Solid-Phase Synthesis of Arylpiperazine Derivatives and Implementation of the Distributed Drug Discovery (D3) Project in the Search for CNS Agents

Abstract
:Abbreviations
| CHCl3 | chloroform |
| DCM | dichloromethane |
| DIC | N,N’-diisopropylcarbodiimide |
| DIEA | diisopropylethylamine |
| DMF | dimethylformamide |
| HBTU | O-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| SOCl2 | thionyl chloride |
| TFA | trifluoroacetic acid |
1. Introduction



2. Results
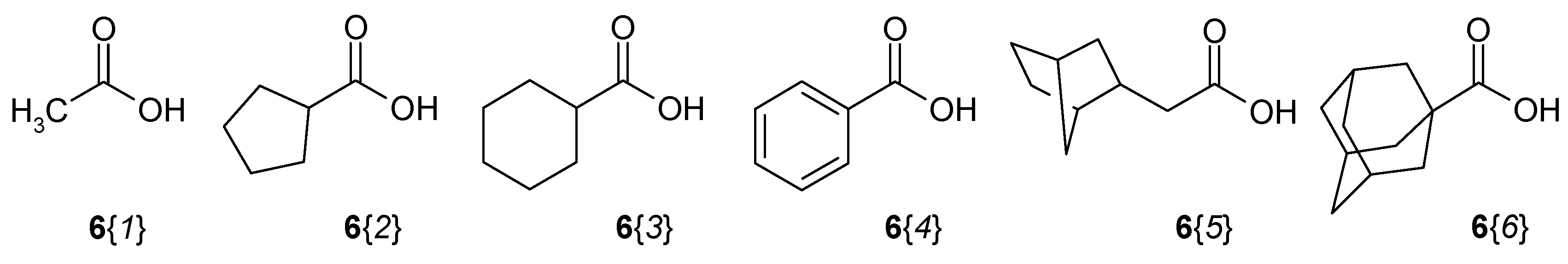
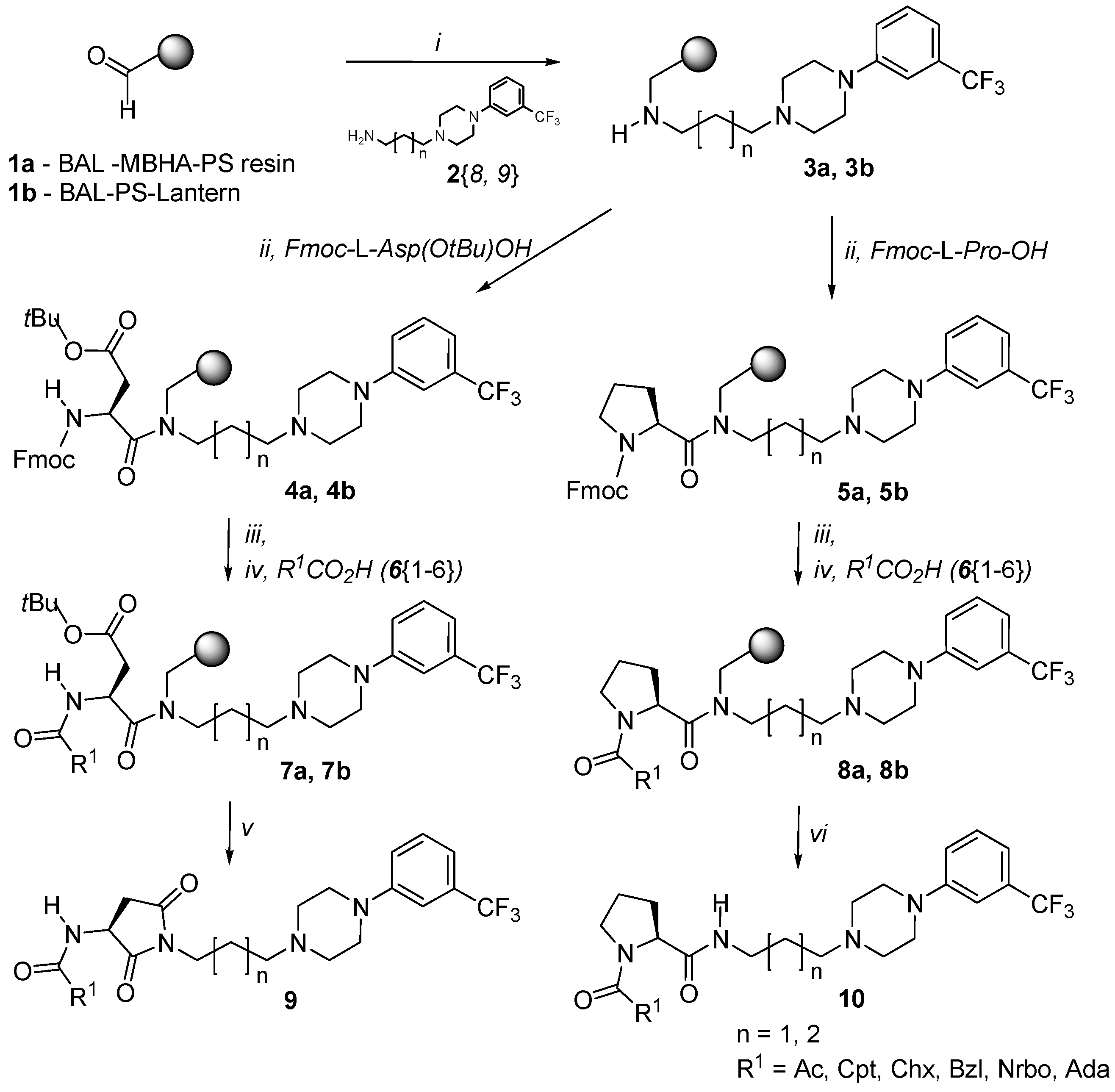
2.1. Library Synthesis




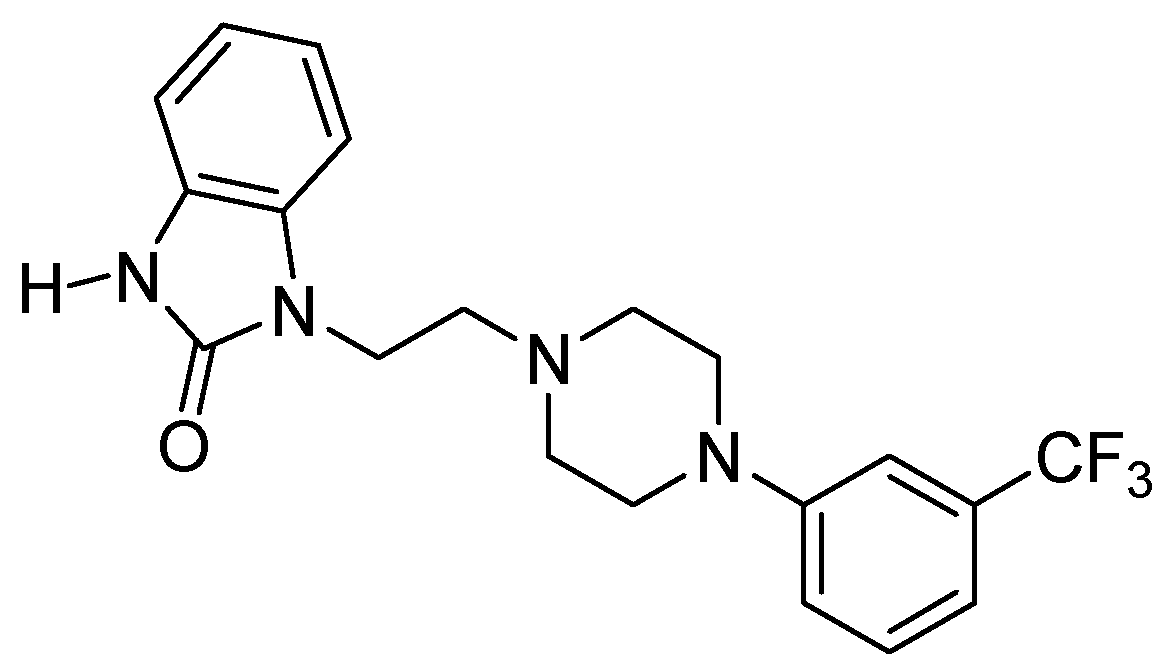{kind=link}
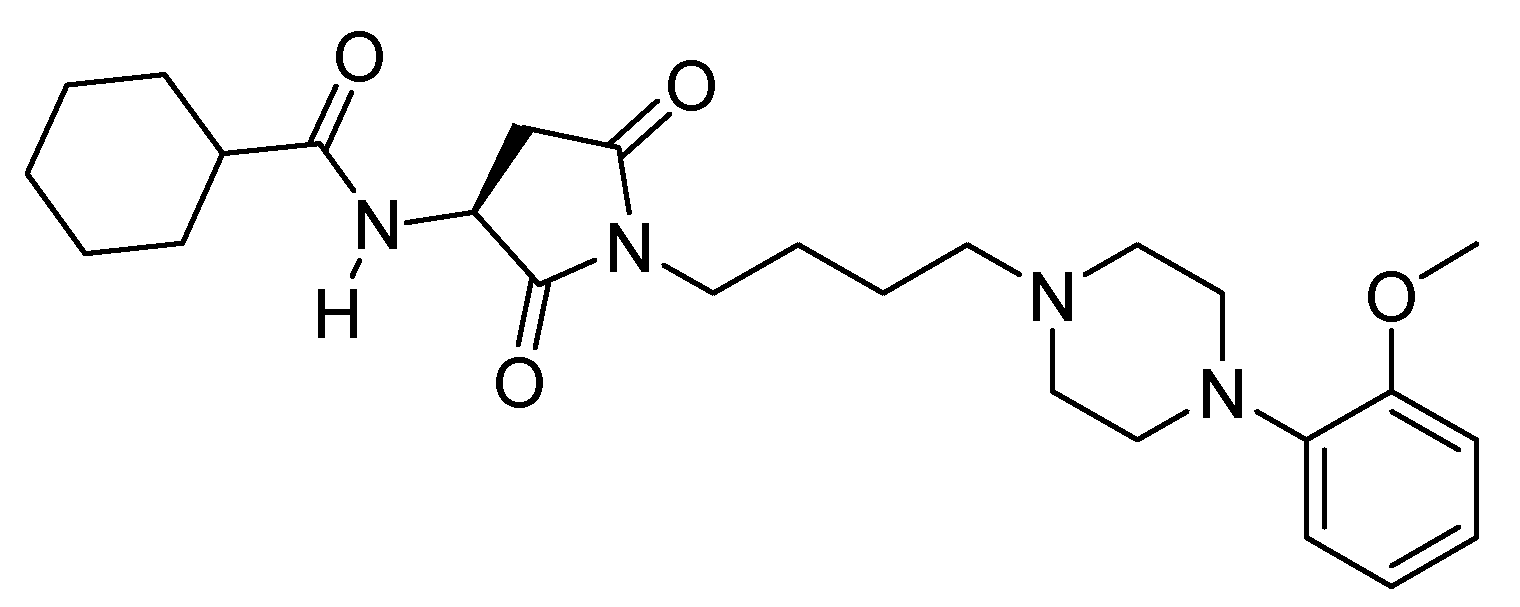{kind=link}
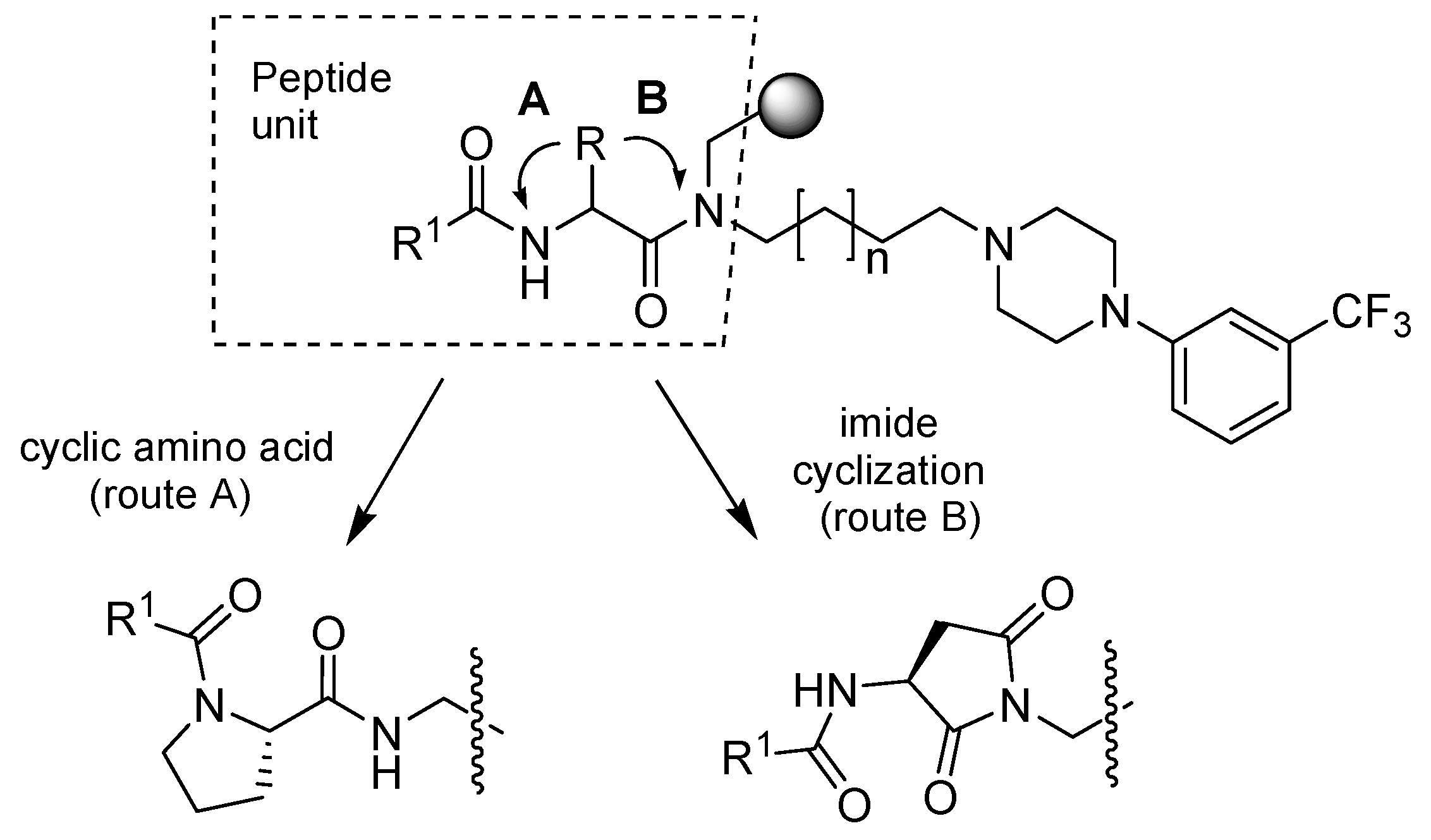{kind=link}
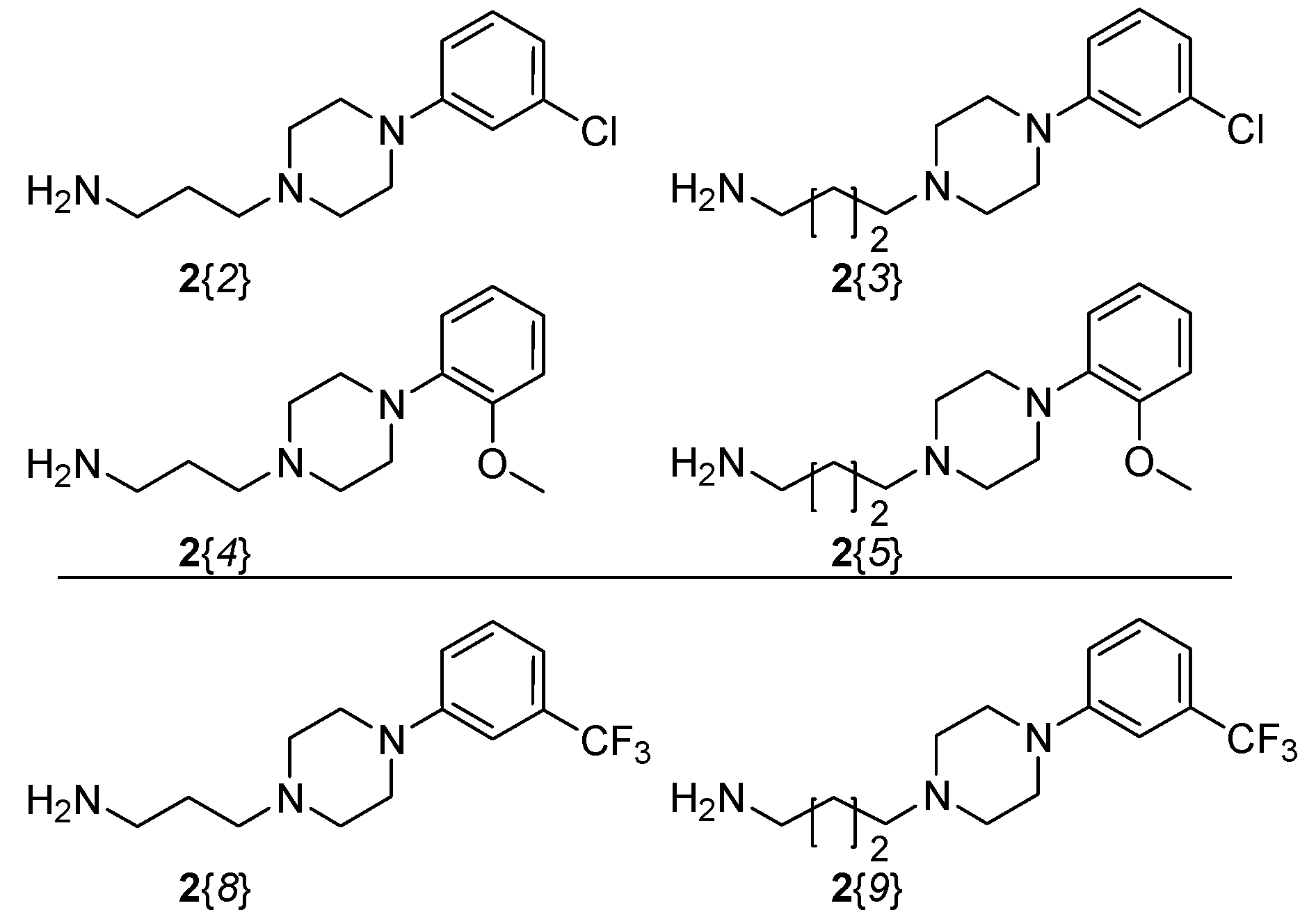{kind=link}
{kind=link}
{kind=link}
| Compound | Purity a | Compound | Purity a | ||
|---|---|---|---|---|---|
| Lantern | Resin | Lantern | Resin | ||
| 9{8,3} | 74 | 61 | 10{8,3} | 96 | 93 |
| 9{8,4} | 70 | 52 | 10{8,4} | 94 | 93 |
| 9{8,5} | 77 | 59 | 10{8,5} | 96 | 87 |
| 9{9,3} | 68 | 62 | 10{9,3} | 97 | 97 |
| 9{9,4} | 64 | 49 | 10{9,4} | 98 | 89 |
| 9{9,6} | 75 | 56 | 10{9,6} | 98 | 86 |
