Synthesis of Quinolin-2-one Alkaloid Derivatives and Their Inhibitory Activities against HIV-1 Reverse Transcriptase

Abstract
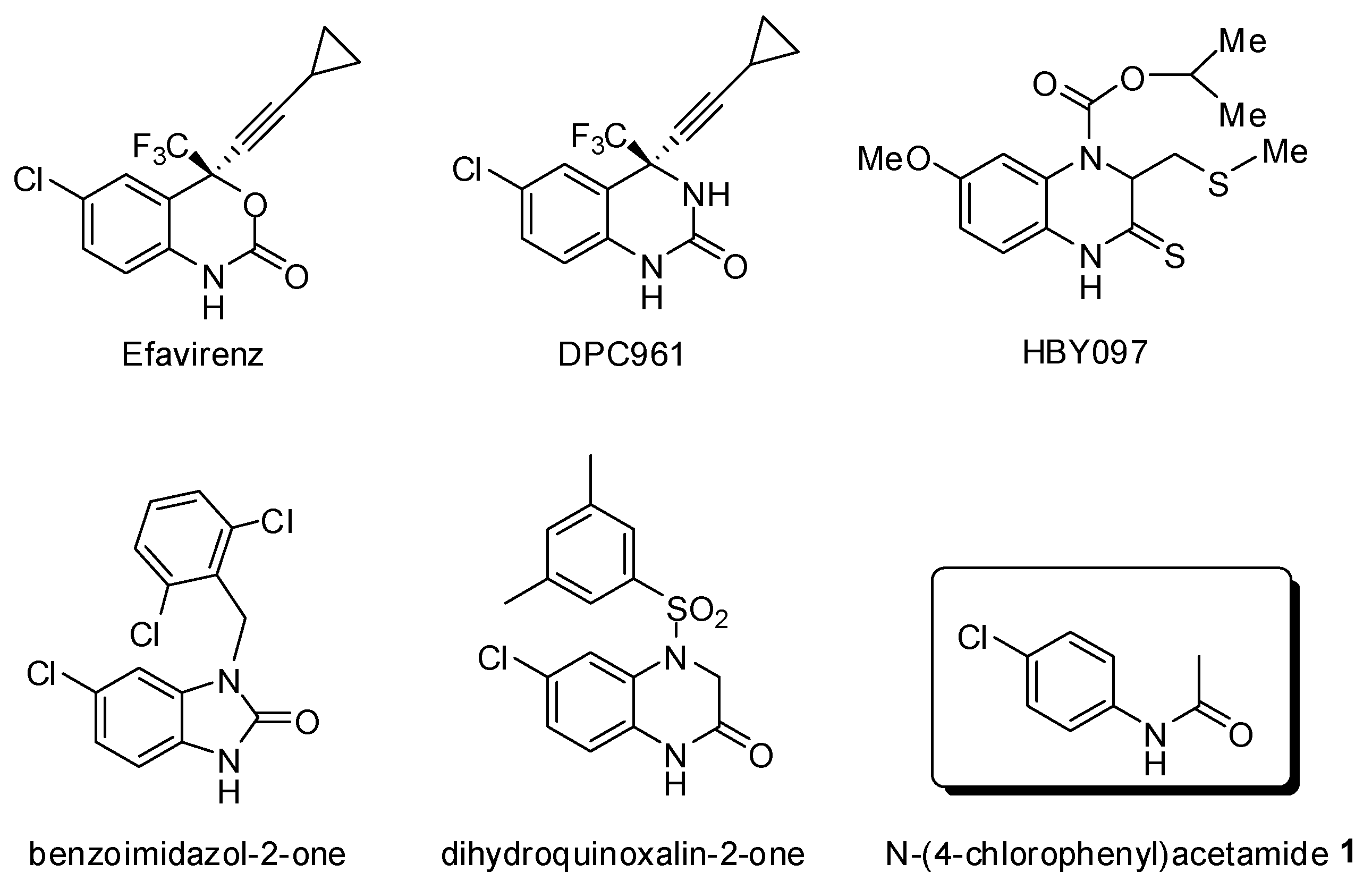
:1. Introduction


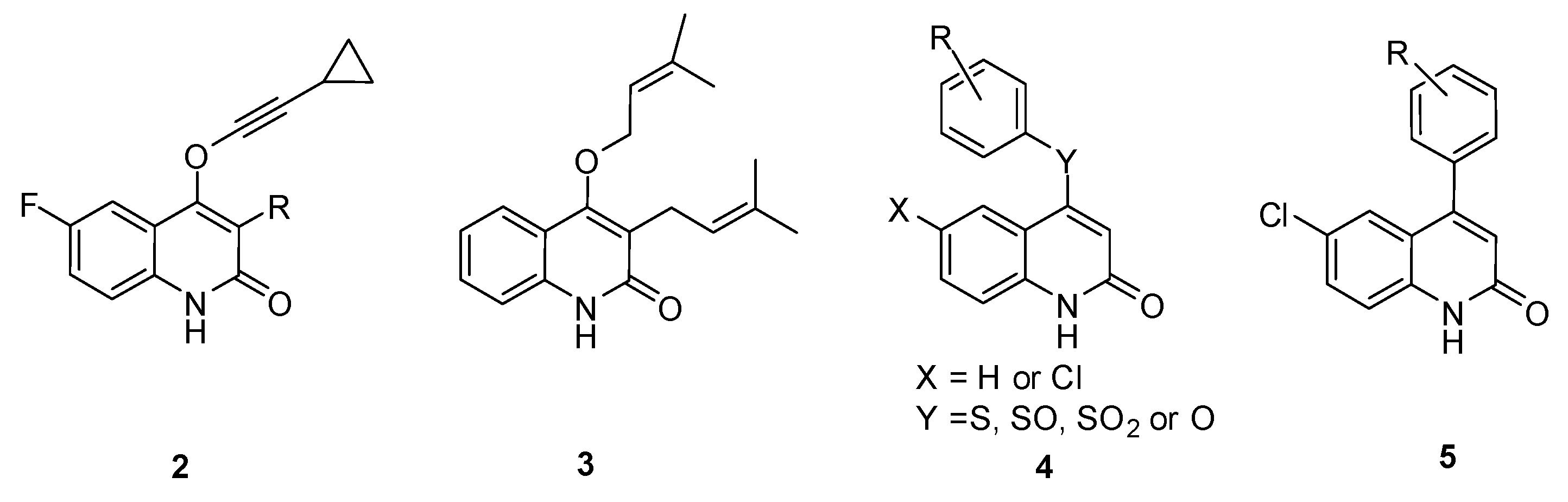
2. Results and Discussion
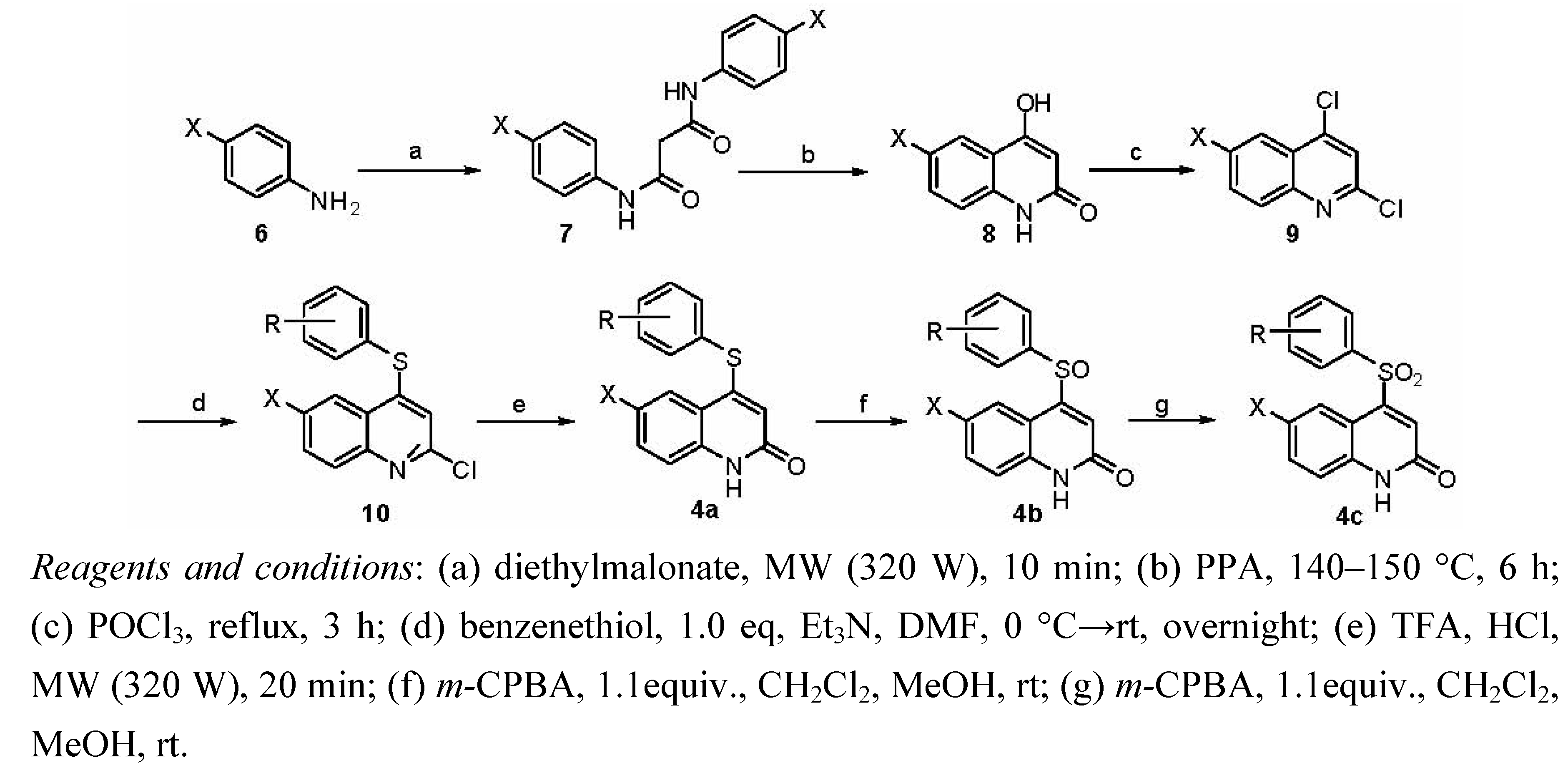
2.1. Chemistry



