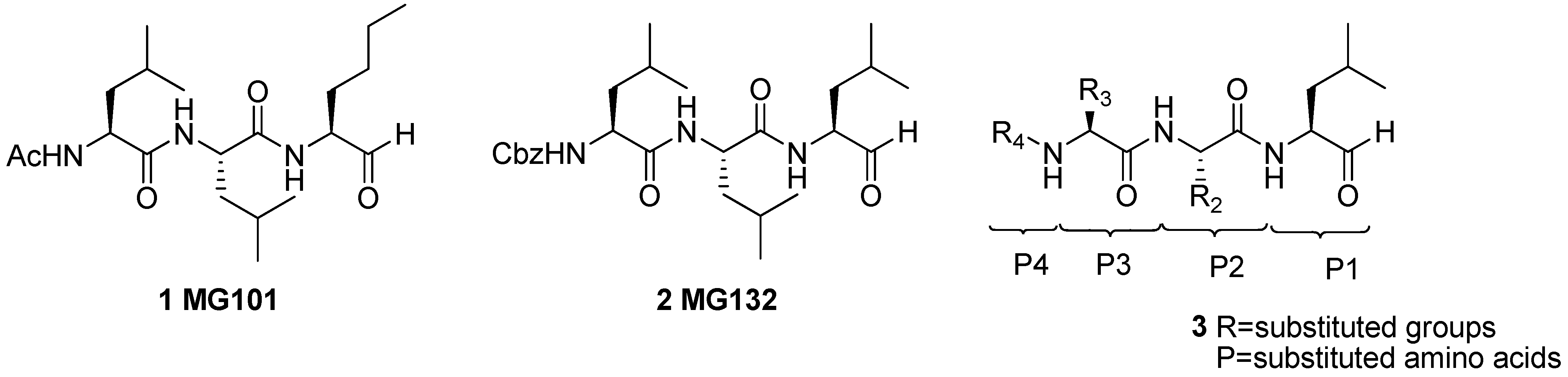
Synthesis and SAR Study of Novel Peptide Aldehydes as Inhibitors of 20S Proteasome


Abstract
:1. Introduction

2. Results and Discussion
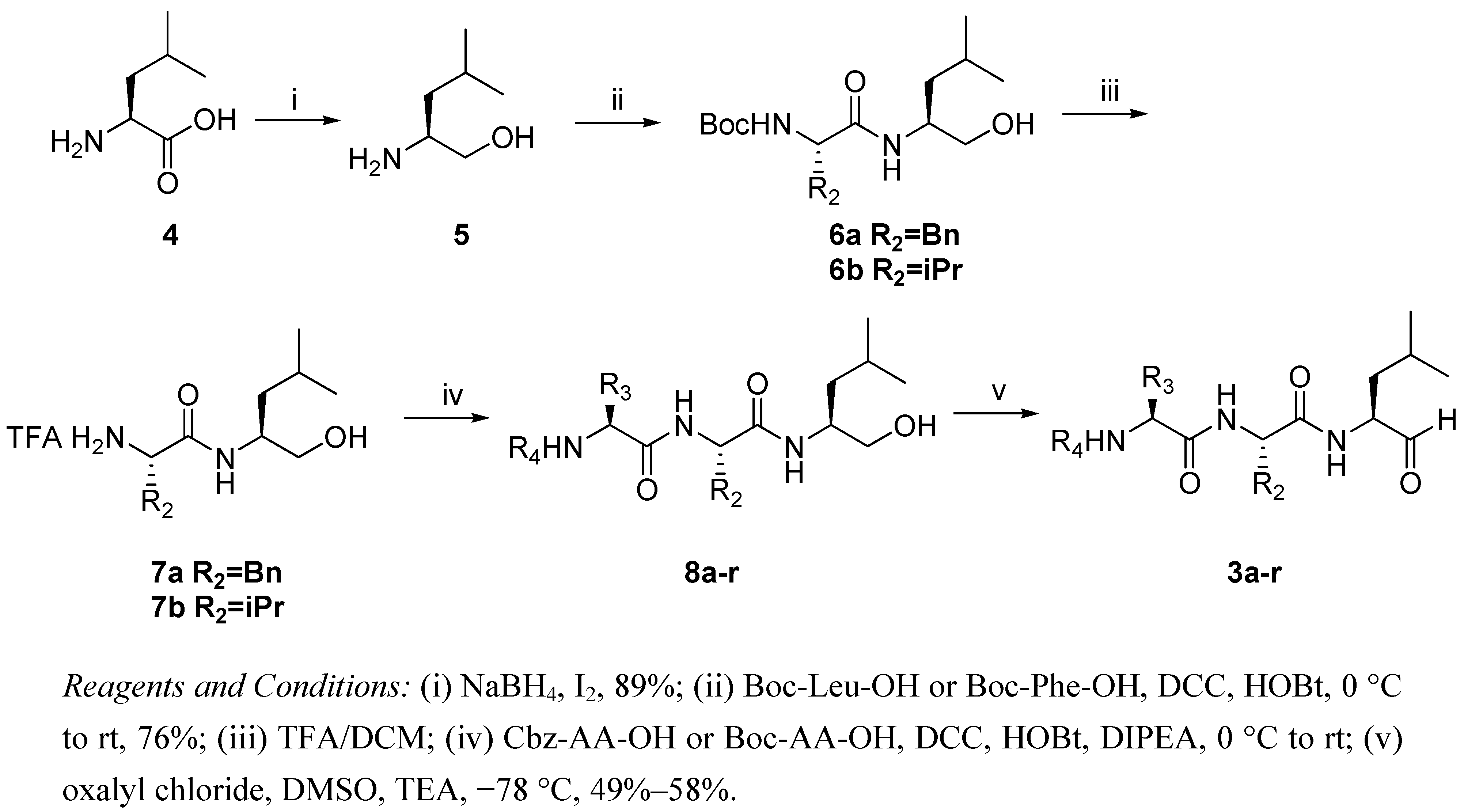
2.1. Synthesis of Peptide Aldehydes 3a-r

2.2. Assays for Proteasome Activities and SAR

{kind=link}
{kind=link}
{kind=link}
| Compounds | R4 | P3 | P2 | ChT-L(IC50,μM) |
|---|---|---|---|---|
| 3a | Cbz | Asp(OtBu) | Phe | 0.21 ± 0.014 |
| 3b | Cbz | Asp(OtBu) | Leu | 0.20 ± 0.035 |
| 3c | Cbz | Glu(OtBu) | Phe | 0.028 ± 0.006 |
| 3d | Cbz | Glu(OtBu) | Leu | 0.089 ± 0.02 |
| 3e | Cbz | Phe | Leu | 0.85 ± 0.047 |
| 3f | Cbz | Arg(NO2) | Leu | >50 |
| 3g | Cbz | Arg(Tos) | Leu | >50 |
| 3h | Cbz | Napa | Leu | 0.41 ± 0.082 |
| 3i | Boc | Asp(OBzl) | Phe | 4.83 ± 2.30 |
| 3j | Boc | Asp(OBzl) | Leu | 20.3 ± 2.05 |
| 3k | Boc | Glu(OBzl) | Phe | 7.14 ± 1.93 |
| 3l | Boc | Glu(OBzl) | Leu | >50 |
| 3m | Boc | Pro | Phe | >50 |
| 3n | Boc | Pro | Leu | >50 |
| 3o | Boc | Ser(OBzl) | Leu | 0.050 ± 0.002 |
| 3p | Boc | Thr(OBzl) | Leu | 0.29 ± 0.021 |
| 3q | Boc | Tyr(OBzl) | Leu | >50 |
| MG132 ( 3r) | Cbz | Leu | Leu | 0.28 ± 0.06 |
3. Experimental
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis
 +4.0 (c 0.3, MeOH). 1H-NMR (500 MHz, CDCl3): δ 0.91 (d, 6H, J = 6.5 Hz), 1.19–1.14 (m, 2H), 1.70–1.65 (m, 1H), 2.04 (br s, 3H, NH2, OH), 3.23 (dd, 1H, J = 3.0, 8.0 Hz), 3.57 (dd, 1H, J = 3.0, 8.0 Hz). MS (ESI-TOF+): 118 [M+H]+. −19.4 (c 0.3, MeOH). 1H-NMR (500 MHz, CDCl3): δ 0.87 (d, 6H, J = 7.2 Hz), 1.26–1.23 (m, 2H), 1.42 (s, 9H), 1.50–1.46 (m, 1H), 1.95 (s, 1H), 3.02–2.98 (m, 1H), 3.11–3.07 (m, 1H), 3.35 (m, 1H), 3.48 (d, 1H, J = 11.0 Hz), 3.95–3.93 (m, 1H), 4.25 (m, 1H), 5.06 (s, 1H), 5.60 (d, 1H), 7.28–7.14 (m, 5H). MS(ESI-TOF+): 345 [M+H]+. −11.6 (c 0.52, MeOH). 1H-NMR (500 MHz, CDCl3): δ 0.93–0.88 (m, 12H), 1.32–1.27 (m, 1H), 1.40–1.36 (m, 1H), 1.42 (s, 9H), 1.64–1.60 (m, 4H), 2.63 (s, 1H), 3.51–3.46 (m, 1H), 3.69–3.63 (m, 1H), 4.01–3.98 (m, 2H), 4.84 (s, 1H), 6.20 (d, 1H, J = 8.0 Hz). MS (ESI-TOF+): 331 [M+H]+. −19.1 (c 0.4, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.86 (d, 6H, J = 6.2 Hz), 1.40 (s, 9H), 1.70–1.53 (m, 3H), 2.73–2.68 (m, 2H), 3.10–3.04 (m, 2H), 4.38–4.30 (m, 2H), 4.71 (d, 1H, J = 7.2 Hz), 5.09 (d, 2H, J = 3.6 Hz), 5.72 (m, 1H), 6.58 (d, 1H, J = 7.5 Hz), 6.82 (t, 1H, J = 8.7 Hz), 7.38–7.19 (m, 10H), 9.43 (d, 1H). MS (ESI-TOF+): 568 [M+H]+. Elemental Anal.Calcd. for C31H41N3O7: C, 65.59; H, 7.28; N, 7.40. found: C, 65.67; H, 7.48; N, 7.32. −21.0 (c 0.5, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.97–0.88 (m, 12H), 1.42 (s, 9H), 1.69–1.57 (m, 6H), 4.48–4.43 (m, 3H), 5.13 (d, 2H, J = 3.6 Hz), 5.88 (s, 1H), 6.67–6.65 (m, 2H), 7.37 (m, 5H), 9.52 (s, 1H). MS (ESI-TOF+): 534 [M+H]+. Elemental Anal.Calcd. for C28H43N3O7: C, 63.02; H, 8.12; N, 7.87. found: C, 63.09; H, 8.19; N, 7.88. −28.1 (c 0.6, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.90 (d, 6H, J = 5.4 Hz), 1.44 (s, 9H), 1.56–1.49 (m, 3H), 1.91–1.88 (m, 2H), 2.25 (t, 2H, J = 6.3 Hz), 3.14 (t, 2H, J = 8.4 Hz), 4.06–3.96 (m, 1H), 4.38–4.33 (m, 1H), 4.75–4.69 (m, 1H), 5.04 (dd, 2H, J = 8.3, 22.2 Hz), 6.02 (s, 1H), 6.70–6.64 (m, 1H), 7.35–7.18 (m, 10H), 9.43 (d, 1H, J = 4.8 Hz). MS (ESI-TOF+): 582 [M+H]+. Elemental Anal.Calcd. for C32H43N3O7: C, 66.07; H, 7.45; N, 7.22. found: C, 65.99; H, 7.45; N, 7.26. −14.4 (c 0.7, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.97–0.90 (m, 6H), 1.44 (s, 9H), 1.74–1.54 (m, 6H), 2.09–1.96 (m, 2H), 2.40 (t, 2H, J = 5.1 Hz), 4.18 (t, 1H, J = 7.2 Hz), 4.49–4.38 (m, 2H), 5.10 (d, 2H, J = 3.3 Hz), 6.14 (s, 1H), 6.62 (s, 1H), 6.96 (s, 1H), 7.35–7.26 (m, 5H), 9.52 (d, 1H, J = 9.3 Hz). MS (ESI-TOF+): 548 [M+H]+. Elemental Anal.Calcd. for C29H45N3O7: C, 63.60; H, 8.28; N, 7.67. found: C, 62.54; H, 8.19; N, 7.70. −16.1 (c 0.3, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.92 (m, 12H), 1.74–1.32 (m, 6H), 4.36–4.33 (m, 1H), 4.54–4.47 (m, 2H ), 5.04–5.00 (d, 2H, J = 12.5 Hz), 5.45–5.40 (m, 1H), 7.00–6.53 (m, 2H), 7.36–7.16 (m, 10H), 9.50 (s, 1H). MS (ESI-TOF+): 544 [M+H]+. Elemental Anal.Calcd. for C32H37N3O5: C, 70.70; H, 6.86; N, 7.73. Found: C, 70.74; H, 6.90; N, 7.65. −67.2 (c 0.1, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.88–0.86 (m, 12H), 1.66–1.38 (m, 8H), 1.80–1.75 (m, 2H), 3.28–3.24 (m, 2H), 4.07 (dd, 1H, J = 4.5, 15.5 Hz), 4.20–4.16( m, 1H), 4.39–4.30 (m, 2H), 5.08 (s, 2H), 5.98 (s, 1H), 6.49–6.33 (m, 1H), 7.35–7.30 (m, 6H), 7.47 (s, 1H), 8.45 (s, 1H), 9.52 (s, 1H). MS (ESI-TOF+): 564 [M+H]+. Elemental Anal.Calcd. for C26H41N7O7, C, 55.40; H, 7.33; N,17.40. Found: C, 55.32; H, 7.42; N, 17.30. −12.3 (c 0.2, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 1.11–0.86 (m, 12H), 1.74–1.31 (m, 8H), 1.95–1.92 (m, 2H), 2.42 (s, 3H), 2.49–2.47 (m, 2H), 4.20–4.15 (m, 1H), 4.32–4.26 (m, 2H), 5.15 (s, 2H), 5.98 (s, 1H), 6.49–6.43 (m, 1H), 7.38–7.25 (m, 6H), 7.81 (d, 1H), 8.11 (s, 1H), 9.47 (s, 1H). MS (ESI-TOF+): 673 [M+H]+. Elemental Anal.Calcd. for C33H48N6O7S, C, 58.91; H, 7.19; N, 12.49. found: C, 58.99; H, 7.12; N,12.36. −51.1 (c 0.7, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.93–0.89 (m, 12H), 1.91–1.46 (m, 6H), 3.22–3.16 (dd, 2H, J = 3.9, 7.5 Hz), 4.48–4.43 (m, H), 4.89–4.75 (m, 2H), 5.12 (d, 2H, J = 4.2 Hz), 6.03 (d, 1H), 6.69–6.58 (m, 2H), 7.38–7.31 (m, 6H), 7.51 (t, 1H, J = 7.5 Hz), 7.58–7.55 (m, 1H), 7.77 (d, 1H, J = 8 Hz), 7.88 (d, 1H, J = 8.0 Hz), 8.26 (d, 1H, J = 9.0 Hz), 9.51 (d, 1H, J = 6.0 Hz). MS (ESI-TOF+): 560 [M+H]+. Elemental Anal.Calcd. for C33H41N3O5: C, 70.82; H, 7.38; N, 7.51. found: C, 70.90; H, 7.42; N, 7.61. −19.5 (c 0.65, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.94–0.88 (m, 6H), 1.40 (s, 9H), 1.60–1.54 (m, 1H), 1.72–1.66 (m, 2H), 3.04–2.77 (m, 3H), 3.26–3.23 (m, 1H), 4.18–4.10 (m, 2H), 4.71–4.67 (m, 1H), 5.11–5.04 (m, 2H), 5.44 (m, 1H), 6.59 (s, 1H), 6.80 (s, 1H), 7.35–7.21 (m, 10H), 9.40 (s, 1H). MS (ESI-TOF+): 568 [M+H]+. Elemental Anal.Calcd. for C31H41N3O7: C, 65.59; H, 7.28; N, 7.40. Found: C, 65.38; H, 7.19; N, 7.47. −44.5 (c 0.7, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.95–0.92 (m, 12H), 1.44 (s, 9H), 1.70–1.59 (m, 6H), 2.92 (t, 