| Compound | Rt (min) | Purity a | MW calc. | [M + H]+ found | Compd | Rt (min) | Puritya | MW calc. | [M + H]+ found |
|---|---|---|---|---|---|---|---|---|---|
| 9{8,1} | 1.35 | 97 | 426.2 | 427.2 | 10{8,1} | 1.34 | 99 | 426.2 | 427.3 |
| 9{8,2} | 1.60 | 95 | 480.2 | 481.4 | 10{8,2} | 1.62 | 99 | 480.3 | 481.0 |
| 9{8,3} | 1.66 | 95 | 494.2 | 495.1 | 10{8,3} | 1.69 | 96 | 494.3 | 495.2 |
| 9{8,4} | 1.60 | 95 | 488.2 | 489.3 | 10{8,4} | 1.58 | 96 | 488.2 | 489.8 |
| 9{8,5} | 1.78 | 95 | 520.2 | 521.6 | 10{8,5} | 1.82 | 96 | 520.3 | 521.5 |
| 9{8,6} | 1.85 | 99 | 546.3 | 547.4 | 10{8,6} | 1.90 | 97 | 546.3 | 547.4 |
| 9{9,1} | 1.38 | 97 | 440.2 | 441.5 | 10{9,1} | 1.37 | 93 | 440.2 | 441.5 |
| 9{9,2} | 1.60 | 99 | 494.2 | 495.5 | 10{9,2} | 1.60 | 96 | 494.3 | 495.1 |
| 9{9,3} | 1.67 | 99 | 508.2 | 509.3 | 10{9,3} | 1.68 | 97 | 508.3 | 509.3 |
| 9{9,4} | 1.62 | 95 | 502.2 | 503.4 | 10{9,4} | 1.58 | 97 | 502.2 | 503.3 |
| 9{9,5} | 1.78 | 96 | 534.3 | 535.4 | 10{9,5} | 1.80 | 98 | 534.3 | 535.6 |
| 9{9,6} | 1.85 | 98 | 560.3 | 561.4 | 10{9,6} | 1.86 | 96 | 560.3 | 561.1 |
2.2. Biological Evaluation
| Compound m-CF3 set | Ki [nM] a | Compound m-Cl set b | Ki [nM] | Compd o-OCH3 set b | Ki [nM] | |||
|---|---|---|---|---|---|---|---|---|
| 5-HT1A | 5-HT2A | 5-HT1A | 5-HT2A | 5-HT1A | 5-HT2A | |||
