Biologically Important Eremophilane Sesquiterpenes from Alaska Cedar Heartwood Essential Oil and Their Semi-Synthetic Derivatives


Abstract
:1. Introduction

2. Results and Discussion
2.1. GC Analysis of the Essential Oil of Alaska Cedar

{kind=link}
{kind=link}
| Compounds in oil | Percentage in oil (% w/w) | Retention time (min) |
|---|---|---|
| 4-terpineol | 2.08 | 8.4 |
| methylcarvacrol | 1.33 | 9.4 |
| carvacrol | 35.37 | 10.8 |
| valencene | 1.50 | 16.4 |
| nootkatene | 20.08 | 17.1 |
| nootkatol | 5.20 | 23.5 |
| valencene-13-ol | 6.35 | 25.3 |
| nootkatone | 17.39 | 26.6 |
| nootkatin | 3.50 | 31.6 |
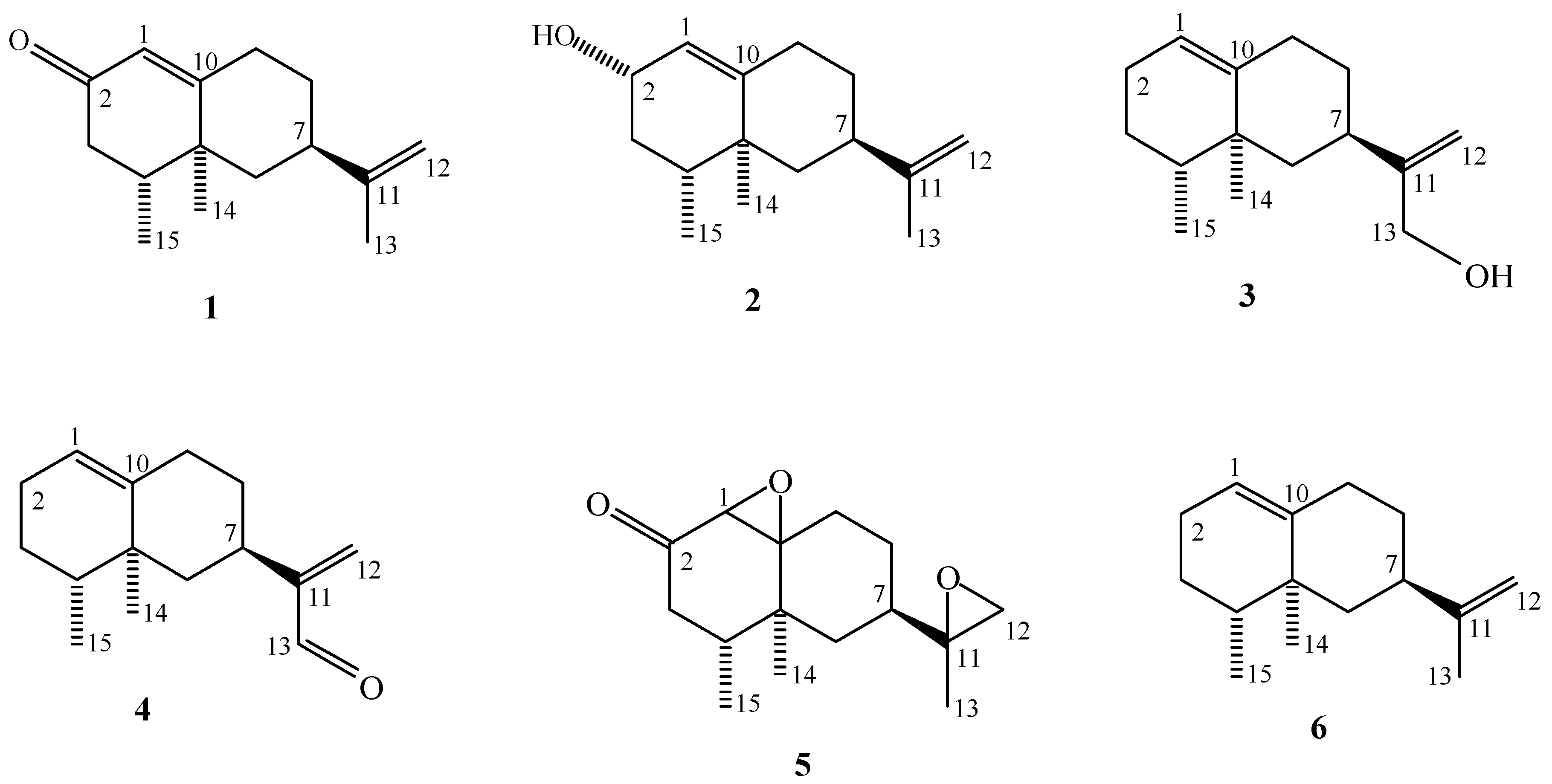
2.2. Isolation and Structure Determination of Isolated Compounds
| Fractions | Main components | Weight (g) | % in total (6.2 g oil) |
|---|---|---|---|
| I | valencene, nootkatene, methyl carvacrol | 1.13 | 18.2 |
| II | carvacrol | 2.06 | 33.2 |
| III | mixture of several minor compounds | 0.05 | 0.8 |
| IV | 3, nootkatone | 1.80 | 29.1 |
| V | mixture of several minor compounds | 0.35 | 5.6 |
| VI | 2, other minor compounds | 0.51 | 8.2 |
| VII | 2 | 0.11 | 1.8 |
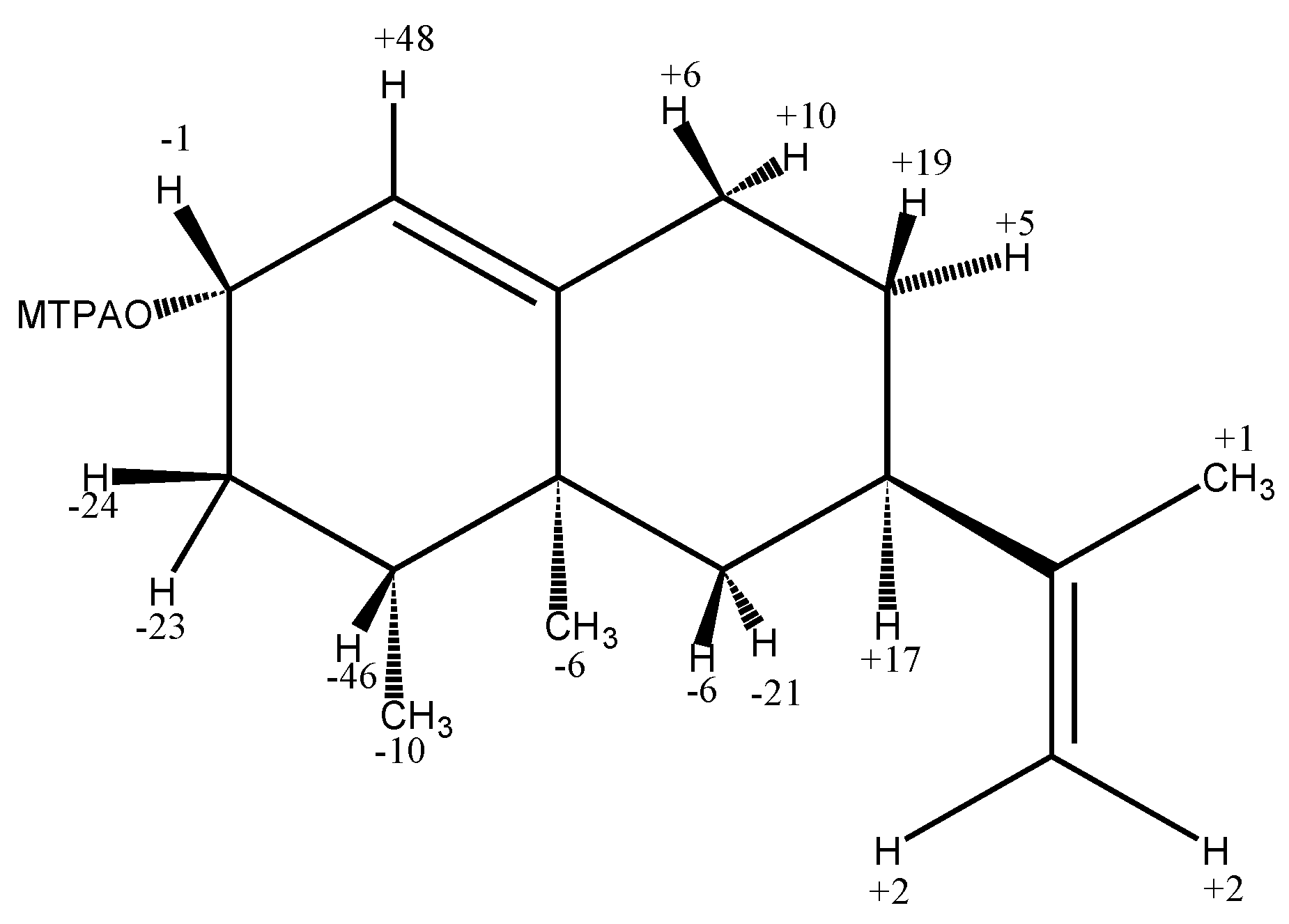
2.2.1. Nootkatol (eremophil-1(10),11-dien-2-ol) (2)
| Carbon | 13C | 1H |
|---|---|---|
| 1 | 124.7 | 5.32, 1H, d, J = 1.6 Hz |
| 2 | 68.4 | 4.25, 1H, m |
| 3 | 37.6 | (a) 1.76, 1H, td, J = 2.0, 6.5 Hz |
| (b) 1.37, 1H, td, J = 12.4, 10.0 Hz | ||
| 4 | 39.7 | 1.51, 1H, br d, J = 2.1 Hz |
| 5 | 38.6 | — |
| 6 | 45.0 | (a) 1.85, 1H, dd, J = 12.6, 2.7 Hz |
| (b) 0.95, 1H, m, J = 2.7 Hz | ||
| 7 | 41.2 | 2.25, 1H, tt, J = 12.4, 3.0 Hz |
| 8 | 32.8 | (a) 2.33, 1H, m |
