Catalytic Synthesis of α-Oxoketene S,S-Acetals in a Wet Ionic Liquid [Bmim]Cl/H2O Homogeneous System

1
Key Laboratory of Forest Plant Ecology, Ministry of Education, Northeast Forestry University, Harbin 150040, China
2
Engineering Research Center of Forest Bio-preparation, Ministry of Education, Northeast Forestry University, Harbin 150040, China
3
Department of Pharmaceutical Biology, Institute of Pharmacy and Biochemistry, University of Mainz, Mainz 55128, Germany
*
Authors to whom correspondence should be addressed.
Molecules 2011, 16(6), 4500-4510; https://doi.org/10.3390/molecules16064500
Submission received: 23 March 2011
/
Revised: 11 May 2011
/
Accepted: 18 May 2011
/
Published: 27 May 2011
Abstract
:A clean, practical and environmentally friendly synthesis in a homogeneous system has been developed for α-oxoketene S,S-acetals. A mixture of [Bmim]Cl and water was used as medium. The best economical and practical molar ratio of [Bmim]Cl to substrate was 4 to 1. With various types of 1,3-dicarbonyl compounds as substrate, the corresponding α-oxoketene S,S-acetals have been prepared under mild reaction conditions with yields of 53–74% after purification with silica gel column. [Bmim]Cl/water can be recycled several times.
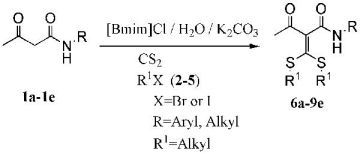
1. Introduction
α-Oxoketene S,S-acetals are known as versatile intermediates for the preparation of various functional groups such as acetylenes [1], substituted furans [2], thiophene derivatives [3] and highly substituted phenols [4,5]. The synthesis of an α-oxoketene S,S-acetal albeit in low yield was first reported by Kelber and co-workers in 1910 using potassium hydroxide as base [6]. In 1988, Choi et al. [7] successfully prepared α-oxoketene S,S-acetals directly from ketones in good yields by choosing potassium carbonate as weak base in N,N-dimethylformamide. However, environmental pollution and post-processing difficulties of N,N-dimethylformamide led to some drawbacks. In 2005, Ouyang et al. [8] successfully prepared α-oxoketene S,S-acetals in good yields using water as medium, and TBAB as phase transfer catalyst in a solid-liquid system. However, uneven mixing is a frequent phenomenon in solid-liquid systems, especially in the amplification reaction. In order to overcome this issue, we have chosen ionic liquids as phase transfer catalyst [9] and cosolvent, water as medium to synthesize α-oxoketene S,S-acetals. The most apparent advantage was that ionic liquids/H2O homogeneous system could efficiently avoid uneven mixing of starting material mixtures. To the best of our knowledge, the present investigation is the first report of such a synthesis method.
Ionic liquids had been generally studied as excellent solvents due to their unique physical and chemical properties [10]. As an imidazolium ionic liquid, [Bmim]Cl has been widely used in many reactions such as elimination reactions [11], one-pot synthesis of α-aminophosphonate derivatives [12], oxidations [13,14], Friedel-Crafts acylations [15], methanolysis of polycarbonates [16], S-methylation reactions [17] and Ferrier rearrangements [18]. Its clean, green, high yield, no vapor pressure and recyclability characteristics were evidenced in these reactions. More interesting is the fact that [Bmim]Cl in some reactions has the feature of decreasing the reaction temperature to room temperature from 100–300 °C [19], promoting the reactivity of metal catalysts [20], greatly reducing the reaction times [21], high selectivity [15,17,18] and less toxicity [17].
Water and ionic liquids mixtures were termed “wet ILs” [22] and are used as single-phase or multi-phase systems. A wide range of applications have been realized in recent years such as Knoevenagel condensation [23], Diels–Alder cycloadditions [24], synthesis of organophosphorus compounds [25] and Biginelli-type reactions [26]. Some studies have indicated that the presence of water has a great impact on ionic liquids [27,28]. These previous achievements encouraged us to establish an environmentally friendly synthesis method for α-oxoketene S,S-acetals in an ionic liquid/water system. The choice of ionic liquid, the ratio of ionic liquid to starting material, recycle times and other factors were investigated and optimized to achieve a green, moderate yield synthesis of α-oxoketene S,S-acetals.
2. Results and Discussion
2.1. Choice of Ionic Liquids
A weak alkaline system is needed for synthesis of α-oxoketene S,S-acetals rather than strong alkali to prevent side reactions. Considering the extensive applications and especially the many favorable features and its lower price [Bmim]Cl has been chosen for this investigation. Another frequently used and cheap pyridine ionic liquid [Epy]BF4 has also been chosen for the investigation. The alkalinity was regulated by addition of the weak base potassium carbonate. Water, the ionic liquids [Epy]BF4, [Bmim]Cl, [Epy]BF4/water and different [Bmim]Cl/water ratios were used as solvents, respectively. Results are shown in Table 1.