2H, J = 5.7 Hz), 4.47–4.40 (m, 3H), 5.22 (s, 2H), 5.48 (d, 1H), 6.70 (d, 1H, J = 6.9 Hz), 6.87 (m, 1H), 7.40–7.36 (m, 5H), 9.50 (s, 1H). MS (ESI-TOF+): 534 [M+H]+. Elemental Anal.Calcd. for C28H43N3O7: C, 63.02; H, 8.12; N, 7.87. found: C, 62.65;H, 7.98;N, 7.95. −18.1 (c 0.7, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.90–0.87 (m, 6H), 1.37 (s, 9H), 1.69–1.54 (m, 3H), 2.07–2.03 (m, 2H), 2.44 (t, 2H, J = 4.2 Hz), 3.21 (dd, 2H, J = 7.2, 14.1 Hz), 4.05–4.01 (m, 1H), 4.38–4.29 (m, 1H), 4.77–4.74 (m, 1H), 5.13 (d, 2H, J = 3.3 Hz), 5.57 (s, 1H), 6.68–6.56 (m, 2H), 7.38–7.20 (m, 10H), 9.41 (d, 1H, J = 5.4 Hz). MS (ESI-TOF+); 582 [M+H]+. Elemental Anal.Calcd. for C32H43N3O7: C, 66.07; H, 7.45; N, 7.22. found: C, 65.91; H, 7.56; N, 7.29. −31.5 (c 0.8, CHCl3). 1H-NMR (300 MHz, CDCl3):δ 0.95–0.90 (m, 12H), 1.43 (s, 9H), 1.97–1.62 (m, 6H), 2.17–2.12 (m, 2H), 2.57–2.48 (m, 2H), 4.11 (t, 1H, J = 5.7 Hz), 4.48–4.43 (m, 2H), 5.13 (s, 2H), 5.49 (d, 1H), 6.83 (s, 1H), 7.37–7.27 (m, 5H), 9.52 (s, 1H). MS (ESI-TOF+): 548 [M+H]+. Elemental Anal.Calcd. for C29H45N3O7: C, 63.60; H, 8.28; N, 7.67. found: C, 63.71; H, 8.19; N, 7.68. −10.0 (c 0.5, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.98–0.88 (m, 6H), 1.43 (s, 9H), 2.08–1.46 (m, 7H), 3.17–3.13 (m, 2H), 3.40–3.35 (m, 2H), 4.45–4.38 (m, 2H), 5.00–4.94 (m, 1H), 5.92 (s, 1H), 6.72 (s, 1H,), 7.40–7.20 (m, 5H), 9.45 (s, 1H). MS (ESI-TOF+): 460 [M+H]+. Elemental Anal.Calcd. for C25H37N3O5: C, 65.34; H, 8.11; N, 9.14. found: C, 65.44; H, 8.19; N, 9.15. −23.3 (c 0.6, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 1.01–0.93 (m, 12H), 1.50–1.32 (m, 14H), 1.75–1.68 (m, 6H), 4.16–4.12 (m, 1H), 4.52–4.45 (m, 1H), 4.91 (m, 1H), 6.70 (s, 1H), 9.56 (d, 1H, J = 2.7 Hz). MS (ESI-TOF+): 426 [M+H]+. Elemental Anal.Calcd. for C22H39N3O5: C, 62.09; H, 9.24; N, 9.87. found: C, 61.18; H, 9.28; N, 9.80. −35.1 (c 0.4, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.92–0.88 (m, 12H), 1.80–1.40 (m, 15H), 3.65–3.59 (m, 1H), 3.91–3.86 (m, 1H), 4.52–4.20 (m, 3H), 4.54 (s, 2H), 5.39 (s, 1H), 6.94–6.52 (m, 2H), 7.38–7.25 (m, 5H), 9.52 (brs, 1H). MS (ESI-TOF+): 506 [M+H]+. Elemental Anal.Calcd. for C27H43N3O6: C, 64.13; H, 8.57; N, 8.31. found: C, 64.10; H, 8.51; N, 8.40. −6.0 (c 0.6, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.87–0.84 (m, 6H), 0.99–0.91 (m, 6H), 1.22–1.16 (m, 3H), 1.45 (s, 9H), 1.77–1.50 (m, 6H), 4.19–4.16 (m, 1H), 4.24–4.20 (m, 1H), 4.36–4.27 (m, 1H), 4.51–4.48 (m, 1H), 4.65 (dd, 1H, J = 4, 12.0 Hz), 5.45 (s, 1H), 6.87–6.54 (m, 2H), 7.35–7.25 (m, 5H), 9.49 (d, 1H, J = 25.0 Hz). MS (ESI-TOF+): 520 [M+H]+. Elemental Anal.Calcd. for C28H45N3O6: C, 64.71; H, 8.73; N, 8.09. found: C, 64.78; H, 8.80; N, 8.01. −4.6 (c 0.3, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.93 (m,12), 1.40 (s, 9H), 1.76–1.45 (m, 6H), 3.03–3.00 (m, 2H), 4.29–4.22 (m, 1H), 4.46–4.40 (m, 1H), 4.89 (d, 1H, J = 4.0 Hz), 5.04 (s, 2H), 6.28 (d, 1H, J = 8.5 Hz, NH), 6.36 (m, 1H), 6.74 (s, 1H), 6.92 (dd, 2H, J = 6.0, 11.5 Hz), 7.11 (d, 2H, J = 8 Hz), 7.43–7.26 (m, 5H), 9.51 (s, 1H). MS (ESI-TOF+): 582 [M+H]+. Elemental Anal.Calcd. for C33H47N3O6: C, 68.13; H, 8.14; N, 7.22. found: C, 68.07; H, 8.19; N, 7.16. −14.4 (c 0.8, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.93–0.88 (m, 18H), 1.65–1.58 (m, 9H), 4.18–4.14 (t, 1H, J = 5.5 Hz), 4.47–4.40 (m, 2H), 5.09–4.89 (m, 2H), 5.44 (d, 1H, J = 22.5 Hz), 6.66 (d, 1H, J = 8.0 Hz), 6.95 (d, 1H, J = 33.5 Hz), 7.28–7.38 (m, 5H), 9.52 (d, 1H). MS (ESI-TOF+): 476 [M+H]+. Elemental Anal.Calcd. for C26H41N3O5: C, 65.66; H, 8.69; N, 8.83. found: C, 65.70; H, 8.62; N, 8.79.
+4.0 (c 0.3, MeOH). 1H-NMR (500 MHz, CDCl3): δ 0.91 (d, 6H, J = 6.5 Hz), 1.19–1.14 (m, 2H), 1.70–1.65 (m, 1H), 2.04 (br s, 3H, NH2, OH), 3.23 (dd, 1H, J = 3.0, 8.0 Hz), 3.57 (dd, 1H, J = 3.0, 8.0 Hz). MS (ESI-TOF+): 118 [M+H]+. −19.4 (c 0.3, MeOH). 1H-NMR (500 MHz, CDCl3): δ 0.87 (d, 6H, J = 7.2 Hz), 1.26–1.23 (m, 2H), 1.42 (s, 9H), 1.50–1.46 (m, 1H), 1.95 (s, 1H), 3.02–2.98 (m, 1H), 3.11–3.07 (m, 1H), 3.35 (m, 1H), 3.48 (d, 1H, J = 11.0 Hz), 3.95–3.93 (m, 1H), 4.25 (m, 1H), 5.06 (s, 1H), 5.60 (d, 1H), 7.28–7.14 (m, 5H). MS(ESI-TOF+): 345 [M+H]+. −11.6 (c 0.52, MeOH). 1H-NMR (500 MHz, CDCl3): δ 0.93–0.88 (m, 12H), 1.32–1.27 (m, 1H), 1.40–1.36 (m, 1H), 1.42 (s, 9H), 1.64–1.60 (m, 4H), 2.63 (s, 1H), 3.51–3.46 (m, 1H), 3.69–3.63 (m, 1H), 4.01–3.98 (m, 2H), 4.84 (s, 1H), 6.20 (d, 1H, J = 8.0 Hz). MS (ESI-TOF+): 331 [M+H]+. −19.1 (c 0.4, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.86 (d, 6H, J = 6.2 Hz), 1.40 (s, 9H), 1.70–1.53 (m, 3H), 2.73–2.68 (m, 2H), 3.10–3.04 (m, 2H), 4.38–4.30 (m, 2H), 4.71 (d, 1H, J = 7.2 Hz), 5.09 (d, 2H, J = 3.6 Hz), 5.72 (m, 1H), 6.58 (d, 1H, J = 7.5 Hz), 6.82 (t, 1H, J = 8.7 Hz), 7.38–7.19 (m, 10H), 9.43 (d, 1H). MS (ESI-TOF+): 568 [M+H]+. Elemental Anal.Calcd. for C31H41N3O7: C, 65.59; H, 7.28; N, 7.40. found: C, 65.67; H, 7.48; N, 7.32. −21.0 (c 0.5, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.97–0.88 (m, 12H), 1.42 (s, 9H), 1.69–1.57 (m, 6H), 4.48–4.43 (m, 3H), 5.13 (d, 2H, J = 3.6 Hz), 5.88 (s, 1H), 6.67–6.65 (m, 2H), 7.37 (m, 5H), 9.52 (s, 1H). MS (ESI-TOF+): 534 [M+H]+. Elemental Anal.Calcd. for C28H43N3O7: C, 63.02; H, 8.12; N, 7.87. found: C, 63.09; H, 8.19; N, 7.88. −28.1 (c 0.6, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.90 (d, 6H, J = 5.4 Hz), 1.44 (s, 9H), 1.56–1.49 (m, 3H), 1.91–1.88 (m, 2H), 2.25 (t, 2H, J = 6.3 Hz), 3.14 (t, 2H, J = 8.4 Hz), 4.06–3.96 (m, 1H), 4.38–4.33 (m, 1H), 4.75–4.69 (m, 1H), 5.04 (dd, 2H, J = 8.3, 22.2 Hz), 6.02 (s, 1H), 6.70–6.64 (m, 1H), 7.35–7.18 (m, 10H), 9.43 (d, 1H, J = 4.8 Hz). MS (ESI-TOF+): 582 [M+H]+. Elemental Anal.Calcd. for C32H43N3O7: C, 66.07; H, 7.45; N, 7.22. found: C, 65.99; H, 7.45; N, 7.26. −14.4 (c 0.7, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.97–0.90 (m, 6H), 1.44 (s, 9H), 1.74–1.54 (m, 6H), 2.09–1.96 (m, 2H), 2.40 (t, 2H, J = 5.1 Hz), 4.18 (t, 1H, J = 7.2 Hz), 4.49–4.38 (m, 2H), 5.10 (d, 2H, J = 3.3 Hz), 6.14 (s, 1H), 6.62 (s, 1H), 6.96 (s, 1H), 7.35–7.26 (m, 5H), 9.52 (d, 1H, J = 9.3 Hz). MS (ESI-TOF+): 548 [M+H]+. Elemental Anal.Calcd. for C29H45N3O7: C, 63.60; H, 8.28; N, 7.67. found: C, 62.54; H, 