| (b) 2.1, 1H, ddd, J = 14.1,4.2, 2.6 Hz | ||
| 9 | 33.3 | (a) 1.79, 1H, dd, J = 2.0, 4.5 Hz |
| (b) 1.20, 1H, dm, J = 4.3 Hz | ||
| 10 | 146.5 | — |
| 11 | 150.6 | — |
| 12 | 108.9 | 4.68, 2H, br s |
| 13 | 21.2 | 1.71, 3H, s |
| 14 | 18.6 | 0.99, 3H, s |
| 15 | 15.8 | 0.89, 3H, d, J = 6.9 Hz |

2.2.2. Eremophil-1(10),11-dien-13-ol (3)
| carbon | 13C (ppm) | 1H (ppm) |
|---|---|---|
| 1 | 120.8 | 5.33, 1H, t, J = 2.4 Hz |
| 2 | 26.3 | 2.01, 2H, m |
| 3 | 27.5 | 1.41-1.47, 3H, m |
| 4 | 41.3 | |
| 5 | 38.3 | — |
| 6 | 45.8 | (a) 1.01, 1H, d, J = 12.6 Hz |
| (b) 1.93, 1H, m | ||
| 7 | 37.1 | 2.31, 1H, m |
| 8 | 33.1 | (a) 2.09, 1H, td, J = 13.4, 3.4 Hz |
| (b) 2.31, 1H, m | ||
| 9 | 34.0 | (a) 1.82, 1H, m |
| (b) 1.21, 1H, dd, J = 4.1, 13.3 Hz | ||
| 10 | 143.2 | — |
| 11 | 154.5 | — |
| 12 | 108.3 | (a) 5.02, 1H, s |
| (b) 4.88, 1H, s | ||
| 13 | 65.7 | 4.12, 2H, s |
| 14 | 18.8 | 0.95, 3H, s |
| 15 | 16.0 | 0.87, 3H, d, J = 6.0 Hz |
2.3. Synthesis
2.3.1. Synthesis of nootkatone-1,10-11,12-diepoxide (5)
2.3.2. Synthesis of valencene-13-aldehyde (4)
3. Experimental
3.1. General
3.2. Plant Material and Essential Oil
3.3. Analysis of Essential Oil
3.4. Chromatographic Fractionation of the Essential Oil
3.5. Synthesis of Nootkatol-(R)-α-methoxy-α-(trifluoromethyl)phenyl acetate and Nootkatol-(S)-α-methoxy-α-(trifluoromethyl)phenyl acetate
3.6. Synthesis of Valencene-13-aldehyde (4)
3.7. Synthesis of Nootkatone-diepoxide (5)
4. Conclusions
References and Notes
- Barton, G.M. A review of yellow cedar (Chamaecyparis nootkatensis [D. Don Spach) extractives and their importance to utilization. Wood Fiber Sci. Fall. 1976, 8, 172–176. [Google Scholar]