No reaction product was detected when using water alone (Entry 1). When using [EPy]BF4, [Bmim]Cl respectively, all materials appeared as a solid phase without further reactivity (Entry 2, 3), indicating that the reaction did not take place in ionic liquids. However, under the systems of ionic liquids [EPy]BF4, [Bmim]Cl and water (wet ILs), the reactions took place with good yields. Acetoacetylaniline 5 mmol (taken as 1 equiv. relatively), potassium carbonate (2.5 equivs.), carbon disulfide (1.2 equivs.) and 1,2-dibromoethane (1 equiv.) in [EPy]BF4/water (10 equivs./15 mL) led to good reactivity and 71% yield (Entry 4). However, the reaction time took up to 15 hours. Then, the reaction of acetylacetanilide (1 equiv.) in [Bmim]Cl/water (10 equivs./15 mL) was attempted in accordance with the experimental procedures. Interestingly, a homogeneous system was obtained giving 73% yield of product after 5 hours (Entry 5). It was reported that [Bmim]Cl plays an important role in accelerating the reaction rate [18,21]. We assume that [Bmim]Cl also played the same role in our reactions.
2.2. The Ratio of [Bmim]Cl to Substrate
To optimize the reaction conditions, several reactions were carried out changing the molar ratio [Bmim]Cl to 1a from 50/5 to 0.1/5. As shown in Table 1 (Entry 5), the effect of using 10 equivs. [Bmim]Cl was excellent. The yields and reaction time were monitored while the amount of [Bmim]Cl was reduced successively to 0.02 equivs. A reduced molar quantity of [Bmim]Cl of 4 equivs. (Entry 8) still resulted in successful dissolution of the starting material. The reaction effect was almost the same as Entry 5. As the molar quantity of [Bmim]Cl was reduced to 2 equivs. (Entry 9), a small amount of insoluble starting material was observed. Nevertheless, no obvious yield decline was measured. With continuous reduction of the amount of [Bmim]Cl, the reaction time was prolonged, accompanied by more amount of unreacted starting material. The explanation is that the amount of [Bmim]Cl as catalyst in the system was relatively low, which consequently led to low reaction rates and conversions. In addition, without enough [Bmim]Cl to dissolve the raw material in water, uneven stirring in solid-liquid system resulted in low conversion. Therefore, the best molar ratio of [Bmim]Cl to substrate was 4 to 1 (Entry 8).
2.3. Recycling of the [Bmim]Cl/water
The feasibility of recycling ionic liquids/H2O represents an important topic and performance criterion of any industrial process. The proportion of [Bmim]Cl and water did not change much after filtering off the products. This prompted us to investigate the recycling of the wet ionic liquid. After isolated the product by filtration, the residual [Bmim]Cl/H2O of Table 1, Entry 8 was collected without any treatment for the reuse. The reaction of acetoacetylaniline (1 equiv.), carbon disulfide (1.2 equivs.) and 1,2-dibromoethane (1 equiv.) while reusing the wet ionic liquid was analyzed. The corresponding α-oxoketene S,S-acetal was produced in a yield of 70% (Table 2, Entry 1). After filtering the product by the same process as described above, the residual [Bmim]Cl/H2O was reused for a third, fourth and fifth time. The results of subsequent studies (Table 2) indicated that the yield did not decline even in the fifth run with reused [Bmim]Cl/water. This finding implied that wet ionic liquid can be recycled, and the catalytic activity of [Bmim]Cl is not decreased.
2.4. The Synthesis of Different α-oxoketene S,S-acetals
To extend the applicability of this protocol, a series of substrates 1a-1e were subjected to the reaction using the optimized conditions (Table 1, Entry 8). The results are summarized in Table 3.
The yields of cyclic α-oxoketene S,S-acetals (e.g., Entry 3) were higher than for acyclic α-oxoketene S,S-acetals (Entries 4, 5). Series 1b, 1d and 1e also follow this regularity. It is possible that acyclic α-oxoketene S,S-acetal molecules were less stable than cyclic ones. It is demonstrated from Table 3 that the reaction status of α-oxoketene S,S-acetals with naphthenic base, methyl and butyl was not as efficient as of α-oxoketene S,S-acetals with phenyl groups. From Table 3, it was found that 1a and 1c bear phenyls while the others do not, and the lone pair electrons on N can form p-π conjugation with the phenyls which disperses the electronic density of the N atom. Without enough electron cloud, the N atom was more resistant to nucleophilic attack by the carbonium ion of halohydrocarbon followed by a Hofmann alkylation reaction mechanism [29,30]. The side reactions of 1a and 1c were reduced while for 1b, 1d and 1e no phenyl p-π conjugation is possible. Using the optimized experimental procedures the results also revealed that the series 1b, 1d and 1e were too sensitive to variations. The reaction always led to poor results, even if the temperature was slightly higher. Therefore, different structural skeletons have different effects on this reaction. Further investigations are necessary to analyze this in more detail.