| 9{8,1} | 129 | 164 | 9{2,1} c | NT e | NT | 9{4,1} d | 480 | NT |
| 9{8,3} | 170 | 276 | 9{2,3} c | 360 | 92 | 9{4,3} d | 230 | NT |
| 9{9,2} | 24 | 79 | 9{3,2} | 28 | 47 | 9{5,2} | 23 | 480 |
| 9{9,3} | 19 | 70 | 9{3,3} | 26 | 6.5 | 9{5,3} | 19 | 183 |
| 9{9,4} | 18 | 61 | 9{3,4} | 52 | 20 | 9{5,4} | 18 | 410 |
| 9{9,5} | 15 | 30 | 9{3,5} | 21 | 4.2 | 9{5,5} | 9 | 47 |
| 9{9,6} | 21 | 39 | 9{3,6} | 47 | 25 | 9{5,6} | 4 | 173 |
| 10{8,4} f | 122 | 898 | 10{2,4} | NT | 875 | 10{4,4} | NT | NT |
| 10{8,6} f | 42 | 1651 | 10{2,6} | 75 | 360 | 10{4,6} | NT | NT |
| 10{9,2} f | 26 | 191 | 10{3,2} c | NT | NT | 10{5,2} | NT | NT |
| 10{9,4} f | 19 | 145 | 10{3,4} | 78 | 128 | 10{5,4} | NT | NT |
| 10{9,5} f | 10 | 82 | 10{3,5} | 37 | 35 | 10{5,5} | 14 | 547 |
| 10{9,6} f | 8 | 699 | 10{3,6} | 13 | 140 | 10{5,6} | 3 | 503 |
3. Discussion
4. Experimental
4.1. General Methods
4.2. General Procedures for Manual Solid-Phase Reactions
4.3. Solid-Phase Synthesis on BAL-MBHA-PS Resin
4.3.1. Procedure for preparation of amine-bound Resin 3a, 3b via reductive amination
4.3.2. Coupling of amine resin 3a, 3b with N-α-Fmoc-Asp(OtBu)-OH and N-α-Fmoc-proline
4.3.3. Standard Fmoc-deprotection protocol
