An Emulsion Based Microarray Method to Detect the Toxin Genes of Toxin-Producing Organisms


Abstract
:1. Introduction
2. Results and Discussion
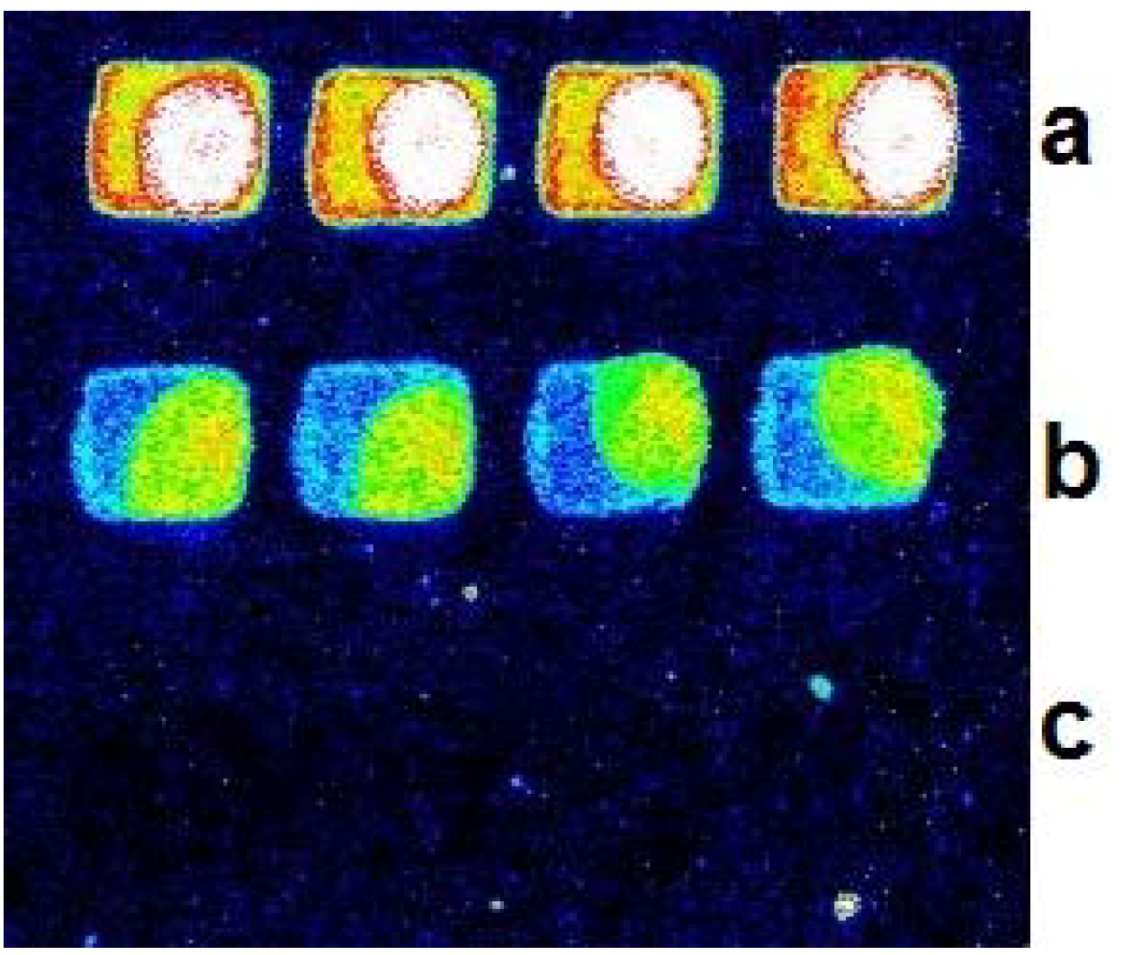
2.1. Comparison of Immobilization Approaches for Primer on Beads Surface

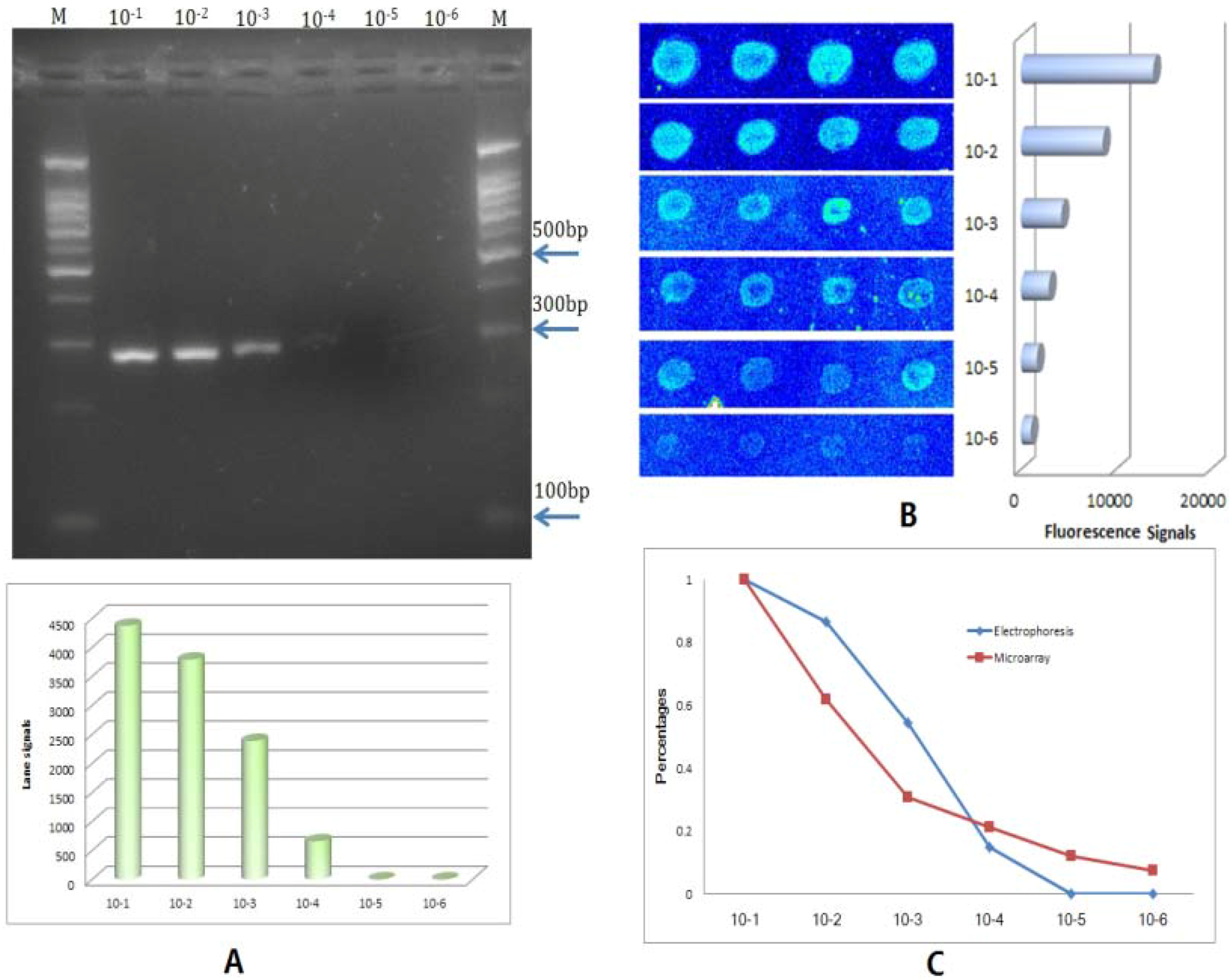
2.2. Reliability and Sensitivity of the BEAMing Based Method


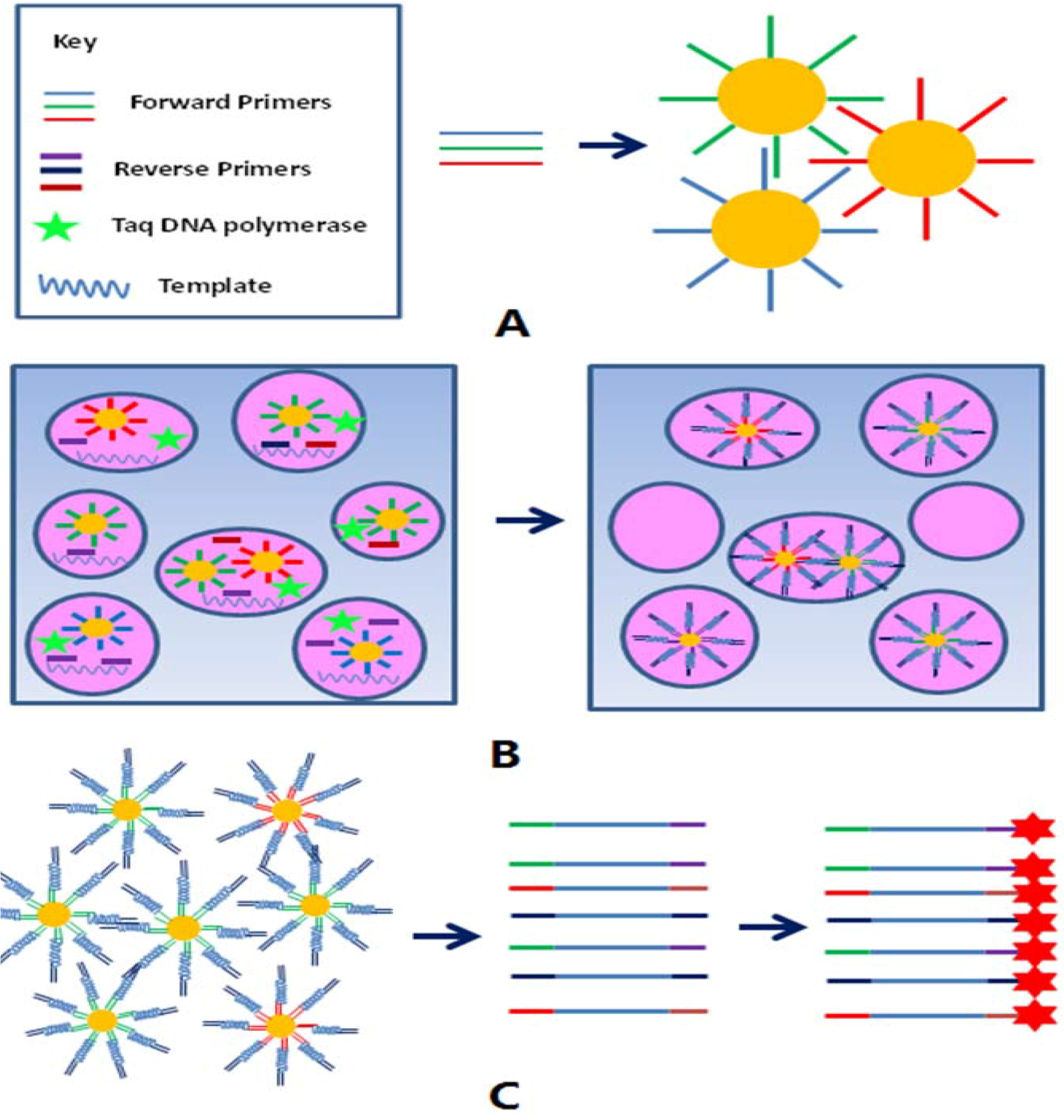
2.3. Detection of the Toxins Producing Bacteria with BEAMing Based Microarray Method

3. Experimental
3.1. Sample Preparation
3.2.Preparation of the Oligonucleotides and Microarray

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene name | Primers/Probes sequence (5’-3’) | Modification | GenBank No. | Toxins | |
|---|---|---|---|---|---|
| PKS4 | (F)gtatcttggagctgccttgc | gi:46562332 | Zearalenone | ||
| (R)ccaccacccaaaagcttaaa | |||||
| (P)ccatcggcactaggagacat | 5’-amino | ||||
| stcS | (F)ggtggtggagctgtgaatct | gi:50058538 | Sterigmatocystin | ||
| (R)ccgatgaggtcgttgttttt | |||||
| (P)gttcctgttctggccttctg | 5’-amino | ||||
| Tri8 | (F)tttgctggaacttgtgttgc | gi:4249355 | Trichothecene | ||
| (R)gtatacagcgccacctggat | |||||
| (P)ctagtcaagtttccaggcgc | 5’-amino | ||||
| Tri7 | (F)tgtttgcctcatcttcaacg | gi:4249355 | Trichothecene | ||
| (R)acattgccacgcaacaataa | |||||
| (P)agcaaagcatctttgtggct | 5’-amino | ||||
| stcA | (F)ggtggaacatgacacactgc | gi:50058538 | Sterigmatocystin | ||
| (R)gctacgtcttgggagtctgc | |||||
| (P)atttcaaggttatcgcgcac | 5’-amino | ||||
| verA | (F)tatggcctgtccctatctcg | gi:50058538 | Sterigmatocystin | ||
| (R)gctgtccaggaggtgaagag | |||||
| (P)tgctgtcctccaaccatgta | 5’-amino | ||||
| pksST | (F)gctacgtcttgggagtctgc | gi:50058538 | Sterigmatocystin | ||
| (R)ggtggaacatgacacactgc | |||||