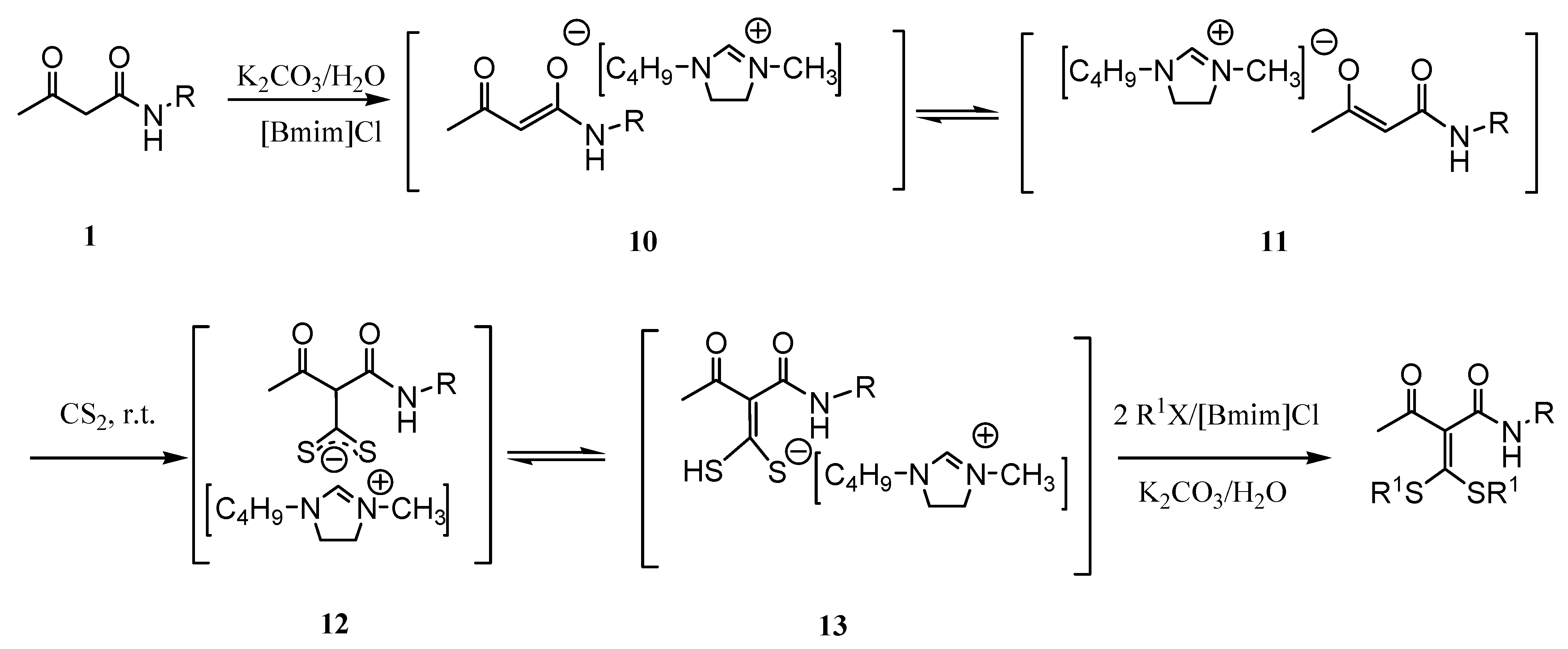
On the basis of these results, in combination with a former study [8], a tentative mechanism of [Bmim]Cl catalytic synthesis of α-oxoketene S,S-acetals was shown in Scheme 1. Enolates 10 and 11 can be derived from deprotonation of 1,3-dicarbonyl compounds in the present of [Bmim]Cl/H2O by a weak base. Then enolates reaction with CS2 resulted in 12 and 12 isomerized to give the unsaturated monothiolate anion 13. Compound 13 undergoes the S-alkylation with the halohydrocarbon to yield the product α-oxoketene S,S-acetal.
3. Experimental
3.1. Preparation of Materials
All reagents used were purchased from the Lanzhou Institute of Chemical Physics, China National Medicines Corporation Ltd and other commercial suppliers and were used as supplied unless otherwise stated. TLC was carried out on a Merck Kieselgel GF254. Purification or separation of product was carried out with flash column chromatography using 300–400 mesh silica gel. 1H-NMR and 13C-NMR spectra were determined using TMS as an internal reference with a Bruker Avance-300 NMR spectrometer operating at 300 MHz and 75 MHz, respectively. MS analysis was carried out on an API-3000 LC-MS-MS instrument.
N-Cyclohexylacetoacetamide/N-Butyl-3-oxo-butanamide was synthesized according to the previous literature method [31]. Diketene (10 mmol) was added dropwise in a mixture of cyclohexylamine/1-butylamine (10 mmol) and benzene or water (20 mL) over 1 hour at 0 °C. Then the resulting mixture was stirred for 2 h at room temperature. After the reaction completed as indicated by TLC, extraction of the resulting mixture and purification the crude product furnished N-cyclohexylacetoacetamide/N-butyl-3-oxo-butanamide in 94%/89% yield.
3.2. General Procedure for the Preparation of α-oxoketene S,S-acetals 6a-7a, 7b-9b and 6c-7c
1,3-Dicarbonyl compounds (5 mmol) and [Bmim]Cl (20 mmol) were added to water (15 mL) at 0 °C. After stirred for 3 min, potassium carbonate (12.5 mmol) was added and then the mixture was stirred for an hour at 0 °C. Then carbon disulfide (6 mmol) was added, and the mixture was stirred for another hour and then halohydrocarbon [5 mmol, 1,3-dibromopropane (6a and 6c), 5 mmol 1,2-dibromoethane (7a, 7b and 7c), 10 mmol bromoethane (8b) or 10 mmol iodomethane (9b)] was added dropwise over 30 min, then the resulting mixture was stirred for about 5-6 hours at room temperature. Because the product was not dissolved in the [Bmim]Cl/H2O system and acted as solid state, the product was filtrated and washed with water and the residual liquid could be reused without any treatment. After drying under vacuum for 5 hours, a crude solid was obtained. Purification by column chromatography afforded the product as a yellowish or white solid in moderate yield. All compounds exhibited spectral data consistent with their structures. 1H-NMR, 13C-NMR and MS spectral data of compounds were shown as below:
N-Phenyl-2-(1,3-dithian-2-ylidene)-3-oxobutanamide (6a): white solid, 1H-NMR (CDCl3) δ: 8.14 (s, 1H, -NH-), 7.65(d, J = 7.9 Hz, 2H, H-Ph), 7.37 (t, J = 7.9 Hz, 2H, H-Ph), 7.16 (t, J = 7.4 Hz, 1H, H-Ph), 2.98 (t, J = 7.0 Hz, 4H, -CH2-), 2.39 (s, 3H, -CH3), 2.32–2.16 (m, 2H, -CH2-); 13C-NMR (CDCl3) δ: 193.88, 164.78, 164.27, 137.86, 132.42, 129.12, 124.71, 119.99, 29.35, 29.17, 28.94, 23.98. MS (ESI): [M+H]+ 294.1.