4.3.4. Acylation with carboxylic acids using HBTU
4.3.5. Succinimide derivatives formation via cleavage and intramolecular ring closure
4.3.6. Cleavage of the proline derivatives off the resin
4.4. Analytical Data for Biologically Tested Library Members Synthesized on MBHA Resin
4.5. Solid-Phase Synthesis on BAL-PS Lanterns
4.5.1. Secondary amine acylation protocol
4.5.2. Standard Fmoc-deprotection protocol
4.5.3. Standard acylation with carboxylic acids
4.5.4. Cleavage/cyclization protocol for succinimide derivatives formation
4.5.5. Cleavage of the proline derivatives off the lantern
4.6. Analytical Data for 12 Library Members Synthesized on SynPhase Lanterns
5. Conclusions
Acknowledgements
References and Notes
- Borsini, F.; Evans, K.; Jason, K.; Rohde, F.; Alexander, B.; Pollentier, S. Pharmacology of Flibanserin. CNS Drug Rev. 2002, 8, 117–142. [Google Scholar]
- Invernizzi, R.W.; Sacchetti, G.; Parini, S.; Acconcia, S.; Samanin, R. Flibanserin, a potential antidepressant drug, lowers 5-HT and raises dopamine and noradrenaline in the rat prefrontal cortex dialysate: Role of 5-HT1A receptors. Br. J. Pharmacol. 2003, 139, 1281–1288. [Google Scholar] [CrossRef]
