One Step Formation of Propene from Ethene or Ethanol through Metathesis on Nickel Ion-loaded Silica

Abstract
:1. Introduction
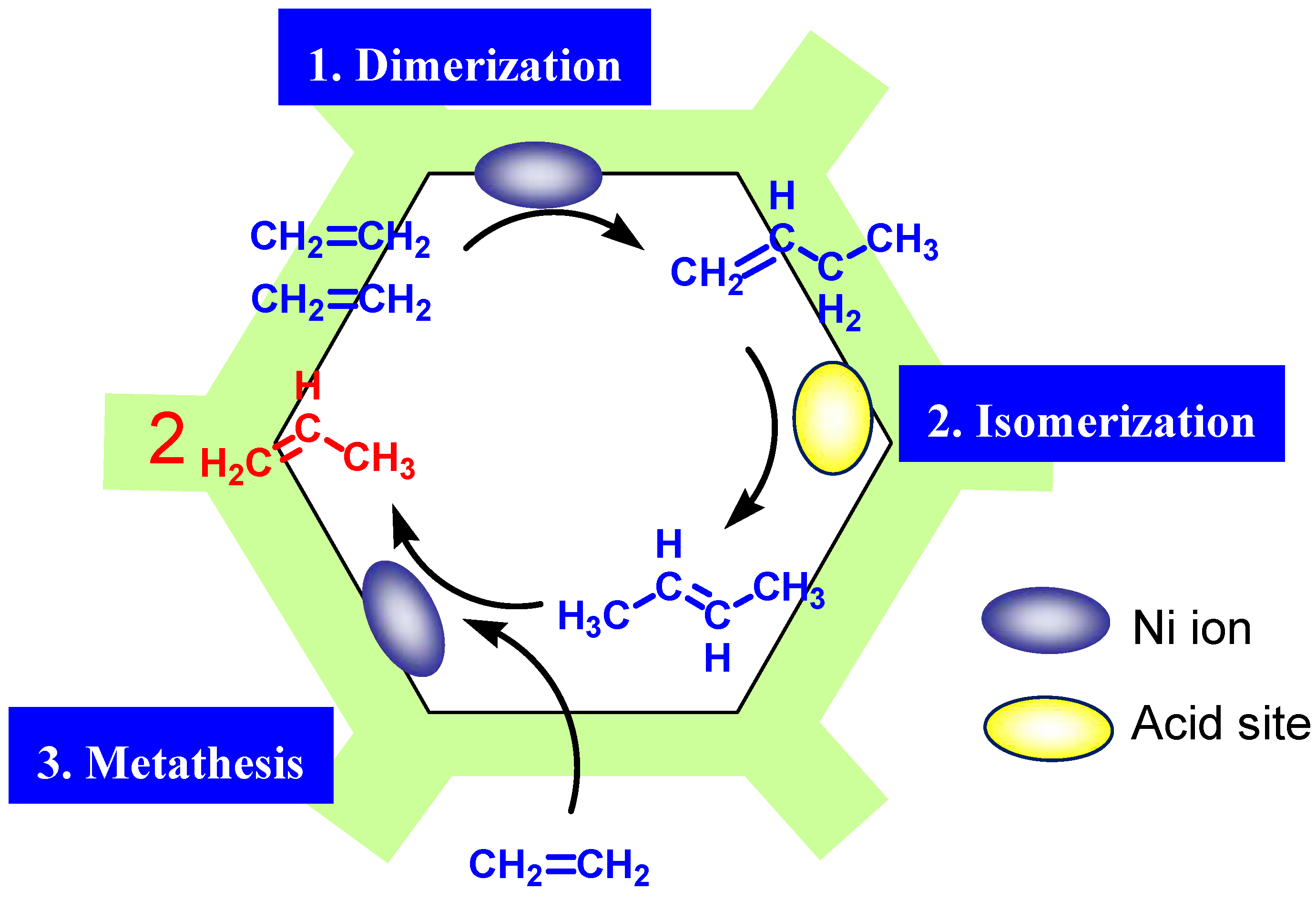