N-Phenyl-2-(1,3-dithiolan-2-ylidene)-3-oxobutanamide (7a): white solid, 1H-NMR (CDCl3) δ: 8.19 (s, 1H, -NH-), 7.65 (d, J = 7.4 Hz, 2H, H-Ph), 7.38 (t, J = 7.9 Hz, 2H, H-Ph), 7.17 (t, J = 7.4 Hz, 1H, H-Ph), 3.48–3.41 (m, 2H, -CH2-), 3.40–3.32 (m, 2H, -CH2-), 2.42 (s, 3H, -CH3); 13C-NMR (CDCl3) δ: 192.91, 170.10, 165.19, 137.68, 129.16, 124.82, 123.77, 120.09, 38.54, 36.62, 28.63. MS (ESI): [M+H]+ 280.0.
N-Cyclohexyl-2-(1,3-dithiolan-2-ylidene)-3-oxobutanamide (7b): white solid, 1H-NMR (CDCl3) δ: 5.84 (d, J = 7.8 Hz, 1H, -NH-), 4.04–3.82 (m, 1H, -CH-), 3.47–3.34 (m, 2H, -CH2-), 3.34–3.21 (m, 2H, -CH2-), 2.28 (s, 3H, -CH3), 2.09–1.93 (m, 2H, -CH2-), 1.82–1.67 (m, 2H, -CH2-), 1.65–1.09 (m, 6H, -CH2-CH2-CH2-); 13C-NMR (CDCl3) δ: 191.76, 167.13, 166.58, 124.19, 48.78, 38.68, 36.29, 32.82, 27.86, 25.53, 24.78. MS (ESI): [M+H]+ 286.1.
N-Cyclohexyl-2-(bis(ethylthio)methylene)-3-oxobutanamide (8b): white solid, 1H-NMR (CDCl3) δ: 6.05 (d, J = 7.1 Hz, 1H, -NH-), 3.91–3.77 (m, 1H, -CH-), 2.88 (q, J = 7.4 Hz, 4H, -S-CH2-), 2.39 (s, 3H, -CO-CH3), 2.07–1.85 (m, 2H, -CH2-), 1.70–0.98 (m, 8H, -CH2-CH2-CH2-CH2-), 1.27 (t, J = 7.4 Hz, 6H, -CH3); 13C-NMR (CDCl3) δ: 197.76, 164.04, 147.26, 142.22, 48.61, 32.78, 30.13, 29.29, 25.53, 24.73, 14.76. MS (ESI): [M+H]+ 316.1.
N-Cyclohexyl-2-(bis(methylthio)methylene)-3-oxobutanamide (9b): white solid, 1H-NMR (CDCl3) δ: 5.99 (d, J = 7.2 Hz, 1H, -NH-), 3.93–3.76 (m, 1H, -CH-), 2.41 (s, 6H, -S-CH3), 2.37 (s, 3H, -CH3), 1.95 (dd, J = 12.8, 3.3 Hz, 2H, -CH2-), 1.78–1.51 (m, 4H, -CH2-CH2-), 1.47–1.02 (m, 6H, -CH2-CH2-CH2-); 13C-NMR (CDCl3) δ: 196.94, 164.33, 152.86, 139.40, 48.67, 32.77, 29.79, 25.53, 24.74, 18.81, 17.81. MS (ESI): [M+H]+ 288.1.
N-(2-Methylphenyl)-2-(1,3-dithian-2-ylidene)-3-oxobutanamide (6c): white solid, 1H-NMR (CDCl3) δ: 8.00 (d, J = 8.3 Hz, 1H, H-Ph), 7.88 (s, 1H, -NH-), 7.26–7.17 (m, 2H, H-Ph), 7.10 (t, J = 7.5 Hz, 1H, H-Ph), 3.00 (t, J = 6.9 Hz, 4H, -CH2-), 2.44 (s, 3H, -CO-CH3), 2.32 (s, 3H, CH3-Ph), 2.31–2.21 (m, 2H, -CH2-); 13C-NMR (CDCl3) δ: 194.75, 164.54, 163.96, 135.55, 132.28, 130.57, 128.92, 126.84, 125.33, 122.61, 29.41, 28.98, 24.08, 18.00. MS (ESI): [M+H]+ 308.1.
N-(2-Methylphenyl)-2-(1,3-dithiolan-2-ylidene)-3-oxobutanamide (7c): white solid, 1H-NMR (CDCl3) δ: 8.08 (s, 1H, H-Ph), 7.96 (d, J = 6.5 Hz, 1H, -NH-), 7.26–7.20 (m, 2H, H-Ph), 7.15–7.04 (m, 1H, H-Ph), 3.52–3.42 (m, 2H, -CH2-), 3.41–3.32 (m, 2H, -CH2-), 2.47 (s, 3H, -CO-CH3), 2.34 (s, 3H, CH3-Ph); 13C-NMR (CDCl3) δ: 193.33, 170.13, 165.16, 135.55, 130.64, 129.36, 126.84, 125.46, 123.68, 122.87, 38.45, 36.71, 28.87, 18.11. MS (ESI): [M+H]+ 294.1.