- Jolly, E.; Clayton, A.; Thorp, J.; Lewis-D’Agostini, G. Design of Phase III pivotal trials of flibanserin in female Hypoactive Sexual Desire Disorder (HSDD). Sexologies 2008, 17, 133–134. [Google Scholar]
- Zajdel, P.; Subra, G.; Bojarski, A.J.; Duszyńska, B.; Tatarczyńska, E.; Nikiforuk, A.; Chojnicka-Wójcik, E.; Pawłowski, M.; Martinez, J. Novel class of arylpiperazines containing N-acylated amino acids: Their synthesis, 5-HT1A, 5-HT2A receptor affinity, and in vivo pharmacological evaluation. Bioorg. Med. Chem. 2007, 15, 2907–2919. [Google Scholar] [CrossRef]
- López-Rodríguez, M.L.; Ayala, D.; Benhamú, B.; Morcillo, M.J.; Viso, A. Arylpiperazine derivatives acting at 5-HT(1A) receptors. Curr Med. Chem. 2002, 9, 443–469. [Google Scholar] [CrossRef]
- Lacivita, E.; Leopoldo, M.; Berardi, F.; Perrone, R. 5-HT1A receptor, an old target for new therapeutic agents. Curr Top Med. Chem. 2008, 8, 1024–1034. [Google Scholar] [CrossRef]
- Leopoldo, M. Serotonin(7) receptors 5-HT(7)Rs and their ligands. Curr. Med. Chem. 2004, 11, 629–661. [Google Scholar] [CrossRef]
- Zajdel, P.; Subra, G.; Verdie, P.; Bojarski, A.J.; Duszyńska, B.; Basista, K.; Obniska, J.; Martinez, J.; Pawłowski, M. The influence of an ethylene spacer on the 5-HT1A and 5-HT2A receptor affinity of arylpiperazine derivatives of amides with N-acylated amino acids and 3-differently substituted pyrrolidine-2,5-diones. Eur. J. Med. Chem. 2009, 44, 800–808. [Google Scholar] [CrossRef]