| (P)gtgcgcgataaccttgaaat | 5’-amino | ||||
| omtA | (F)ctcctctaccagtggcttcg | gi:169775554 | Aflatoxin | ||
| (R)aacctccgagttggaatgtg | |||||
| (P)ccgcccatacctagatcaaa | 5’-amino | ||||
| Nor1 | (F)cacttagccagcacgatcaa | gi:169775554 | Aflatoxin | ||
| (R)tttgggacgttggagaaaag | |||||
| (P)ccgaggtacggtctatcgaa | 5’-amino | ||||
| Ver1 | (F)tccccaatggtgagactttc | gi:169775554 | Aflatoxin | ||
| (R)caccccaatgatctttccac | |||||
| (P)ccccataaactgcgtcttgt | 5’-amino | ||||
| pksA | (F)gaacgtaccggatgaagcat | gi:169775554 | Aflatoxin | ||
| (R)atgctgcagagcatgaacac | |||||
| (P)gaggcacactagagcggttc | 5’-amino | ||||
| PKS13 | (F)tgggcgcttaagactgagat | gi:46562332 | Zearalenone | ||
| (R)atttccccaccaaacatgaa | |||||
| (P)ttgaatcctggatccgaaag | 5’-amino | ||||
| UGT | (F)acgagaagctgatcgtggac | gi:21240774 | Deoxynivalenol | ||
| (R)ttacatgccagagccttcct | |||||
| (P)gttgaaggggaggacatgag | 5’-amino | ||||
| SRY | (F)acctgttgtccagttgcact | gi:4507224 | -- | ||
| (R)actgaaagctgtaactctaagta | |||||
| (P)tgaagcgacccatgaacgcattca | 5’-amino | ||||
3.3. Strategy of the Emulsion Based Detection Method

3.4. BEAMing
3.5. Separation and Labeling of the Target Molecules
3.6. Hybridization and Scanning
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Rasooly, L.; Rasooly, A. Real time biosensor analysis of staphylococcal enterotoxin A in food. Int. J. Food Microbiol. 1999, 49, 119–127. [Google Scholar] [CrossRef]
- Becker, K.; Roth, R.; Peters, G. Rapid and specific detection of toxigenic Staphylococcus aureus: Use of two multiplex PCR enzyme immunoassays for amplification and hybridization of staphylococcal enterotoxin genes, exfoliative toxin genes, and toxic shock syndrome toxin 1 gene. J. Clin. Microbiol. 1998, 36, 2548–2553. [Google Scholar]