3.3. General Procedure for the Preparation of α-oxoketene S,S-acetals 7d-9d and 7e-9e
1,3-Dicarbonyl compounds (5 mmol) and [Bmim]Cl (20 mmol) were added into water (15 mL) at 0 °C. After stirring for 3 min, potassium carbonate (12.5 mmol) was added and then the mixture was stirred for another hour at 0 °C. Then carbon disulfide (6 mmol) was added, and the mixture was stirred for another hour and then halohydrocarbon [5 mmol, 1,2-dibromoethane (7d and 7e), 10 mmol bromoethane (8d and 8e) or 10 mmol iodomethane (9d and 9e)] was added dropwise over 30 min, then the resulting mixture was stirred for about 6 hours at room temperature. Because the product was dissolved in the [Bmim]Cl/H2O system and acted as liquid state, the product was extracted by ether for four times and the residual liquid could be reused without any treatment. Purification with column chromatography afforded the product as a yellowish solid or yellow oil in moderate yield.
N-Methyl-2-(1,3-dithiolan-2-ylidene)-3-oxobutanamide (7d): yellowish solid, 1H-NMR (CDCl3) δ: 6.14 (s, 1H, -NH-), 3.47–3.26 (m, 4H, -CH2-), 2.97 (d, J = 4.9 Hz, 3H, CH3-N), 2.32 (s, 3H, -CH3); 13C-NMR (CDCl3) δ: 192.22, 168.17, 123.73, 38.62, 36.35, 28.15, 26.71. MS (ESI): [M+H]+ 218.0.
N-Methyl-2-(bis(ethylthio)methylene)-3-oxobutanamide (8d): yellow solid, 1H-NMR (CDCl3) δ: 6.31 (s, 1H, -NH-), 2.90 (q, J = 7.4 Hz, 4H, -CH2-), 2.89 (d, J = 4.9 Hz, 3H, CH3-N), 2.43 (s, 3H, -CO-CH3), 1.29 (t, J = 7.4 Hz, 6H, -CH3); 13C-NMR (CDCl3) δ: 198.52, 165.28, 147.45, 141.76, 30.40, 29.21, 26.56, 14.69. MS (ESI): [M+H]+ 248.1.
N-Methyl-2-(bis(methylthio)methylene)-3-oxobutanamide (9d): yellow solid, 1H-NMR (CDCl3) δ: 6.25 (s, 1H, -NH-), 2.88 (d, J = 4.9 Hz, 3H, N-CH3), 2.40 (s, 3H, -S-CH3), 2.40(s, 3H, -S-CH3), 2.39 (s, 3H, -CO-CH3); 13C-NMR (CDCl3) δ: 197.78, 165.54, 152.96, 138.93, 30.07, 26.62, 18.17. MS (ESI): [M+H]+ 220.0.
N-(n-Butyl)-2-(1,3-dithiolan-2-ylidene)-3-oxobutanamide (7e): yellowish solid, 1H-NMR (CDCl3) δ: 6.09 (s, 1H, -NH-), 3.48–3.23 (m, 6H, -CH2-), 2.28 (s, 3H, -CO-CH3), 1.65–1.52 (m, 2H, -CH2-), 1.49–1.29 (m, 2H, -CH2-), 0.93 (t, J = 7.3 Hz, 3H, -CH3); 13C-NMR (CDCl3) δ: 192.00, 167.61, 167.50, 124.01, 39.80, 38.65, 36.33, 31.44, 28.01, 20.23, 13.75. MS (ESI): [M+H]+ 260.1.
N-(n-Butyl)-2-(bis(ethylthio)methylene)-3-oxobutanamide (8e): yellow oil, 1H-NMR (CDCl3) δ: 6.24 (s, 1H, -NH-), 2.07 (q, J = 5.9 Hz, 2H, N-CH2-), 2.95–2.80 (m, 4H, -S-CH2-), 2.40 (s, 3H, -CO-CH3), 1.53–1.45 (m, 2H, -CH2-), 1.38–1.31 (m, 2H, -CH2-), 1.30–1.21 (m, 6H, -CH2-), 0.91 (t, J = 6.9 Hz, 3H, -CH3); 13C-NMR (CDCl3) δ: 198.30, 164.67, 147.07, 142.12, 39.63, 31.37, 30.31, 29.19, 20.15, 14.72, 13.71. MS (ESI): [M+H]+ 290.1.
N-(n-Butyl)-2-(bis(methylthio)methylene)-3-oxobutanamide (9e): yellow oil, 1H-NMR (CDCl3) δ: 6.22 (s, 1H, -NH-), 3.34–3.30 (m, 2H, N-CH2-), 2.40–2.36 (m, 9H, -CO-CH3, -S-CH3), 1.53–1.48 (m, 2H, -CH2-), 1.37–1.32 (m, 2H, -CH2-), 0.94–0.87 (m, 3H, -CH3); 13C-NMR (CDCl3) δ: 197.44, 165.02, 152.80, 139.23, 39.69, 31.32, 29.95, 20.15, 18.19, 13.72. MS (ESI): [M+H]+ 262.1.