2.2. Enzymatic Activities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | X | Y | R | IC50 (μM) b,c |
|---|---|---|---|---|
| 4a1 | Cl | S | H | 5.6 |
| 4a2 | Cl | S | 3,5-CH3 | 0.21 |
| 4a3 | H | S | 3,5-CH3 | 18 |
| 4b1 | Cl | SO | H | 8.9 |
| 4b2 | Cl | SO | 3,5-CH3 | 2.6 |
| 4c1 | Cl | SO2 | H | 49 |
| 4c2 | Cl | SO2 | 3,5-CH3 | 10 |
| 4d1 | Cl | O | H | 3.0 |
| 4d2 | Cl | O | 3,5-CH3 | 0.15 |
| 5a | Cl | / | H | >100 |
| 5b | Cl | / | 2-F | >100 |
| 5c | Cl | / | 2-Cl | >100 |
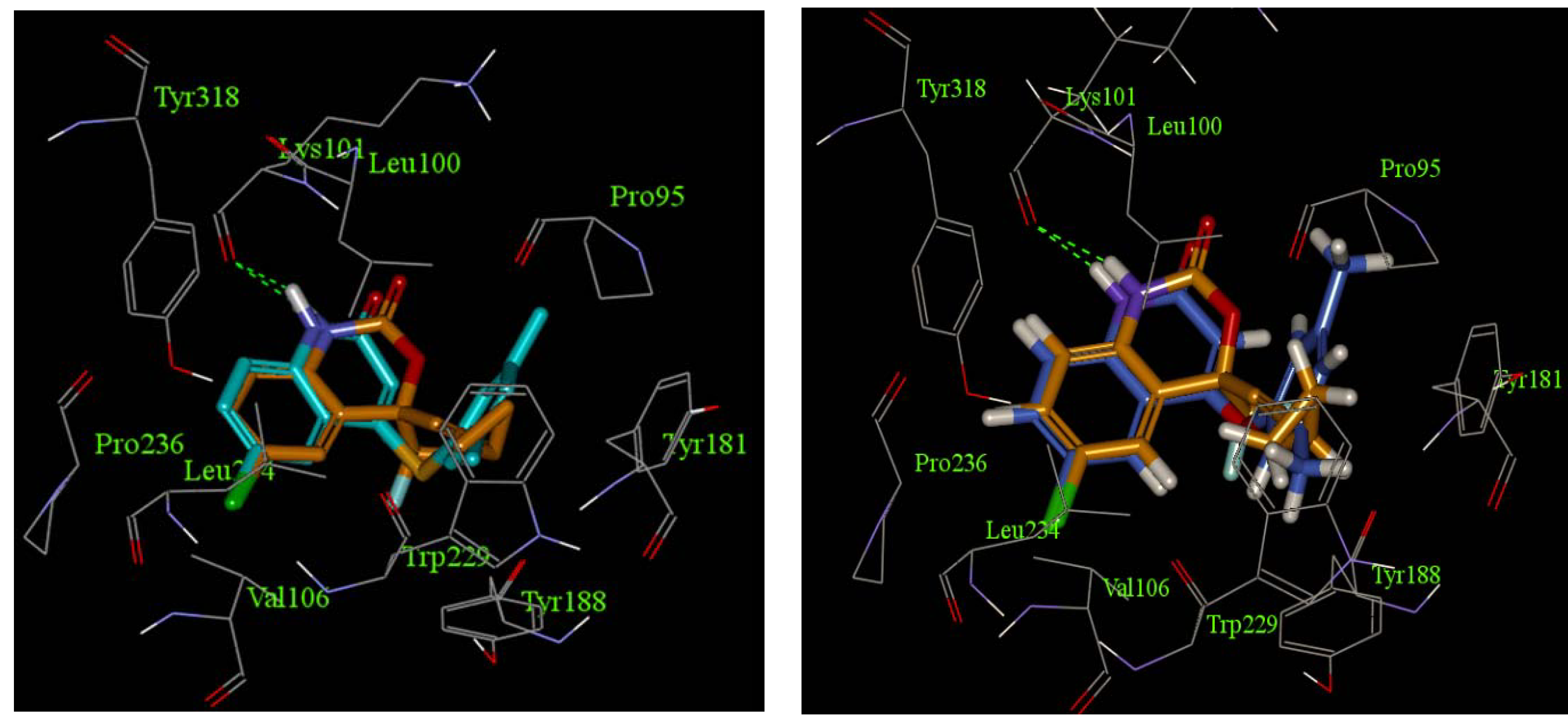
2.3. Docking Studies

3. Experimental
3.1. Chemistry
3.1.1. Instruments and Materials
3.1.2. General Procedure for Preparation of Compounds 4
3.1.2.1. Synthesis of Intermediates 7
3.1.2.2. Synthesis of Intermediates 8