- Yim, S.H.; Hee, S.S. Genotoxicity of nicotine and cotinine in the bacterial luminescence test. Mutat. Res. 1995, 335, 275–283. [Google Scholar] [CrossRef]
- Ueno, Y.; Nagata, S.; Tsutsumi, T.; Hasegawa, A.; Watanabe, M.F.; Park, H.D.; Chen, G.C.; Chen, G.; Yu, S.Z. Detection of microcystins, a blue-green algal hepatotoxin, in drinking water sampled in Haimen and Fusui, endemic areas of primary liver cancer in China, by highly sensitive immunoassay. Carcinogenesis 1996, 17, 1317–1321. [Google Scholar] [CrossRef]
- Nedelkov, D.; Rasooly, A.; Nelson, R.W. Multitoxin biosensor-mass spectrometry analysis: A new approach for rapid, real-time, sensitive analysis of staphylococcal toxins in food. Int. J. Food Microbiol. 2000, 60, 1–13. [Google Scholar] [CrossRef]
- Sloan, L.M.; Duresko, B.J.; Gustafson, D.R.; Rosenblatt, J.E. Comparison of real-time PCR for detection of the tcdC gene with four toxin immunoassays and culture in diagnosis of Clostridium difficile infection. J. Clin. Microbiol. 2008, 46, 1996–2001. [Google Scholar] [CrossRef]
- de Boer, E.; Beumer, R.R. Methodology for detection and typing of foodborne microorganisms. Int. J. Food Microbiol. 1999, 50, 119–130. [Google Scholar] [CrossRef]
- Lockhart, D.J.; Dong, H.; Byrne, M.C.; Follettie, M.T.; Gallo, M.V.; Chee, M.S.; Mittmann, M.; Wang, C.; Kobayashi, M.; Horton, H.; Brown, E.L. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol. 1996, 14, 1675–1680. [Google Scholar] [CrossRef]
- Roth, F.P.; Hughes, J.D.; Estep, P.W.; Church, G.M. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat. Biotechnol. 1998, 16, 939–945. [Google Scholar] [CrossRef]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar]