4. Conclusions
In conclusion, a clean, environmentally friendly method has been developed for the synthesis of α-oxoketene S,S-acetals in moderate yields based on the reaction of 1,3-dicarbonyl compounds with carbon disulfide and halohydrocarbon in the presence of potassium carbonate in a [Bmim]Cl/water homogeneous system. A simple procedure, mild conditions, moderate yields, multiple recycling of wet ionic liquid and environmental tolerability make the present protocol highly attractive for academic research and practical applications.
Acknowledgments
We gratefully acknowledge the financial supports by Key Program for Science and Technology Development of Harbin (2009AA3BS083), Heilongjiang Province Science Foundation for Excellent Youths (JC200704), Agricultural Science and Technology Achievements Transformation Fund Program (2009GB23600514), Project for Distinguished Teacher Abroad, Chinese Ministry of Education (MS2010DBLY031), Fundamental Research Funds for the Central Universities (DL09EA04), the Special Fund of Forestry Industrial Research for Public Welfare of China (201004040) and Prof. Qun Liu for the guidance.
References
- Kumar, S.; Peruncheralathan, S.; Ila, H.; Junjappa, H. A Novel Anionic Domino Process for the Synthesis of o-Cyanoaryl-Methylthio/Alkyl/Aryl/Heteroaryl Acetylenes. Org. Lett. 2008, 10, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Sasikala, K.A.; Kalesh, K.A.; Anabha, E.R.; Pillai, P.M.; Asokan, C.V.; Devaky, K.S. Synthesis of 2,3,5-trisubstituted furans from [alpha]-formylaroylketene dithioacetals. Tetrahedron Lett. 2011, 52, 1667–1669. [Google Scholar] [CrossRef]
- Li, Y.; Liang, F.; Bi, X.; Liu, Q. Intramolecular Thia-anti-Michael Addition of a Sulfur Anion to Enones: A Regiospecific Approach to Multisubstituted Thiophene Derivatives. J. Org. Chem. 2006, 71, 8006–8010. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Dong, D.; Liu, Q.; Pan, W.; Zhao, L.; Li, B. [5 + 1] Annulation: A Synthetic Strategy for Highly Substituted Phenols and Cyclohexenones. J. Am. Chem. Soc. 2005, 127, 4578–4579. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fu, Z.; Feng, H.; Dong, Y.; Liu, J.; Liu, Q. Tandem [4 + 1 + 1] annulation and metal-free aerobic oxidative aromatization: straightforward synthesis of highly substituted phenols from one aldehyde and two ketones. Chem. Commun. 2010, 46, 9061–9063. [Google Scholar] [CrossRef] [PubMed]
- Kelber, C.; Dtsch, B. Über die Einwirkung von Schwefelkohlenstoff und Ätzkali auf Acetophenon. Chem. Ges. 1910, 43, 1252–1259. [Google Scholar] [CrossRef]
- Choi, E.B.; Youn, I.K.; Pak, C.S. Preparation of Protected β- Keto Esters via Selective Reduction of Acyl(alkoxycarbonyl)ketene Dithioacetals. Synthesis 1988, 792–794. [Google Scholar] [CrossRef]
- Ouyang, Y.; Dong, D.; Yu, H.; Liang, Y.; Liu, Q. A Clean, Facile and Practical Synthesis of α-Oxoketene S,S-Acetals in Water. Adv. Synth. Catal. 2006, 348, 206–210. [Google Scholar] [CrossRef]
- Muthusamy, S.; Gnanaprakasam, B. Imidazolium salts as phase transfer catalysts for the dialkylation and cycloalkylation of active methylene compounds. Tetrahedron Lett. 2005, 46, 635–638. [Google Scholar] [CrossRef]
- Welton, T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhou, Q.; He, X.; Wei, S.; Wang, L.; van Kasteren, J.M.N.; Wang, Y.Z. A highly efficient approach for dehydrochlorinating polyvinyl chloride: Catalysis by 1-butyl-3-methylimidazolium chloride. Green Chem. 2010, 12, 1062–1065. [Google Scholar] [CrossRef]
- Bai, S.; Song, B.; Bhadury, P.S.; Yang, S.; Hu, D.; Xue, W. [BMIM]Cl Catalyzed One-Pot Synthesis of α-Aminophosphonate Derivatives Containing a 4-Phenoxyquinazoline Moiety under Microwave Irradiation. Chin. J. Chem. 2011, 29, 109–117. [Google Scholar] [CrossRef]
- Sun, H.; Li, X.; Sundermeyer, J. Aerobic oxidation of phenol to quinone with copper chloride as catalyst in ionic liquid. J. Mol. Catal. A Chem. 2005, 240, 119–122. [Google Scholar] [CrossRef]
- Li, H.; Zhu, W.; Wang, Y.; Zhang, J.; Lu, J.; Yan, Y. Deep oxidative desulfurization of fuels in redox ionic liquids based on iron chloride. Green Chem. 2009, 11, 810–815. [Google Scholar] [CrossRef]