3.1.2.3. Synthesis of Intermediates 9
3.1.2.4. Synthesis of Intermediates 10
3.1.2.5. Synthesis of Target Compounds 4a
3.1.2.6. Synthesis of Target Compounds 4b
3.1.2.7. Synthesis of Target Compounds 4c
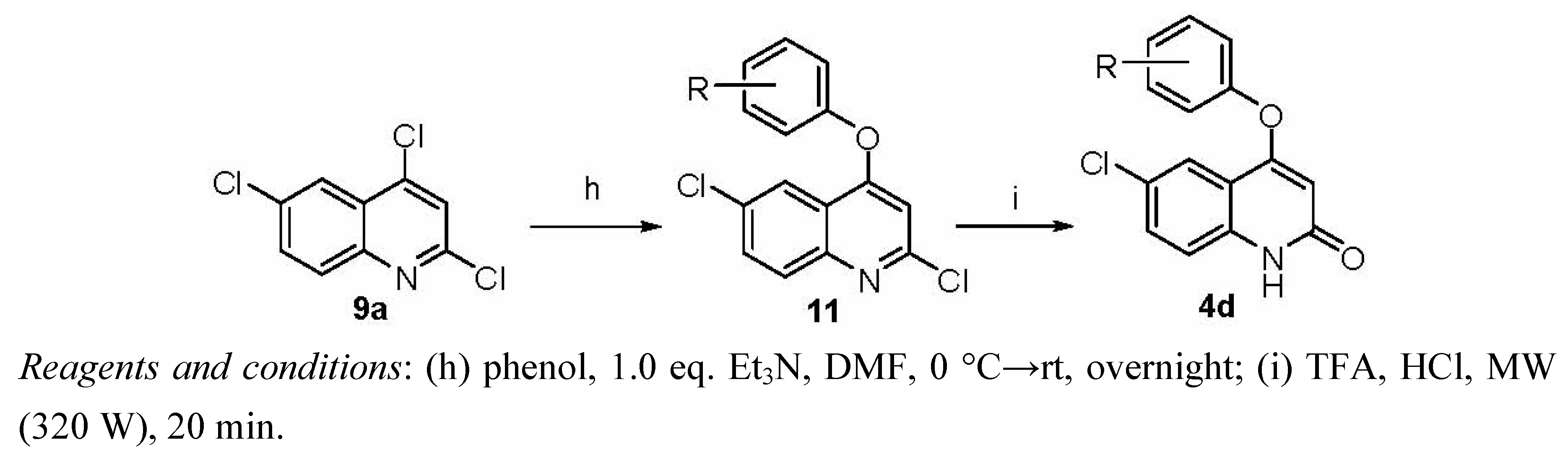
3.1.2.8. Synthesis of Target Compounds 4d
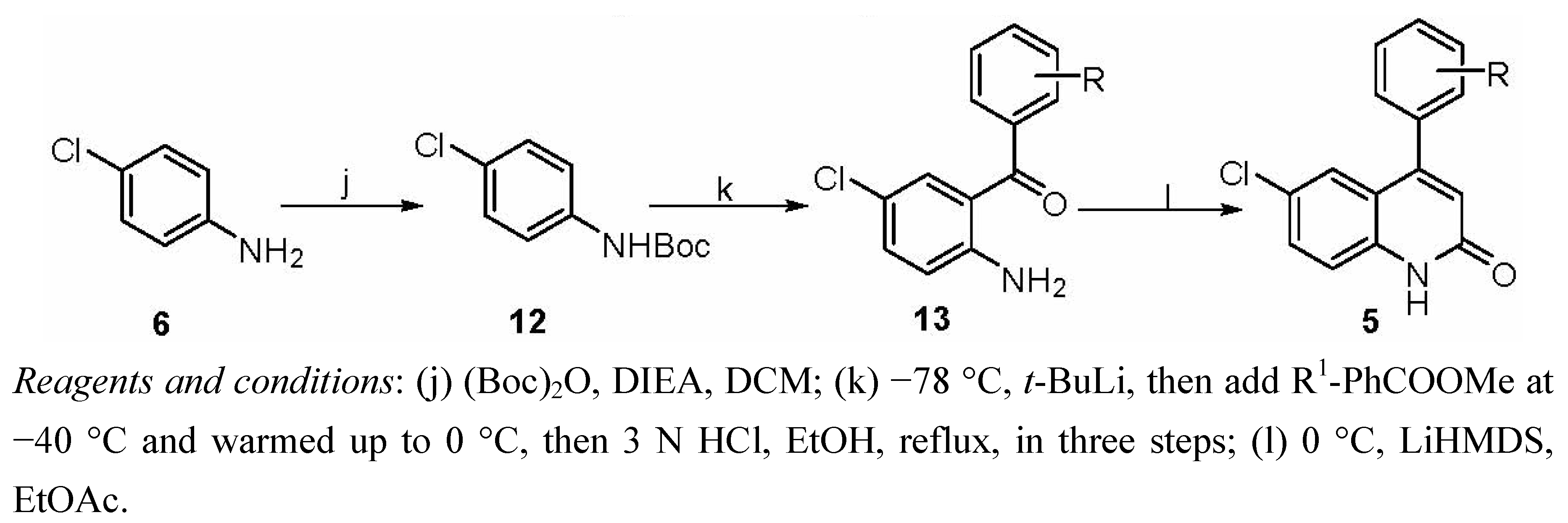
3.1.3. General Procedure for Preparation of Compounds 5
3.1.3.1. Synthesis of Intermediate 12
3.1.3.2. Synthesis of Compound 13
3.1.3.3. Synthesis of Compounds 5
3.2. General Procedure for HIV-1 RT Inhibitory Assay
3.3. General Procedures for Docking Study
4. Conclusions
Acknowledgments
- Samples Availability: Samples of the all compounds are available from the authors.