- Gunderson, K.L.; Huang, X.C.; Morris, M.S.; Lipshutz, R.J.; Lockhart, D.J.; Chee, M.S. Mutation detection by ligation to complete n-mer DNA arrays. Genome Res. 1998, 8, 1142–1153. [Google Scholar]
- Wang, D.G.; Fan, J.B.; Siao, C.J.; Berno, A.; Young, P.; Sapolsky, R.; Ghandour, G.; Perkins, N.; Winchester, E.; Spencer, J.; Kruglyak, L.; Stein, L.; Hsie, L.; Topaloglou, T.; Hubbell, E.; Robinson, E.; Mittmann, M.; Morris, M.S.; Shen, N.; Kilburn, D.; Rioux, J.; Nusbaum, C.; Rozen, S.; Hudson, T.J.; Lipshutz, R.; Chee, M.; Lander, E.S. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 1998, 280, 1077–1082. [Google Scholar]
- Brownie, J.; Shawcross, S.; Theaker, J.; Whitcombe, D.; Ferrie, R.; Newton, C.; Little, S. The elimination of primer-dimer accumulation in PCR. Nucl. Acid. Res. 1997, 25, 3235–3241. [Google Scholar] [CrossRef]
- Vandamme, P.; Pot, B.; Gillis, M.; de Vos, P.; Kersters, K.; Swings, J. Polyphasic taxonomy, a consensus approach to bacterial systematics. Microbiol. Rev. 1996, 60, 407–438. [Google Scholar]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; Dewell, S.B.; Du, L.; Fierro, J.M.; Gomes, X.V.; Godwin, B.C.; He, W.; Helgesen, S.; Ho, C.H.; Irzyk, G.P.; Jando, S.C.; Alenquer, M.L.; Jarvie, T.P.; Jirage, K.B.; Kim, J.B.; Knight, J.R.; Lanza, J.R.; Leamon, J.H.; Lefkowitz, S.M.; Lei, M.; Li, J.; Lohman, K.L.; Lu, H.; Makhijani, V.B.; McDade, K.E.; McKenna, M.P.; Myers, E.W.; Nickerson, E.; Nobile, J.R.; Plant, R.; Puc, B.P.; Ronan, M.T.; Roth, G.T.; Sarkis, G.J.; Simons, J.F.; Simpson, J.W.; Srinivasan, M.; Tartaro, K.R.; Tomasz, A.; Vogt, K.A.; Volkmer, G.A.; Wang, S.H.; Wang, Y.; Weiner, M.P.; Yu, P.; Begley, R.F.; Rothberg, J.M. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar]