2. Results and Discussion
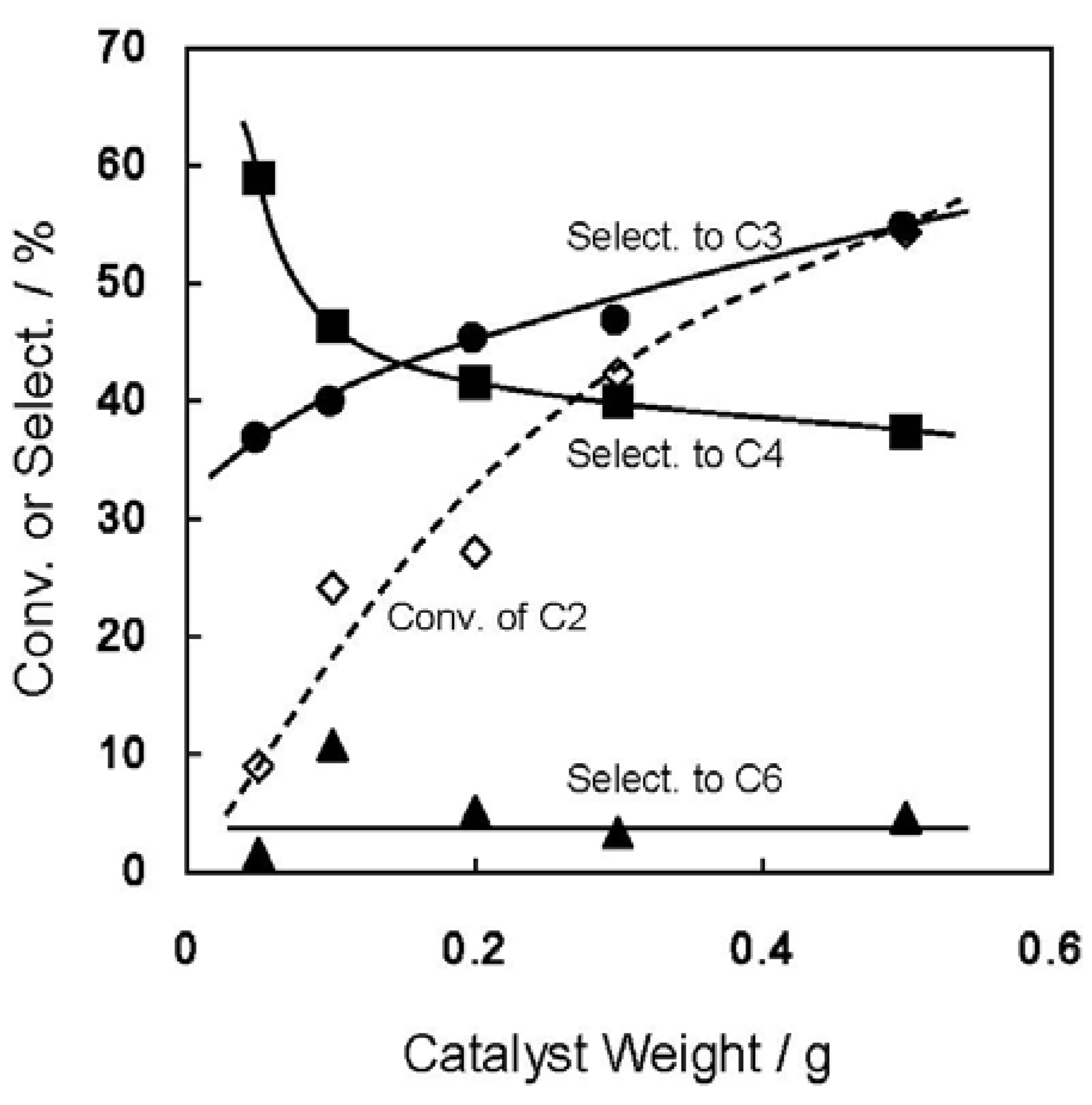