References
- Mehellou, Y.; Clercq, E.D. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef]
- Ragno, R.; Coluccia, A.; Regina, G.L.; Martino, G.D.; Piscitelli, F.; Lavecchia, A.; Novellino, E.; Bergamini, A.; Ciaprini, C.; Sinistro, A.; et al. Design, molecular modeling, synthesis, and anti-HIV-1 activity of new indolyl aryl sulfones. Novel derivatives of the indole-2-carboxamide. J. Med. Chem. 2006, 49, 3172–3184. [Google Scholar] [CrossRef]
- Barreca, M.L.; Rao, A.; Luca, L.D.; Iraci, N.; Monforte, A.; Maga, G.; Clercq, E.D.; Pannecouque, C.; Balzarini, J.; Chimirri, A. Discovery of novel benzimidazolones as potent non-nucleoside reverse transcriptase inhibitors active against wild-type and mutant HIV-1 strains. Bioorg. Med. Chem. Lett. 2007, 17, 1956–1960. [Google Scholar] [CrossRef]
- Regina, G.L.; Coluccia, A.; Piscitelli, F.; Bergamini, A.; Sinistro, A.; Cavazza, A.; Maga, G.; Samuele, A.; Zanoli, S.; Novellino, E.; et al. Indolyl aryl sulfones as HIV-1 non-nucleoside reverse transcriptase inhibitors: Role of two halogen atoms at the indole ring in developing new analogues with improved antiviral activity. J. Med. Chem. 2007, 50, 5034–5038. [Google Scholar] [CrossRef]
- Jones, L.H.; Allan, G.; Barba, O.; Burt, C.; Corbau, R.; Dupont, T.; Knochel, T.; Irving, S.; Middleton, D. S.; Mowbray, C. E.; et al. Novel indazole non-nucleoside reverse transcriptase inhibitors using molecular hybridization based on crystallographic overlays. J. Med. Chem. 2009, 52, 1219–1923. [Google Scholar]
- Freeman, G.A.; Andrews, C.W., III.; Hopkins, A.L.; Lowell, G.S.; Schaller, L.T.; Cowan, J.R.; Gonzales, S.S.; Koszalka, G.W.; Hazen, R.J.; Boone, L.R.; et al. Design of non-nucleoside inhibitors of HIV-1 reverse transcriptase with improved drug resistance properties. 2. J. Med. Chem. 2004, 47, 5923–5936. [Google Scholar]
- Barreca, M.L.; Rao, A.; Luca, L.D.; Zappalà, M.; Monforte, A.; Maga, G.; Pannecouque, C.; Balzarini, J.; Clercq, E.D.; Chimirri, A.; Monforte, P. Computational strategies in discovering novel non-nucleoside inhibitors of HIV-1 RT. J. Med. Chem. 2005, 48, 3433–3437. [Google Scholar] [CrossRef]
- Monforte, A.; Logoteta, P.; Ferro, S.; Luca, L.D.; Iraci, N.; Maga, G.; Clercq, E.D.; Pannecouque, C.; Chimirri, A. Design, synthesis, and structure-activity relationships of 1,3-dihydrobenzimidazol-2-one analogues as anti-HIV agents. Bioorg. Med. Chem. Lett. 2009, 17, 5962–5967. [Google Scholar] [CrossRef]
- Mccormick, J.L.; McKee, T.C.; Cardellina, J.H., II.; Boyd, M.R. HIV inhibitory natural products. 26. quinoline alkaloids from Euodia roxburghiana. J. Nat. Prod. 1996, 59, 469–471. [Google Scholar] [CrossRef]
- Guillemont, J.; benjahad, A.; Oumouch, S.; Decrane, L.; Palandjian, P.; Vernier, D.; Queguiner, L.; Andries, K.; Béthune, M.; Hertogs, K.; et al. Synthesis and biological evaluation of C-5 methyl substituted 4-arylthio and 4-aryloxy-3-Iodopyridin-2(1H)-one type anti-HIV agents. J. Med. Chem. 2009, 52, 7473–7487. [Google Scholar] [CrossRef]