- Williams, R.; Peisajovich, S.G.; Miller, O.J.; Magdassi, S.; Tawfik, D.S.; Griffiths, A.D. Amplification of complex gene libraries by emulsion PCR. Nat. Methods 2006, 3, 545–550. [Google Scholar] [CrossRef]
- Kojima, T.; Takei, Y.; Ohtsuka, M.; Kawarasaki, Y.; Yamane, T.; Nakano, H. PCR amplification from single DNA molecules on magnetic beads in emulsion: Application for high-throughput screening of transcription factor targets. Nucl. Acid. Res. 2005, 33, e150. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Ge, Q.; Liu, Z.; Bai, Y.; Zhang, D.; Yu, P.; Lu, Z. Emulsion PCR-based method to detect Y chromosome microdeletions. Anal. Biochem. 2007, 367, 173–178. [Google Scholar]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef]
- Li, M.; Diehl, F.; Dressman, D.; Vogelstein, B.; Kinzler, K.W. BEAMing up for detection and quantification of rare sequence variants. Nat. Methods 2006, 3, 95–97. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar]
- Leamon, J.H.; Link, D.R.; Egholm, M.; Rothberg, J.M. Overview: Methods and applications for droplet compartmentalization of biology. Nat. Methods 2006, 3, 541–543. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Ansorge, W.J. Next-generation DNA sequencing techniques. N. Biotechnol. 2009, 25, 195–203. [Google Scholar]
- Vannuffel, P.; Gigi, J.; Ezzedine, H.; Vandercam, B.; Delmee, M.; Wauters, G.; Gala, J.L. Specific detection of methicillin-resistant Staphylococcus species by multiplex PCR. J. Clin. Microbiol. 1995, 33, 2864–2867. [Google Scholar]
- Cebula, T.A.; Payne, W.L.; Feng, P. Simultaneous identification of strains of Escherichia coli serotype O157:H7 and their Shiga-like toxin type by mismatch amplification mutation assay-multiplex PCR. J. Clin. Microbiol. 1995, 33, 248–250. [Google Scholar]