- Yuan, X.; Chen, M.; Dai, Q.; Cheng, X. Friedel-Crafts acylation of anthracene with oxalyl chloride catalyzed by ionic liquid of [bmim]Cl/AlCl3. Chem. Eng. J. 2009, 146, 266–269. [Google Scholar]
- Liu, F.; Li, Z.; Yu, S.; Cui, X.; Ge, X. Environmentally benign methanolysis of polycarbonate to recover bisphenol A and dimethyl carbonate in ionic liquids. J. Hazard. Mater. 2010, 174, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wu, C.; Christopher, B.W.; Quan, J.; Zhu, L. Ionic Liquids—Promoted S-Methylation of Thiols Utilizing Dimethyl Carbonate. Phosphorus, Sulfur Silicon Relat. Elem. 2011, 186, 31–37. [Google Scholar] [CrossRef]
- Tilve, R.D.; Alexander, M.V.; Khandekar, A.C.; Samant, S.D.; Kanetkar, V.R. Synthesis of 2,3-unsaturated glycopyranosides by Ferrier rearrangement in FeCl3 based ionic liquid. J. Mol. Catal. A: Chem. 2004, 223, 237–240. [Google Scholar] [CrossRef]
- Qi, X.; Watanabe, M.; Aida, T.M.; Smith, R.L. Efficient Catalytic Conversion of Fructose into 5-Hydroxymethylfurfural in Ionic Liquids at Room Temperature. ChemSusChem 2009, 2, 944–946. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, S.J.; Yu, C.S.; Ma, Y.C.; Li, K.L.; Lin, L.W. Direct conversion of methane to methanol over nano-[Au/SiO2] in [Bmim]Cl ionic liquid. Appl. Catal. A Gen. 2011, 398, 150–154. [Google Scholar] [CrossRef]
- Qi, X.; Watanabe, M.; Aida, T.M.; Smith, J.R.L. Efficient process for conversion of fructose to 5-hydroxymethylfurfural with ionic liquids. Green Chem. 2009, 11, 1327–1331. [Google Scholar] [CrossRef]
- Brown, R.A.; Pollet, P.; McKoon, E.; Eckert, C.A.; Liotta, C.L.; Jessop, P.G. Asymmetric Hydrogenation and Catalyst Recycling Using Ionic Liquid and Supercritical Carbon Dioxide. J. Am. Chem. Soc. 2001, 123, 1254–1255. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liu, Y.; Shi, S.; Wang, Y. A simple, efficient and green procedure for Knoevenagel condensation catalyzed by [C4dabco][BF4] ionic liquid in water. Green Chem. 2010, 12, 514–517. [Google Scholar] [CrossRef]
- De Nino, A.; Bortolini, O.; Maiuolo, L.; Garofalo, A.; Russo, B.; Sindona, G. A sustainable procedure for highly enantioselective organocatalyzed Diels-Alder cycloadditions in homogeneous ionic liquid/water phase. Tetrahedron Lett. 2011, 52, 1415–1417. [Google Scholar] [CrossRef]
- Odinets, I.L.; Matveeva, E.V. Ionic Liquids and Water as “Green” Solvents in Organophosphorus Synthesis. Curr. Org. Chem. 2010, 14, 1171–1184. [Google Scholar] [CrossRef]
- Fang, D.; Zhang, D.; Liu, Z. One-pot three-component Biginelli-type reaction catalyzed by ionic liquids in aqueous media. Monatsh. Chem. 2010, 141, 419–423. [Google Scholar] [CrossRef]
- Gutowski, K.E.; Broker, G.A.; Willauer, H.D.; Huddleston, J.G.; Swatloski, R.P.; Holbrey, J.D.; Rogers, R.D. Controlling the Aqueous Miscibility of Ionic Liquids: Aqueous Biphasic Systems of Water-Miscible Ionic Liquids and Water-Structuring Salts for Recycle, Metathesis, and Separations. J. Am. Chem. Soc. 2003, 125, 6632–6633. [Google Scholar] [CrossRef] [PubMed]
- Mele, A.; Tran, C.D.; De Paoli Lacerda, S.H. The Structure of a Room-Temperature Ionic Liquid with and without Trace Amounts of Water: The Role of C H O and C H F Interactions in 1-n-Butyl-3-Methylimidazolium Tetrafluoroborate. Angew. Chem. Int. Ed. 2003, 42, 4364–4366. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.W. Ueber zwei neue Reihen flüchtiger organischer Basen. Justus Liebigs Annalen der Chemie 1850, 73, 91–92. [Google Scholar] [CrossRef]
- Hofmann, A.W. Beiträge zur Kenntniss der flüchtigen organischen Basen. Justus Liebigs Annalen der Chemie 1851, 79, 11–39. [Google Scholar] [CrossRef]
- Raleigh, E.W.; Wilmington, D. Preparation of 1-(carbamoyl)-N-(carbamoyloxy)thioformimidates from acetoacetamides. U.S. Patent 3,557,089, 19 January 1971. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |
Scheme 1.
A tentative mechanism of [Bmim]Cl catalytic synthesis of α-oxoketene S,S-acetals.


{kind=link}
{kind=link}
Table 1.
Reaction of 1a with carbon disulfide and 1,2-dibromoethane in ionic liquids/water under different conditions.
Table 1.
Reaction of 1a with carbon disulfide and 1,2-dibromoethane in ionic liquids/water under different conditions.