- Khasawneh, M.A.; Karchesy, J.J. Terpenoids of the Heartwood of Chamaecyparis nootkatensis. Nat. Prod. Comm. 2011. submitted. [Google Scholar]
- Panella, N.A.; Dolan, M.C.; Karchesy, J.J.; Xiong, Y.; Peralta-Cruz, J.; Khasawneh, M.; Montenieri, J.A.; Maupin, G.O. Use of novel compounds for pest control: Insecticidal and acaricidal activity of essential oil components from heartwood of Alaska yellow cedar (Chamaecyparis nootkatensis). J. Med. Entomol. 2005, 42, 352–358. [Google Scholar] [CrossRef]
- Dietrich, G.; Dolan, M.C.; Peralta-Cruz, J.; Schmidt, J.; Piesman, J.; Eisen, R.J.; Karchesy, J.J. Repellent activity of fractionated compounds from Chamaecyparis nootkatensis essential oil against Nymphal Ixodes scapularis (Acari: Ixodidae). J. Med. Entomol. 2006, 43, 957–961. [Google Scholar] [CrossRef]
- de Kraker, J.-W.; Schurink, M.; Franssen, M.C.R.; König, W.A.; de Groot, A.; Bouwmeester, H.J. Hydroxylation of sesquiterpenes by enzymes from chicory (Cichorium intybus L.) roots. Tetrahedron 2003, 59, 409–418. [Google Scholar] [CrossRef]
- Davies, A.G.; Davison, I.E.G. The rearrangements of allylic hydroperoxides derived from (+)-valencene. J. Chem. Soc. Perkin Trans. II 1989, 825–830. [Google Scholar]
- Shoji, N.; Umeyama, A.; Asakawa, Y.; Takemoto, T.; Nomoto, K.; Ohizumi, Y.J. Structural determination of nootkatol, a new sesquiterpene isolated from Alpinia oxyphylla miquel possessing calcium-antagonistic activity. J. Pharm. Sci. 1984, 73, 843–844. [Google Scholar] [CrossRef]