8.19; N, 7.70. −16.1 (c 0.3, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.92 (m, 12H), 1.74–1.32 (m, 6H), 4.36–4.33 (m, 1H), 4.54–4.47 (m, 2H ), 5.04–5.00 (d, 2H, J = 12.5 Hz), 5.45–5.40 (m, 1H), 7.00–6.53 (m, 2H), 7.36–7.16 (m, 10H), 9.50 (s, 1H). MS (ESI-TOF+): 544 [M+H]+. Elemental Anal.Calcd. for C32H37N3O5: C, 70.70; H, 6.86; N, 7.73. Found: C, 70.74; H, 6.90; N, 7.65. −67.2 (c 0.1, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.88–0.86 (m, 12H), 1.66–1.38 (m, 8H), 1.80–1.75 (m, 2H), 3.28–3.24 (m, 2H), 4.07 (dd, 1H, J = 4.5, 15.5 Hz), 4.20–4.16( m, 1H), 4.39–4.30 (m, 2H), 5.08 (s, 2H), 5.98 (s, 1H), 6.49–6.33 (m, 1H), 7.35–7.30 (m, 6H), 7.47 (s, 1H), 8.45 (s, 1H), 9.52 (s, 1H). MS (ESI-TOF+): 564 [M+H]+. Elemental Anal.Calcd. for C26H41N7O7, C, 55.40; H, 7.33; N,17.40. Found: C, 55.32; H, 7.42; N, 17.30. −12.3 (c 0.2, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 1.11–0.86 (m, 12H), 1.74–1.31 (m, 8H), 1.95–1.92 (m, 2H), 2.42 (s, 3H), 2.49–2.47 (m, 2H), 4.20–4.15 (m, 1H), 4.32–4.26 (m, 2H), 5.15 (s, 2H), 5.98 (s, 1H), 6.49–6.43 (m, 1H), 7.38–7.25 (m, 6H), 7.81 (d, 1H), 8.11 (s, 1H), 9.47 (s, 1H). MS (ESI-TOF+): 673 [M+H]+. Elemental Anal.Calcd. for C33H48N6O7S, C, 58.91; H, 7.19; N, 12.49. found: C, 58.99; H, 7.12; N,12.36. −51.1 (c 0.7, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.93–0.89 (m, 12H), 1.91–1.46 (m, 6H), 3.22–3.16 (dd, 2H, J = 3.9, 7.5 Hz), 4.48–4.43 (m, H), 4.89–4.75 (m, 2H), 5.12 (d, 2H, J = 4.2 Hz), 6.03 (d, 1H), 6.69–6.58 (m, 2H), 7.38–7.31 (m, 6H), 7.51 (t, 1H, J = 7.5 Hz), 7.58–7.55 (m, 1H), 7.77 (d, 1H, J = 8 Hz), 7.88 (d, 1H, J = 8.0 Hz), 8.26 (d, 1H, J = 9.0 Hz), 9.51 (d, 1H, J = 6.0 Hz). MS (ESI-TOF+): 560 [M+H]+. Elemental Anal.Calcd. for C33H41N3O5: C, 70.82; H, 7.38; N, 7.51. found: C, 70.90; H, 7.42; N, 7.61. −19.5 (c 0.65, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.94–0.88 (m, 6H), 1.40 (s, 9H), 1.60–1.54 (m, 1H), 1.72–1.66 (m, 2H), 3.04–2.77 (m, 3H), 3.26–3.23 (m, 1H), 4.18–4.10 (m, 2H), 4.71–4.67 (m, 1H), 5.11–5.04 (m, 2H), 5.44 (m, 1H), 6.59 (s, 1H), 6.80 (s, 1H), 7.35–7.21 (m, 10H), 9.40 (s, 1H). MS (ESI-TOF+): 568 [M+H]+. Elemental Anal.Calcd. for C31H41N3O7: C, 65.59; H, 7.28; N, 7.40. Found: C, 65.38; H, 7.19; N, 7.47. −44.5 (c 0.7, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.95–0.92 (m, 12H), 1.44 (s, 9H), 1.70–1.59 (m, 6H), 2.92 (t, 2H, J = 5.7 Hz), 4.47–4.40 (m, 3H), 5.22 (s, 2H), 5.48 (d, 1H), 6.70 (d, 1H, J = 6.9 Hz), 6.87 (m, 1H), 7.40–7.36 (m, 5H), 9.50 (s, 1H). MS (ESI-TOF+): 534 [M+H]+. Elemental Anal.Calcd. for C28H43N3O7: C, 63.02; H, 8.12; N, 7.87. found: C, 62.65;H, 7.98;N, 7.95. −18.1 (c 0.7, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.90–0.87 (m, 6H), 1.37 (s, 9H), 1.69–1.54 (m, 3H), 2.07–2.03 (m, 2H), 2.44 (t, 2H, J = 4.2 Hz), 3.21 (dd, 2H, J = 7.2, 14.1 Hz), 4.05–4.01 (m, 1H), 4.38–4.29 (m, 1H), 4.77–4.74 (m, 1H), 5.13 (d, 2H, J = 3.3 Hz), 5.57 (s, 1H), 6.68–6.56 (m, 2H), 7.38–7.20 (m, 10H), 9.41 (d, 1H, J = 5.4 Hz). MS (ESI-TOF+); 582 [M+H]+. Elemental Anal.Calcd. for C32H43N3O7: C, 66.07; H, 7.45; N, 7.22. found: C, 65.91; H, 7.56; N, 7.29. −31.5 (c 0.8, CHCl3). 