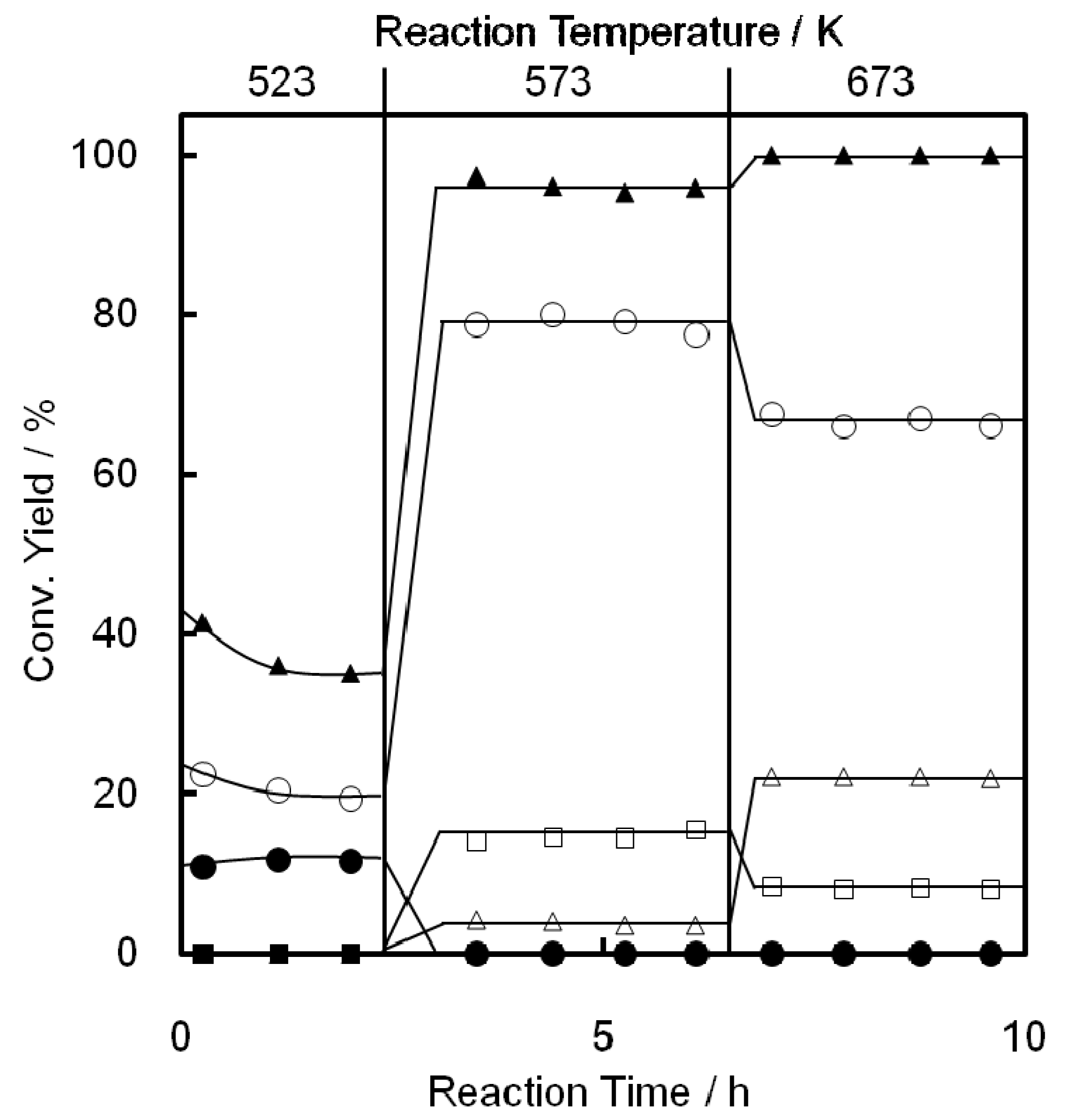
2.1. Conversion of Ethene to Propene on Ni-M41 Catalysts



