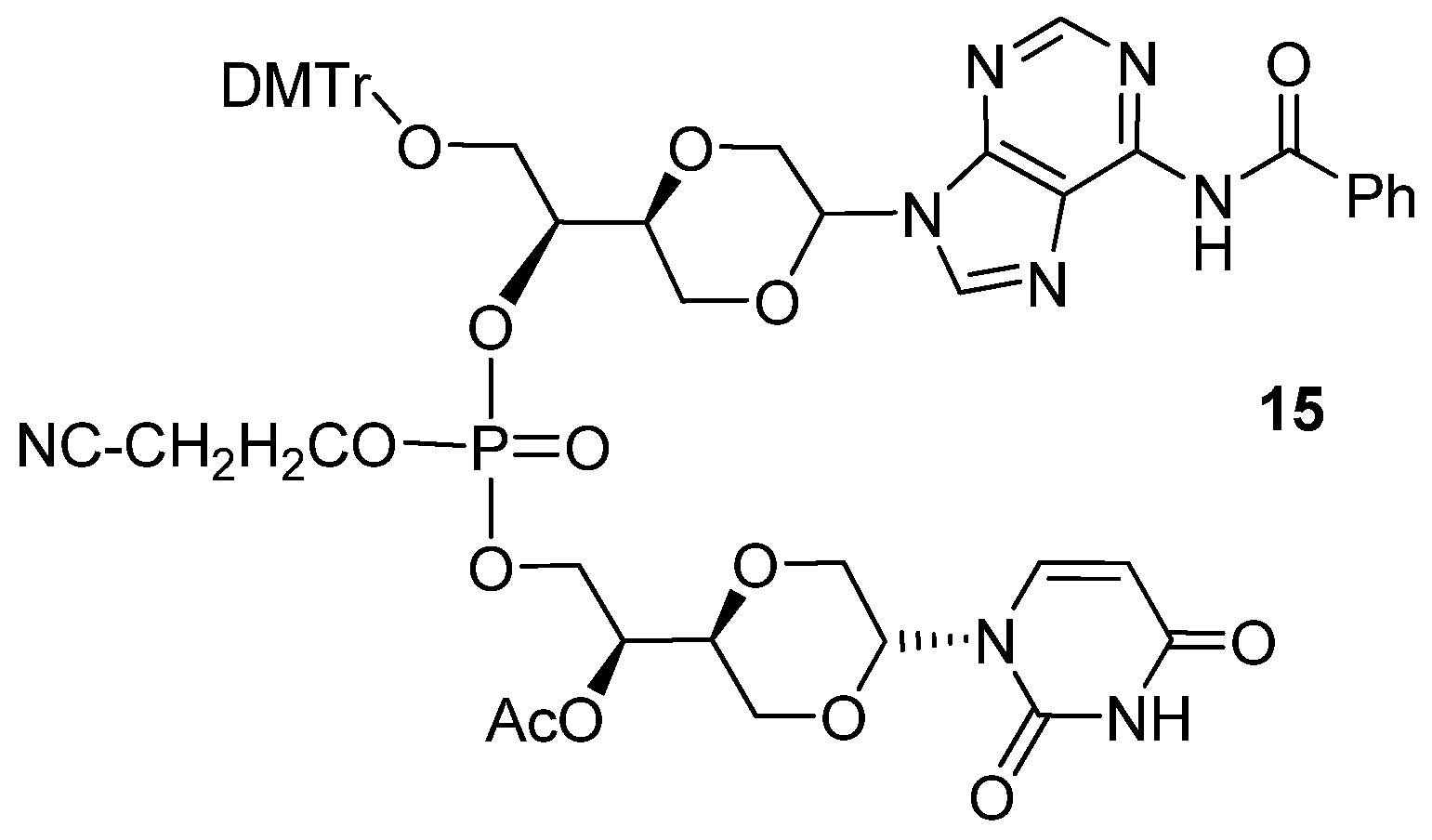
N6-Benzoyladenine uracil dinucleotide (
15). Compound
13a (30 mg, 0.039 mmol) and
14a (8 mg, 0.027 mmol) in a reaction flask with a magnetic stirring bar were dried under high vacuum for 4 hours. Then a mixture 1-
H tetrazole (0.3 mL, 0.45 M, 0.135 mmol) in dry acetonitrile (6 mL) was added under a nitrogen atmosphere. The solution was stirred overnight at room temperature. Then was added a few drops of iodine in THF (1M solution), 2,6-lutidine and H
2O (2:2:1) until an orange color persisted. The solution was then quenched with saturated sodium thiosulfate solution (4 mL). Then the two phases were treated with saturated NaHCO
3 solution (4 mL). The separated aqueous phase was extracted with 4 × 8 mL of dichloromethane. The combined organic phase was dried over anhydrous Na
2SO
4 and filtered and the solvent evaporated. Flash chromatographic purification, using mixtures of dichloromethane and methanol in the ratio from 25:1 to 15:1 as gradient solvents, afforded a product (16 mg, 61.5%), which was assigned structure
15 (
Figure 9) based on the following spectroscopic properties:
1H-NMR (CDCl
3, 400 MHz): δ 2.02 (s, 3H, C
H3COO), 2.09 (s, 3H, C
H3COO), 2.51–2.62 (m, 2H, C
H2CN), 2.71–2.74 (m, 2H, C
H2CN), 3.15 (dd, 1H,
J = 11.2 Hz, 9.6Hz), 3.21–3.28 (m), 3.38 (dd,
J = 11.4 Hz, 9.8Hz), 3.45–3.58 (m), 3.62–3.96 (m), 4.01 (dd, 1H,
J = 11.2 Hz, 2.8Hz), 4.03–4.13 (m), 4.14–4.35 (m), 4.37–4.47 (m), 4.63–4.74 (m), 5.58 (dd, 1H,
J = 9.8 Hz, 3 Hz, anomeric proton), 5.66–5.71 (m, 3H, two protons in uracil and one anomeric proton), 6.62–6.68 (m), 6.84–6.89 (m, aromatic protons), 7.20 (d,
J = 8 Hz, 1H, a proton in uracil), 7.22–7.28 (m, aromatic protons), 7.30–7.37 (m, aromatic protons and one proton in uracil), 7.42–7.49 (m, aromatic protons), 7.52–7.58 (m, aromatic protons), 8.143 (s, 1H, a proton in adenine), 8.148 (s, 1H, a proton in adenine), 8.23–8.25 (m, aromatic protons), 8.33 (br. 1H, NH), 8.63 (s, a proton in adenine), 8.64 (s, a proton in adenine) ppm.