- Zajdel, P.; Subra, G.; Bojarski, A.J.; Duszyńska, B.; Pawłowski, M.; Martinez, J. A new class of arylpiperazine derivatives: The library synthesis on SynPhase lanterns and biological evaluation on serotonin 5-HT1A and 5-HT2A receptors. J. Comb. Chem. 2004, 6, 761–767. [Google Scholar] [CrossRef]
- Scott, W.L.; O'Donnell, M.J. Distributed Drug Discovery, Part 1: Linking academia and combinatorial chemistry to find drug leads for developing world diseases. J. Comb. Chem. 2009, 11, 3–13. [Google Scholar] [CrossRef]
- Scott, W.L.; Alsina, J.; Audu, C.O.; Babaev, E.; Cook, L.; Dage, J.L.; Goodwin, L.A.; Martynow, J.G.; Matosiuk, D.; Royo, M.; et al. Distributed Drug Discovery, Part 2: Global rehearsal of alkylating agents for the synthesis of resin-bound unnatural amino acids and virtual D(3) catalog construction. J. Comb. Chem. 2009, 11, 14–33. [Google Scholar] [CrossRef]
- Scott, W.L.; Audu, C.O.; Dage, J.L.; Goodwin, L.A.; Martynow, J.G.; Platt, L.K.; Smith, J.G.; Strong, A.T.; Wickizer, K.; Woerly, E.M.; et al. Distributed Drug Discovery, Part 3: Using D(3) methodology to synthesize analogs of an anti-melanoma compound. J. Comb. Chem. 2009, 11, 34–43. [Google Scholar] [CrossRef]