- Mowbray, C.E.; Burt, C.; Corbau, R.; Perros, M.; Tran, I.; Stupple, P.A.; Webster, R.; Wood, A. Pyrazole NNRTIs 1: Design and initial optimisation of a novel template. Bioorg. Med. Chem. Lett. 2009, 19, 5599–5602. [Google Scholar] [CrossRef]
- Xu., B.; Sun., Y.; Guo, Y.; Cao, Y.; Yu, T. Synthesis and biological evaluation of N4-(hetero)arylsulfonylquinoxalinones as HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. 2009, 17, 2767–2774. [Google Scholar] [CrossRef]
- Ren, J.; Chamberlain, P.P.; Stamp, A.; Short, S.A.; Weaver, K.L.; Romines, K.R.; Hazen, R.; Freeman, A.; Ferris, R.G.; Andrew, C.W.; et al. Structural basis for the improved drug resistance profile of new generation benzophenone non-nucleoside HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 2008, 51, 5000–5008. [Google Scholar] [CrossRef]
- O’llah, E.; Rufchahi, M. Synthesis of 6-chloro and 6-fluoro-4-hydroxyl-2-quinolone and their azo disperse dyes. Chin. Chem. Lett. 2010, 21, 542–546. [Google Scholar] [CrossRef]
- Zhu, Q.; Reza, F.; Yang, Z.; Anthony, A.; Liu, Y.; Joon, C.H.; Richard, W. 4-Thio substituted quinoline and naphthyridine compounds. Patent EP 2,061,466, 2009. [Google Scholar]
- Whisler, M.C.; MacNeil, S.; Snieckus, V.; Beak, P. Beyond thermodynamic acidity: A perspective on the complex-induced proximity effect (CIPE) in deprotonation reactions. Angew. Chem. Int. Ed. Engl. 2004, 43, 2206–2225. [Google Scholar] [CrossRef]
- Hewawasam, P.; Fan, W.; Knipe, J.; Moon, S.L.; Boissard, C.G.; Gribkoff, V.K.; Starrett, J.E. The synthesis and structure-activity relationships of 4-aryl-3-aminoquinolin-2-ones: A new class of calcium-dependent, large conductance, potassium (maxi-K) channel openers targeted for post-stroke neuroprotection. Bioorg. Med. Chem. Lett. 2002, 12, 1779–1783. [Google Scholar] [CrossRef]
- Ren, J.; Milton, J.; Weaver, K.L.; Short, S.A.; Stuart, D.I. Structural basis for the resilience of efavirenz (DMP-266) to drug resistance mutations in HIV-1 reverse transcriptase. Structure 2000, 8, 1089–1094. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cheng, P.; Gu, Q.; Liu, W.; Zou, J.-F.; Ou, Y.-Y.; Luo, Z.-Y.; Zeng, J.-G. Synthesis of Quinolin-2-one Alkaloid Derivatives and Their Inhibitory Activities against HIV-1 Reverse Transcriptase. Molecules 2011, 16, 7649-7661. https://doi.org/10.3390/molecules16097649
Cheng P, Gu Q, Liu W, Zou J-F, Ou Y-Y, Luo Z-Y, Zeng J-G. Synthesis of Quinolin-2-one Alkaloid Derivatives and Their Inhibitory Activities against HIV-1 Reverse Transcriptase. Molecules. 2011; 16(9):7649-7661. https://doi.org/10.3390/molecules16097649
Chicago/Turabian StyleCheng, Pi, Qiong Gu, Wei Liu, Jian-Feng Zou, Yang-Yong Ou, Zhong-Yong Luo, and Jian-Guo Zeng. 2011. "Synthesis of Quinolin-2-one Alkaloid Derivatives and Their Inhibitory Activities against HIV-1 Reverse Transcriptase" Molecules 16, no. 9: 7649-7661. https://doi.org/10.3390/molecules16097649