| Entry | Reaction Medium | Amount a (mmol)/(mL) | K2CO3 (mmol) | 1a (mmol) | Time b (h) | Yield c (%) |
|---|---|---|---|---|---|---|
| 1 | H2O | 15 (mL) | 12.5 | 5 | 20 | 0 |
| 2 | [EPy]BF4 | 50 (mmol) | 12.5 | 5 | 20 | trace |
| 3 | [Bmim]Cl | 50 (mmol) | 12.5 | 5 | 20 | trace |
| 4 | [EPy]BF4/H2O | 50/15 | 12.5 | 5 | 15 | 71 |
| 5 | [Bmim]Cl/H2O | 50/15 | 12.5 | 5 | 5 | 73 |
| 6 | [Bmim]Cl/H2O | 40/15 | 12.5 | 5 | 5 | 74 |
| 7 | [Bmim]Cl/H2O | 30/15 | 12.5 | 5 | 5 | 73 |
| 8 | [Bmim]Cl/H2O | 20/15 | 12.5 | 5 | 5 | 74 |
| 9 | [Bmim]Cl/H2O | 10/15 | 12.5 | 5 | 8 | 65 |
| 10 | [Bmim]Cl/H2O | 5/15 | 12.5 | 5 | 11 | 61 |
| 11 | [Bmim]Cl/H2O | 0.1/15 | 12.5 | 5 | 20 | 43 |
a Amount of reaction medium; b The reaction time was recorded after the addition of 1,2-dibromoethane; c Yields of 7a were obtained by purification with column chromatography.
Table 2.
Recycling of [Bmim]Cl/water in synthesis of 7a.
| Entry | Reuse times | Reaction medium | 1a (mmol) | CS2 (mmol) | BrCH2CH2Br (mmol) | Time a (h) | Yield b (%) |
|---|---|---|---|---|---|---|---|
| 1 | 2 | [Bmim]Cl/H2O c | 5 | 6 | 5 | 5 | 70 |
| 2 | 3 | [Bmim]Cl/H2O d | 5 | 6 | 5 | 6 | 65 |
| 3 | 4 | [Bmim]Cl/H2O e | 5 | 6 | 5 | 6 | 65 |
| 4 | 5 | [Bmim]Cl/H2O f | 5 | 6 | 5 | 7 | 66 |
a The reaction time was recorded after the addition of 1,2-dibromoethane; b Yields of 7a were obtained by purification with column chromatography; c [Bmim]Cl/H2O was obtained after filtration the product of Table 1 Entry 8 and without any treatment; d [Bmim]Cl/H2O was obtained after filtration the product of Table 2 Entry 1 and without any treatment; e [Bmim]Cl/H2O was obtained after filtration the product of Table 2 Entry 2 and without any treatment; f [Bmim]Cl/H2O was obtained after filtration the product of Table 2 Entry 3 and without any treatment.
Table 3.
Preparation of α-oxoketene S,S-acetals 6a-9e from 1,3-dicarbonyl compounds 1 in [Bmim]Cl/H2O.
Table 3.
Preparation of α-oxoketene S,S-acetals 6a-9e from 1,3-dicarbonyl compounds 1 in [Bmim]Cl/H2O.

| Entry | Substrate | Substrate | Time a (h) | Product | Yield b (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 1 R | 2–5 | X R1 | R1 | ||||||
| 1 | 1a | C6H5 | 2 | Br | -(CH2)3- | 5 | 6a | 73 | |
| 2 | 1a | C6H5 | 3 | Br | -(CH2)2- | 5 | 7a | 74 | |
| 3 | 1b | CH(CH2)5 | 3 | Br | -(CH2)2- | 6 | 7b | 60 | |
| 4 | 1b | CH(CH2)5 | 4 | Br | -C2H5 | -C2H5 | 6 | 8b | 55 |
| 5 | 1b | CH(CH2)5 | 5 | I | -CH3 | -CH3 | 6 | 9b | 53 |
| 6 | 1c | o-Methylphenyl | 2 | Br | -(CH2)3- | 6 | 6c | 70 | |
| 7 | 1c | o-Methylphenyl | 3 | Br | -(CH2)2- | 6 | 7c | 72 | |
| 8 | 1d | -CH3 | 3 | Br | -(CH2)2- | 6 | 7d | 65 | |
| 9 | 1d | -CH3 | 4 | Br | -C2H5 | -C2H5 | 6 | 8d | 61 |
| 10 | 1d | -CH3 | 5 | I | -CH3 | -CH3 | 6 | 9d | 60 |
| 11 | 1e | -(CH2)3CH3 | 3 | Br | -(CH2)2- | 6 | 7e | 66 | |
| 12 | 1e | -(CH2)3CH3 | 4 | Br | -C2H5 | -C2H5 | 6 | 8e | 55 |
| 13 | 1e | -(CH2)3CH3 | 5 | I | -CH3 | -CH3 | 6 | 9e | 58 |
a The reaction time was recorded after the addition of halohydrocarbon; b Yields of 6a-9e obtained by purification with column chromatography.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Yu, P.; Zu, Y.; Fu, Y.; Efferth, T. Catalytic Synthesis of α-Oxoketene S,S-Acetals in a Wet Ionic Liquid [Bmim]Cl/H2O Homogeneous System. Molecules 2011, 16, 4500-4510. https://doi.org/10.3390/molecules16064500
AMA Style
Yu P, Zu Y, Fu Y, Efferth T. Catalytic Synthesis of α-Oxoketene S,S-Acetals in a Wet Ionic Liquid [Bmim]Cl/H2O Homogeneous System. Molecules. 2011; 16(6):4500-4510. https://doi.org/10.3390/molecules16064500
Chicago/Turabian StyleYu, Ping, Yuangang Zu, Yujie Fu, and Thomas Efferth. 2011. "Catalytic Synthesis of α-Oxoketene S,S-Acetals in a Wet Ionic Liquid [Bmim]Cl/H2O Homogeneous System" Molecules 16, no. 6: 4500-4510. https://doi.org/10.3390/molecules16064500