- Available from Leads Metal Products, PO Box 441186, Indianapolis, IN, 46244-1186 ([email protected]).
- Maclean, D.; Martin, E.J. On the representation of combinatorial libraries. J. Comb. Chem. 2004, 6, 1–11. [Google Scholar] [CrossRef]
- Zajdel, P.; Subra, G.; Bojarski, A.J.; Duszyńska, B.; Pawłowski, M.; Martinez, J. Arylpiperazines with N-acylated amino acids as 5-HT1A receptor ligands. Bioorg. Med. Chem. Lett. 2006, 16, 3406–3410. [Google Scholar]
- Zajdel, P.; Subra, G.; Bojarski, A.J.; Duszyńska, B.; Pawłowski, M.; Martinez, J. Parallel solid-phase synthesis and characterization of new sulfonamide and carboxamide proline derivatives as potential CNS agents. Bioorg. Med. Chem. 2005, 13, 3029–3035. [Google Scholar] [CrossRef]
- Available online: http://www.mimotopes.com (accessed on 26 January 2011).
- Bojarski, A.J.; Cegła, M.T.; Charakchieva-Minol, S.; Mokrosz, M.J.; Maćkowiak, M.; Mokrosz, J.L. Structure-activity relationship studies of CNS agents. Part 9: 5-HT1A and 5-HT2 receptor affinity of some 2- and 3-substituted 1,2,3,4-tetrahydro-beta-carbolines. Pharmazie 1993, 48, 289–294. [Google Scholar]