- Panicker, G.; Call, D.R.; Krug, M.J.; Bej, A.K. Detection of pathogenic Vibrio spp. in shellfish by using multiplex PCR and DNA microarrays. Appl. Environ. Microbiol. 2004, 70, 7436–7444. [Google Scholar] [CrossRef]
- Henegariu, O.; Heerema, N.A.; Dlouhy, S.R.; Vance, G.H.; Vogt, P.H. Multiplex PCR: Critical parameters and step-by-step protocol. Biotechniques 1997, 23, 504–511. [Google Scholar]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar]
- Gaffoor, I.; Trail, F. Characterization of two polyketide synthase genes involved in zearalenone biosynthesis in Gibberella zeae. Appl. Environ. Microbiol. 2006, 72, 1793–1799. [Google Scholar] [CrossRef]
- Holden, M.J.; Blasic, J.R., Jr.; Bussjaeger, L.; Kao, C.; Shokere, L.A.; Kendall, D.C.; Freese, L.; Jenkins, G.R. Evaluation of extraction methodologies for corn kernel (Zea mays) DNA for detection of trace amounts of biotechnology-derived DNA. J. Agric. Food Chem. 2003, 51, 2468–2474. [Google Scholar]
- Rogers, H.W.; Callery, M.P.; Deck, B.; Unanue, E.R. Listeria monocytogenes induces apoptosis of infected hepatocytes. J. Immunol. 1996, 156, 679–684. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, Y.; Lu, J.; Yang, Q.; Bai, Y.; Ge, Q. An Emulsion Based Microarray Method to Detect the Toxin Genes of Toxin-Producing Organisms. Molecules 2011, 16, 7365-7376. https://doi.org/10.3390/molecules16097365
Wang Y, Lu J, Yang Q, Bai Y, Ge Q. An Emulsion Based Microarray Method to Detect the Toxin Genes of Toxin-Producing Organisms. Molecules. 2011; 16(9):7365-7376. https://doi.org/10.3390/molecules16097365
Chicago/Turabian StyleWang, Ying, Jiafeng Lu, Qi Yang, Yunfei Bai, and Qinyu Ge. 2011. "An Emulsion Based Microarray Method to Detect the Toxin Genes of Toxin-Producing Organisms" Molecules 16, no. 9: 7365-7376. https://doi.org/10.3390/molecules16097365