1H-NMR (300 MHz, CDCl3):δ 0.95–0.90 (m, 12H), 1.43 (s, 9H), 1.97–1.62 (m, 6H), 2.17–2.12 (m, 2H), 2.57–2.48 (m, 2H), 4.11 (t, 1H, J = 5.7 Hz), 4.48–4.43 (m, 2H), 5.13 (s, 2H), 5.49 (d, 1H), 6.83 (s, 1H), 7.37–7.27 (m, 5H), 9.52 (s, 1H). MS (ESI-TOF+): 548 [M+H]+. Elemental Anal.Calcd. for C29H45N3O7: C, 63.60; H, 8.28; N, 7.67. found: C, 63.71; H, 8.19; N, 7.68. −10.0 (c 0.5, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.98–0.88 (m, 6H), 1.43 (s, 9H), 2.08–1.46 (m, 7H), 3.17–3.13 (m, 2H), 3.40–3.35 (m, 2H), 4.45–4.38 (m, 2H), 5.00–4.94 (m, 1H), 5.92 (s, 1H), 6.72 (s, 1H,), 7.40–7.20 (m, 5H), 9.45 (s, 1H). MS (ESI-TOF+): 460 [M+H]+. Elemental Anal.Calcd. for C25H37N3O5: C, 65.34; H, 8.11; N, 9.14. found: C, 65.44; H, 8.19; N, 9.15. −23.3 (c 0.6, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 1.01–0.93 (m, 12H), 1.50–1.32 (m, 14H), 1.75–1.68 (m, 6H), 4.16–4.12 (m, 1H), 4.52–4.45 (m, 1H), 4.91 (m, 1H), 6.70 (s, 1H), 9.56 (d, 1H, J = 2.7 Hz). MS (ESI-TOF+): 426 [M+H]+. Elemental Anal.Calcd. for C22H39N3O5: C, 62.09; H, 9.24; N, 9.87. found: C, 61.18; H, 9.28; N, 9.80. −35.1 (c 0.4, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.92–0.88 (m, 12H), 1.80–1.40 (m, 15H), 3.65–3.59 (m, 1H), 3.91–3.86 (m, 1H), 4.52–4.20 (m, 3H), 4.54 (s, 2H), 5.39 (s, 1H), 6.94–6.52 (m, 2H), 7.38–7.25 (m, 5H), 9.52 (brs, 1H). MS (ESI-TOF+): 506 [M+H]+. Elemental Anal.Calcd. for C27H43N3O6: C, 64.13; H, 8.57; N, 8.31. found: C, 64.10; H, 8.51; N, 8.40. −6.0 (c 0.6, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.87–0.84 (m, 6H), 0.99–0.91 (m, 6H), 1.22–1.16 (m, 3H), 1.45 (s, 9H), 1.77–1.50 (m, 6H), 4.19–4.16 (m, 1H), 4.24–4.20 (m, 1H), 4.36–4.27 (m, 1H), 4.51–4.48 (m, 1H), 4.65 (dd, 1H, J = 4, 12.0 Hz), 5.45 (s, 1H), 6.87–6.54 (m, 2H), 7.35–7.25 (m, 5H), 9.49 (d, 1H, J = 25.0 Hz). MS (ESI-TOF+): 520 [M+H]+. Elemental Anal.Calcd. for C28H45N3O6: C, 64.71; H, 8.73; N, 8.09. found: C, 64.78; H, 8.80; N, 8.01. −4.6 (c 0.3, CHCl3). 1H-NMR (500 MHz, CDCl3): δ 0.93 (m,12), 1.40 (s, 9H), 1.76–1.45 (m, 6H), 3.03–3.00 (m, 2H), 4.29–4.22 (m, 1H), 4.46–4.40 (m, 1H), 4.89 (d, 1H, J = 4.0 Hz), 5.04 (s, 2H), 6.28 (d, 1H, J = 8.5 Hz, NH), 6.36 (m, 1H), 6.74 (s, 1H), 6.92 (dd, 2H, J = 6.0, 11.5 Hz), 7.11 (d, 2H, J = 8 Hz), 7.43–7.26 (m, 5H), 9.51 (s, 1H). MS (ESI-TOF+): 582 [M+H]+. Elemental Anal.Calcd. for C33H47N3O6: C, 68.13; H, 8.14; N, 7.22. found: C, 68.07; H, 8.19; N, 7.16. −14.4 (c 0.8, CHCl3). 1H-NMR (300 MHz, CDCl3): δ 0.93–0.88 (m, 18H), 1.65–1.58 (m, 9H), 4.18–4.14 (t, 1H, J = 5.5 Hz), 4.47–4.40 (m, 2H), 5.09–4.89 (m, 2H), 5.44 (d, 1H, J = 22.5 Hz), 6.66 (d, 1H, J = 8.0 Hz), 6.95 (d, 1H, J = 33.5 Hz), 7.28–7.38 (m, 5H), 9.52 (d, 1H). MS (ESI-TOF+): 476 [M+H]+. Elemental Anal.Calcd. for C26H41N3O5: C, 65.66; H, 8.69; N, 8.83. found: C, 65.70; H, 8.62; N, 8.79.3.2. Biological Testing
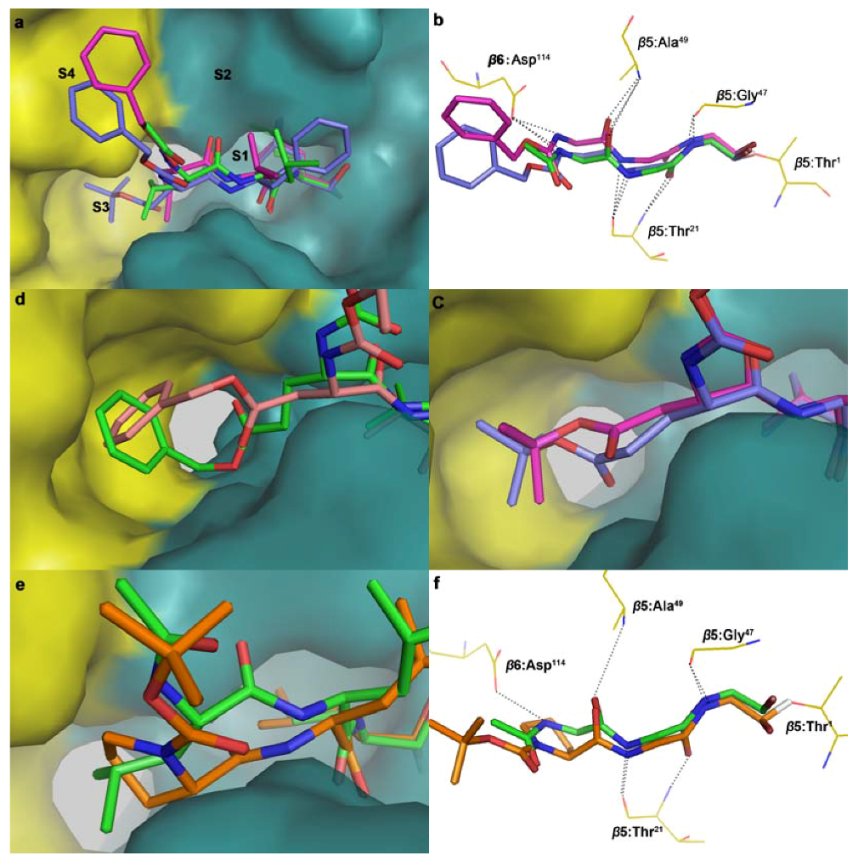
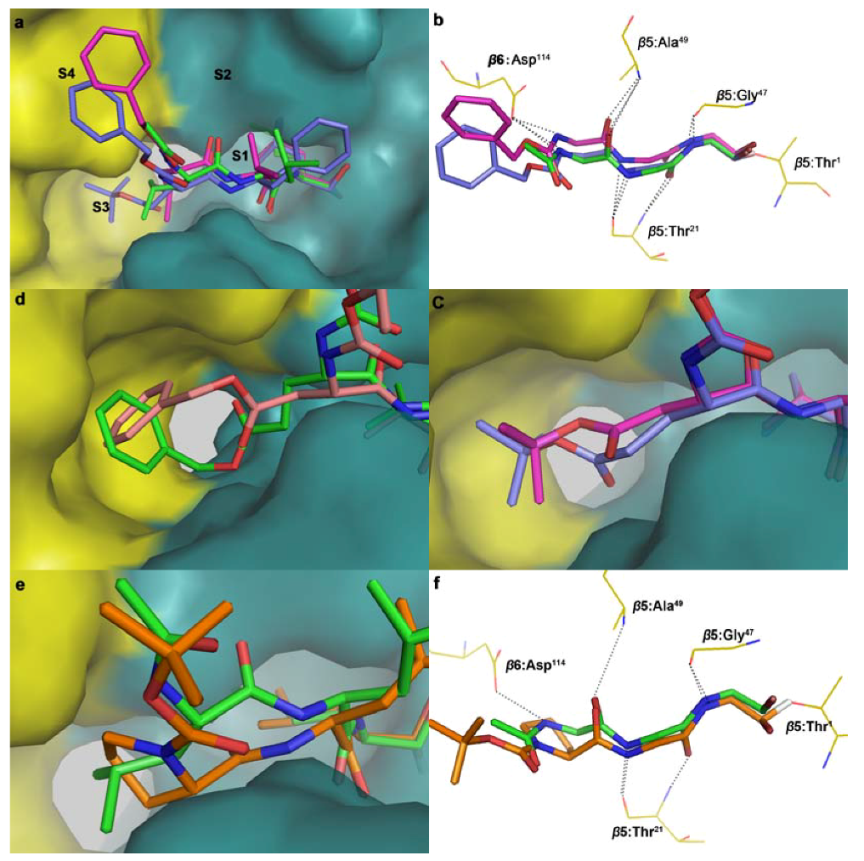
3.3. Molecular Docking
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the compounds 3a-3r are available from the authors.