- Sample Availability: Contact the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zajdel, P.; Król, J.; Grychowska, K.; Pawłowski, M.; Subra, G.; Nomezine, G.; Martinez, J.; Satała, G.; Bojarski, A.J.; Zhou, Z.; et al. Solid-Phase Synthesis of Arylpiperazine Derivatives and Implementation of the Distributed Drug Discovery (D3) Project in the Search for CNS Agents. Molecules 2011, 16, 4104-4121. https://doi.org/10.3390/molecules16054104
Zajdel P, Król J, Grychowska K, Pawłowski M, Subra G, Nomezine G, Martinez J, Satała G, Bojarski AJ, Zhou Z, et al. Solid-Phase Synthesis of Arylpiperazine Derivatives and Implementation of the Distributed Drug Discovery (D3) Project in the Search for CNS Agents. Molecules. 2011; 16(5):4104-4121. https://doi.org/10.3390/molecules16054104
Chicago/Turabian StyleZajdel, Paweł, Joanna Król, Katarzyna Grychowska, Maciej Pawłowski, Gilles Subra, Gaël Nomezine, Jean Martinez, Grzegorz Satała, Andrzej J. Bojarski, Ziniu Zhou, and et al. 2011. "Solid-Phase Synthesis of Arylpiperazine Derivatives and Implementation of the Distributed Drug Discovery (D3) Project in the Search for CNS Agents" Molecules 16, no. 5: 4104-4121. https://doi.org/10.3390/molecules16054104