13C-NMR (CDCl
3, 100 MHz): δ 20.76, 20.82, 55.3, 62.0–62.5 (m), 67.6, 68.1, 68.4, 69.4, 73.2, 74.4, 74.8, 78.4, 78.5, 80.9, 86.8, 86.9, 102.7, 102.8, 128.06, 128.10, 128.3, 129.9, 130.0, 132.4, 134.98, 135.02, 135.2, 137.1, 139.2, 139.3, 141.9, 142.9, 148.4, 149.5, 157.2, 158.8, 162.1, 170.4, 175.6 ppm.
31P-NMR (CDCl
3): −1.28, −1.49 ppm. HRMS (ESI): Calcd. for C
54H
55N
8O
16P [M+Na]
+ 1125.3372, Found 1125.3354.
Figure 9.
Structure of compound 15.
Figure 9.
Structure of compound 15.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
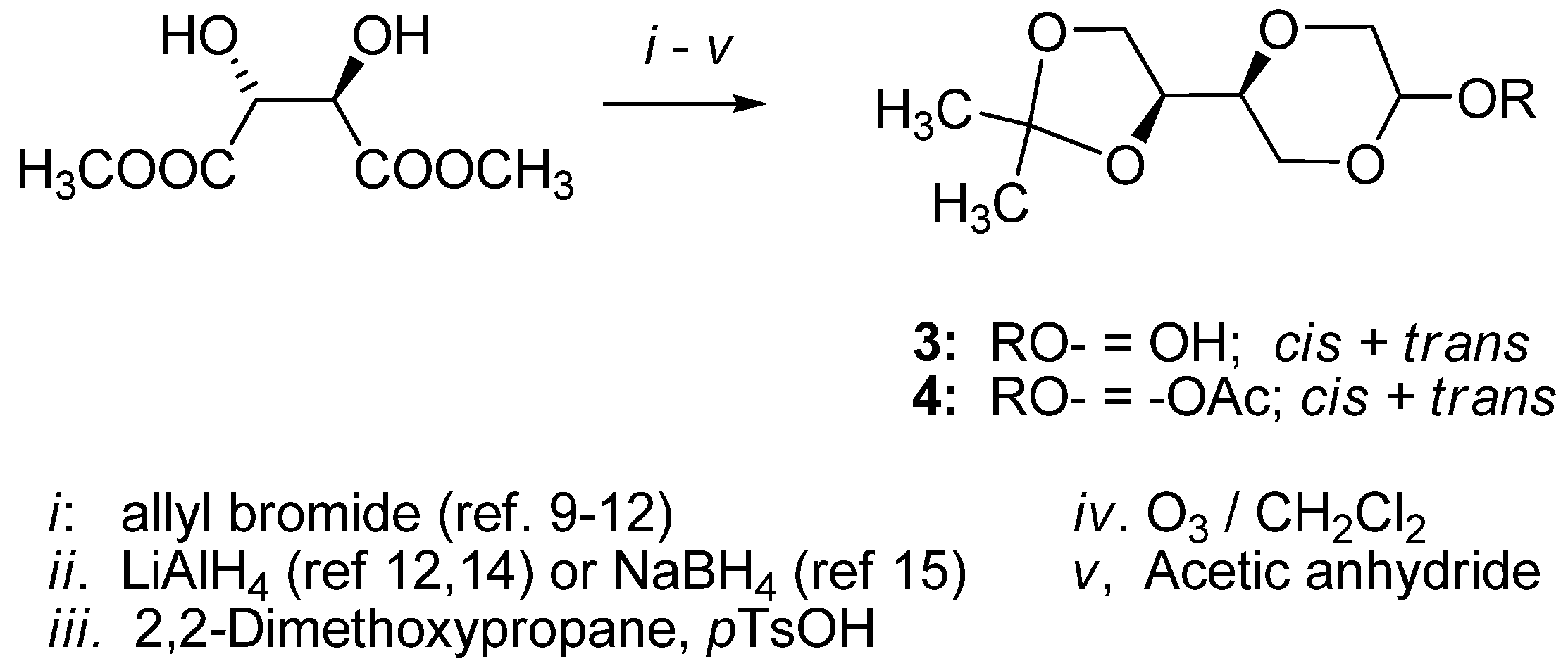{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