References
- Etlinger, J.D.; Goldberg, A.L. Soluble Atp-dependent prooteolytic system resonsible for degradation of abnormal proteins in reticulocytes. Proc. Natl. Acad. Sci. USA 1977, 74, 54–58. [Google Scholar] [CrossRef]
- Hershko, A. Lessons from the discovery of the ubiquitin system. Trends Biochem. Sci. 1996, 21, 445–449. [Google Scholar] [CrossRef]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell-proteins and the generation of peptides presented on Mhc Class-I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Osaki, T.; Kimura, T.; Tatemoto, Y.; Lu, D.P.; Yoneda, K.; Yamamoto, T. Diffuse mode of tumor cell invasion and expression of mutant p53 protein but not of p21 protein are correlated with treatment failure in oral carcinomas and their metastatic foci. Oncology 2000, 59, 36–43. [Google Scholar] [CrossRef]
- Pagano, M.; Tam, S.W.; Theodoras, A.M.; Beerromero, P.; Delsal, G.; Chau, V.; Yew, P.R.; Draetta, G.F.; Rolfe, M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995, 269, 682–685. [Google Scholar]
- Alves-Rodrigues, A.; Gregori, L.; Figueiredo-Pereira, M.E. Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci. 1998, 21, 516–520. [Google Scholar] [CrossRef]
- Merforth, S.; Kuehn, L.; Osmers, A.; Dahmann, B. Alteration of 20S proteasome-subtypes and proteasome activator PA28 in skeletal muscle of rat after induction of diabetes mellitus. Int. J. Biochem. Cell Biol. 2003, 35, 740–748. [Google Scholar] [CrossRef]
- Sun, J.Z.; Nam, S.K.; Lee, C.S.; Li, B.Y.; Coppola, D.; Hamilton, A.D.; Dou, Q.P.; Sebti, S.M. CEP1612, a dipeptidyl proteasome inhibitor, induces p21(WAF1) and p27(KIP1) expression and apoptosis and inhibits the growth of the human lung adenocarcinoma A-549 in nude mice. Cancer Res. 2001, 61, 1280–1284. [Google Scholar]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef]
- Groll, M.; Schellenberg, B.; Bachmann, A.S.; Archer, C.R.; Huber, R.; Powell, T.K.; Lindow, S.; Kaiser, M.; Dudler, R. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 2008, 452, 755–758. [Google Scholar]
- Pirrung, M.C.; Biswas, G.; Ibarra-Rivera, T.R. Total Synthesis of Syringolin A and B. Org. Lett. 2010, 12, 2402–2405. [Google Scholar] [CrossRef]
- Borissenko, L.; Groll, M. 20S proteasome and its inhibitors: Crystallographic knowledge for drug development. Chem. Rev. 2007, 107, 687–717. [Google Scholar] [CrossRef]
- Marques, A.J.; Palanimurugan, R.; Matias, A.C.; Ramos, P.C.; Dohmen, R.J. Catalytic mechanism and assembly of the proteasome. Chem. Rev. 2009, 109, 1509–1536. [Google Scholar] [CrossRef]
- Genin, E.; Reboud-Ravaux, M.; Vidal, J. Proteasome inhibitors: Recent advances and new perspectives in medicinal chemistry. Curr. Top. Med. Chem. 2010, 10, 232–256. [Google Scholar] [CrossRef]
- Arrigo, A.P.; Tanaka, K.; Goldberg, A.L.; Welch, W.J. Identity of the 19s prosome particle with the large multifunctional protease complex of mammalian-cells. Nature 1988, 331, 192–194. [Google Scholar] [CrossRef]
- Lowe, J.; Stock, D.; Jap, R.; Zwickl, P.; Baumeister, W.; Huber, R. Crystal-structure of the 20s proteasome from the archaeon T-acidophilum at 3.4-Angstrom resolution. Science 1995, 268, 533–539. [Google Scholar]
- Groll, M.; Ditzel, L.; Lowe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 angstrom resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. The structure of the mammalian 20S proteasome at 2.75 angstrom resolution. Structure 2002, 10, 609–618. [Google Scholar] [CrossRef]
- Groll, M.; Larionov, O.V.; Huber, R.; de Meijere, A. Inhibitor-binding mode of homobelactosin C to proteasomes: New insights into class I MHC ligand generation. Proc. Natl. Acad. Sci. USA 2006, 103, 4576–4579. [Google Scholar]
- Kaiser, M.; Groll, M.; Siciliano, C.; Assfalg-Machleidt, I.; Weyher, E.; Kohno, J.; Milbradt, A.G.; Renner, C.; Huber, R.; Moroder, L. Binding mode of TMC-95A analogues to eukaryotic 20S proteasome. ChemBioChem 2004, 5, 1256–1266. [Google Scholar] [CrossRef]
- Groll, M.; Gotz, M.; Kaiser, M.; Weyher, E.; Moroder, L. TMC-95-based inhibitor design provides evidence for the catalytic versatility of the proteasom. Chem. Biol. 2006, 13, 607–614. [Google Scholar] [CrossRef]
- Clerc, J.; Florea, B.I.; Kraus, M.; Groll, M.; Huber, R.; Bachman, A.; Dudler, R.; Driessen, C.; Overkleeft, H.S.; Kaiser, M.C. Syringolin A selectively labels the 20S proteasome in murine EL4 and wild-type and bortezomib-adapted leukaemic cell lines. ChemBioChem 2009, 10, 2638–2643. [Google Scholar] [CrossRef]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef]
- Meng, L.; Mohan, R.; Kwok, B.H.; Elofsson, M.; Sin, N.; Crews, C.M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo anti-inflammatory activity. Proc. Natl. Acad. Sci. USA 1999, 96, 10403–10408. [Google Scholar] [CrossRef]
- Palmer, J.T.; Rasnick, D.; Klaus, J.L.; Bromme, D. Vinyl sulfones as mechanism-based cysteineprotease inhbibitors. J. Med. Chem. 1995, 38, 3193–3196. [Google Scholar] [CrossRef]