- Moshonas, M.G.; Shaw, P.E.J. Composition of aqueous essence and essence oil from Citrus temple. J.Agric. Food Chem. 1983, 31, 334–336. [Google Scholar] [CrossRef]
- Shaffer, G.W.; Eschinasi, E.H.; Purzycki, K.L.; Doerr, A.B. Oxidations of valencene. J. Org. Chem. 1975, 40, 2181–2185. [Google Scholar] [CrossRef]
- Miyazawa, M.; Nakamura, Y.; Ishikawa, Y. Insecticidal sesquiterpene from Alpinia oxyphylla against Drosophila melanogaster. J. Agric. Food Chem. 2000, 48, 3639–3641. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
- Sullivan, G.R.; Dale, J.A.; Mosher, H.S. Correlation of configuration and fluorine-19 chemical shifts of α-methoxy-α-trifluoromethylphenyl acetate derivatives. J. Org. Chem. 1973, 38, 2143–2147. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the isolated compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khasawneh, M.A.; Xiong, Y.; Peralta-Cruz, J.; Karchesy, J.J. Biologically Important Eremophilane Sesquiterpenes from Alaska Cedar Heartwood Essential Oil and Their Semi-Synthetic Derivatives. Molecules 2011, 16, 4775-4785. https://doi.org/10.3390/molecules16064775
Khasawneh MA, Xiong Y, Peralta-Cruz J, Karchesy JJ. Biologically Important Eremophilane Sesquiterpenes from Alaska Cedar Heartwood Essential Oil and Their Semi-Synthetic Derivatives. Molecules. 2011; 16(6):4775-4785. https://doi.org/10.3390/molecules16064775
Chicago/Turabian StyleKhasawneh, Mohammad A., Yeping Xiong, Javier Peralta-Cruz, and Joe J. Karchesy. 2011. "Biologically Important Eremophilane Sesquiterpenes from Alaska Cedar Heartwood Essential Oil and Their Semi-Synthetic Derivatives" Molecules 16, no. 6: 4775-4785. https://doi.org/10.3390/molecules16064775