- Omura, S.; Matsuzaki, K.; Fujimoto, T.; Kosuge, K.; Furuya, T.; Fujita, S.; Nakagawa, A. Structure of lactacystin, a new microbial metabolite which induces differentiation of neuroblastoma cells. J. Antibiot. 1991, 44, 117–118. [Google Scholar] [CrossRef]
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Chois, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activites and subunit-specific amino-terminal threonine modification by lactacystin. Scinece 1995, 268, 726–731. [Google Scholar]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Groll, M.; Huber, R.; Potts, B.C.M. Crystal structures of salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of beta-lactone ring opening and a mechanism for irreversible binding. J. Am. Chem. Soc. 2006, 128, 5136–5141. [Google Scholar] [CrossRef]
- Vinitsky, A.; Michaud, C.; Powers, J.C.; Orlowski, M. Inhibition of the chymotrypsin-Like activity of the pituitary multicatalytic proteinase complex. Biochemistry 1992, 31, 9421–9428. [Google Scholar] [CrossRef]
- Momose, I.; Umezawa, Y.; Hirosawa, S.; Iinuma, H.; Ikeda, D. Structure-based design of derivatives of tyropeptin A as the potent and selective inhibitors of mammalian 20S proteasome. Bioorg. Med. Chem. Lett. 2005, 15, 1867–1871. [Google Scholar] [CrossRef]
- Loidl, G.; Groll, M.; Musiol, H.J.; Ditzel, L.; Huber, R.; Moroder, L. Bifunctional inhibitors of the trypsin-like activity of eukaryotic proteasomes. Chem. Biol. 1999, 6, 197–204. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, B.; Liu, D.; Yang, Y.; Xiong, Z.; Zeng, J.; Dong, Y. MG132 treatment attenuates cardiac remodeling and dysfunction followingaortic banding in rats via the NF-κB/TGFβ1 pathway. Biochem. Pharmacol. 2011, 81, 1228–1236. [Google Scholar] [CrossRef]
- Donkor, I.O. A survey of calpain inhibitors. Curr. Med. Chem. 2000, 7, 1171–1188. [Google Scholar] [CrossRef]
- Tsubuki, S.; Saito, Y.; Tomioka, M.; Ito, H.; Kawashima, S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J. Biochem. 1996, 119, 572–576. [Google Scholar] [CrossRef]
- Zhang, X.C.; Kumar, S.; Chen, J.H.; Teplyakov, A.V. Covalent attachment of shape-restricted DNA molecules on amine-functionalized Si(III) surface. Surface Sci. 2009, 603, 2445–2457. [Google Scholar] [CrossRef]
- Zhu, Y.Q.; Pei, J.F.; Liu, Z.M.; Lai, L.H.; Cui, J.R.; Li, R.T. 3D-QSAR studies on tripeptide aldehyde inhibitors of proteasome using CoMFA and CoMSIA methods. Bioorg. Med. Chem. 2006, 14, 1483–1496. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, H.; Xiao, Z.; Wang, F.; Wang, X.; Wang, Y. Combined 3D-QSAR, molecular focking and molecular fynamics dtudy on ferivatives of peptide epoxyketone and tyropeptin-boronic acid as inhibitors against the β5 subunit of Human 20S proteasome. Int. J. Mol. Sci. 2011, 12, 1807–1835. [Google Scholar] [CrossRef]
- Mckennon, M.J.; Meyers, A.I.; Drauz, K.; Schwarm, M. A Convenient reduction of amino-acids and their derivatives. J. Org. Chem. 1993, 58, 3568–3571. [Google Scholar] [CrossRef]
- Drag, M.; Latajka, R.; Gumienna-Kontecka, E.; Kozlowski, H.; Kafarski, P. Stereoselective synthesis, solution structure and metal complexes of (1S,2S)-2-amino-1-hydroxyalkylphosphonic acids. Tetrahedron Asymmetry 2003, 14, 1837–1845. [Google Scholar] [CrossRef]
- Fu, Y.Q.; Xu, B.; Zou, X.M.; Ma, C.; Yang, X.M.; Mou, K.; Fu, G.; Lu, Y.; Xu, P. Design and synthesis of a novel class of furan-based molecules as potential 20S proteasome inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1102–1106. [Google Scholar]
- Zhu, Y.Q.; Zhao, X.; Zhu, X.R.; Wu, G.; Li, Y.J.; Ma, Y.H.; Yuan, Y.X.; Yang, J.; Hu, Y.; Ai, L.; Gao, Q.Z. Design, synthesis, biological evaluation, and structure-activity relationship (SAR) discussion of dipeptidyl boronate proteasome inhibitors, Part I: Comprehensive understanding of the SAR of α-amino acid boronates. J. Med. Chem. 2009, 52, 4192–4199. [Google Scholar] [CrossRef]
- Rydzewski, R.M.; Burrill, L.; Mendonca, R.; Palmer, J.T.; Rice, M.; Tahilramani, R.; Bass, K.E.; Leung, L.; Gjerstad, E.; Janc, J.W.; Pan, L. Optimization of subsite binding to the 5 subunit of the human 20S proteasome using vinyl sulfones and 2-keto-1, 3, 4-oxadiazoles: Syntheses and cellular properties of potent, selective proteasome inhibitors. J. Med. Chem. 2006, 49, 2953–296. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ma, Y.; Xu, B.; Fang, Y.; Yang, Z.; Cui, J.; Zhang, L.; Zhang, L. Synthesis and SAR Study of Novel Peptide Aldehydes as Inhibitors of 20S Proteasome. Molecules 2011, 16, 7551-7564. https://doi.org/10.3390/molecules16097551
Ma Y, Xu B, Fang Y, Yang Z, Cui J, Zhang L, Zhang L. Synthesis and SAR Study of Novel Peptide Aldehydes as Inhibitors of 20S Proteasome. Molecules. 2011; 16(9):7551-7564. https://doi.org/10.3390/molecules16097551
Chicago/Turabian StyleMa, Yuheng, Bo Xu, Yuan Fang, Zhenjun Yang, Jingrong Cui, Liangren Zhang, and Lihe Zhang. 2011. "Synthesis and SAR Study of Novel Peptide Aldehydes as Inhibitors of 20S Proteasome" Molecules 16, no. 9: 7551-7564. https://doi.org/10.3390/molecules16097551