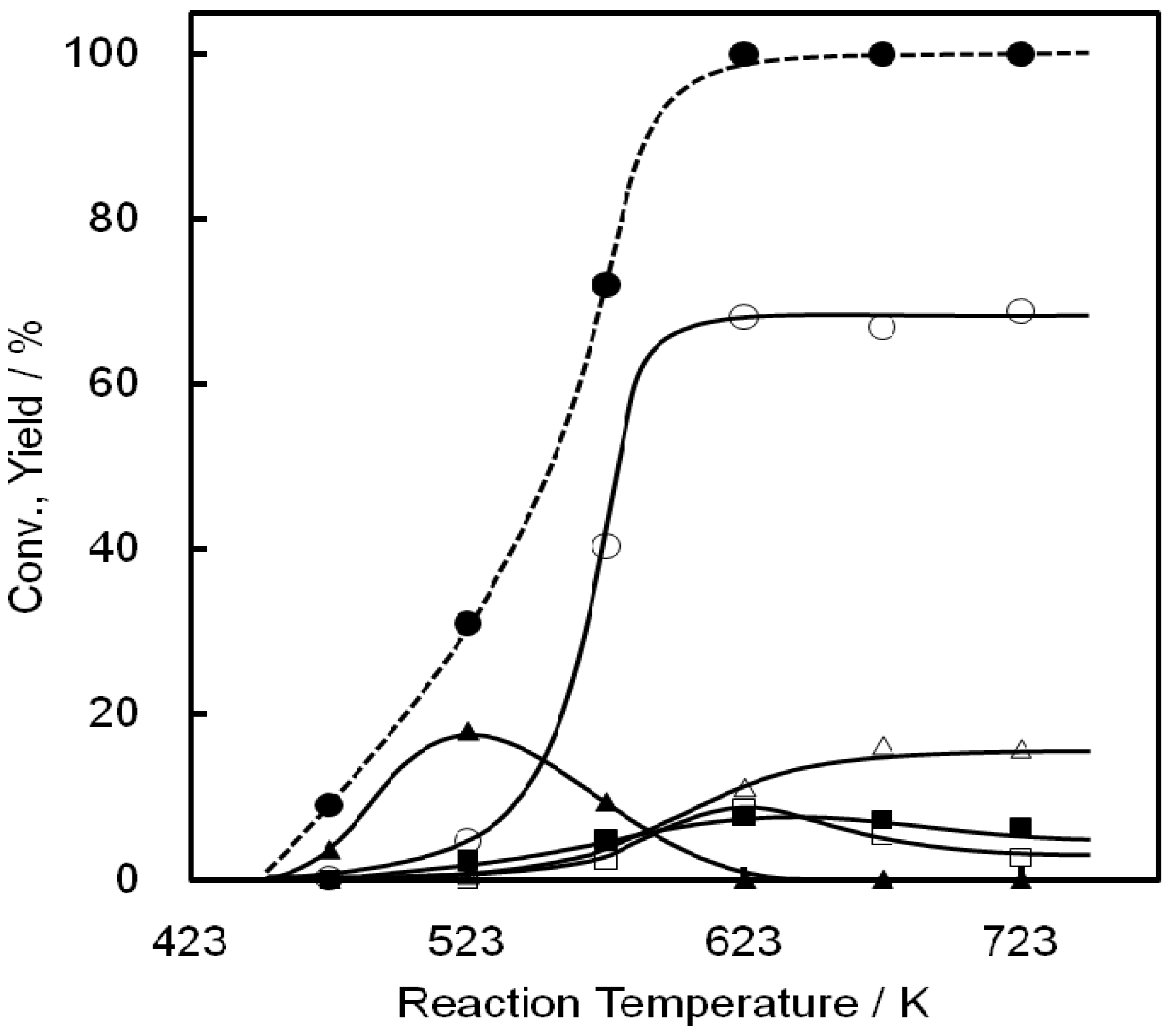
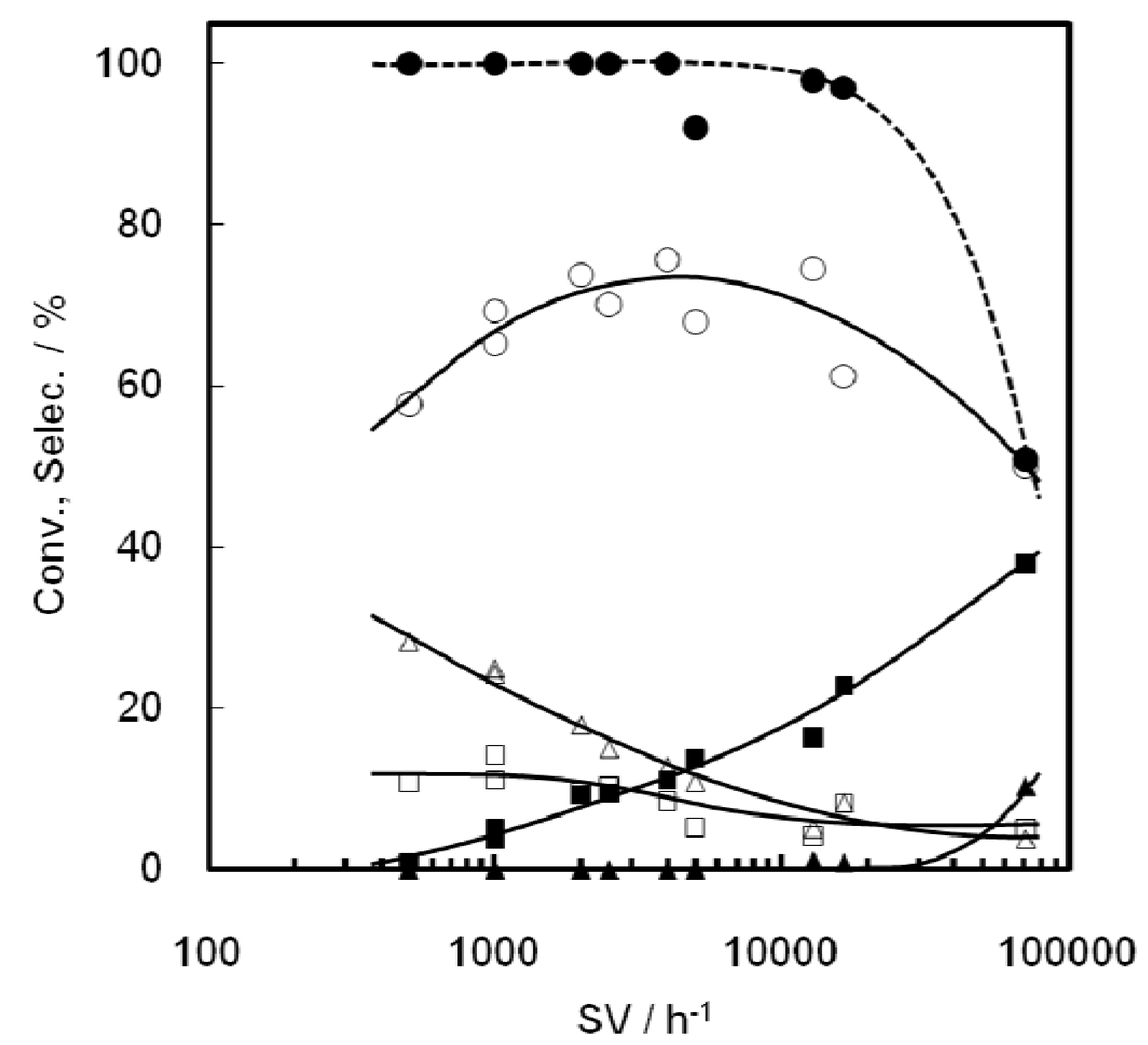
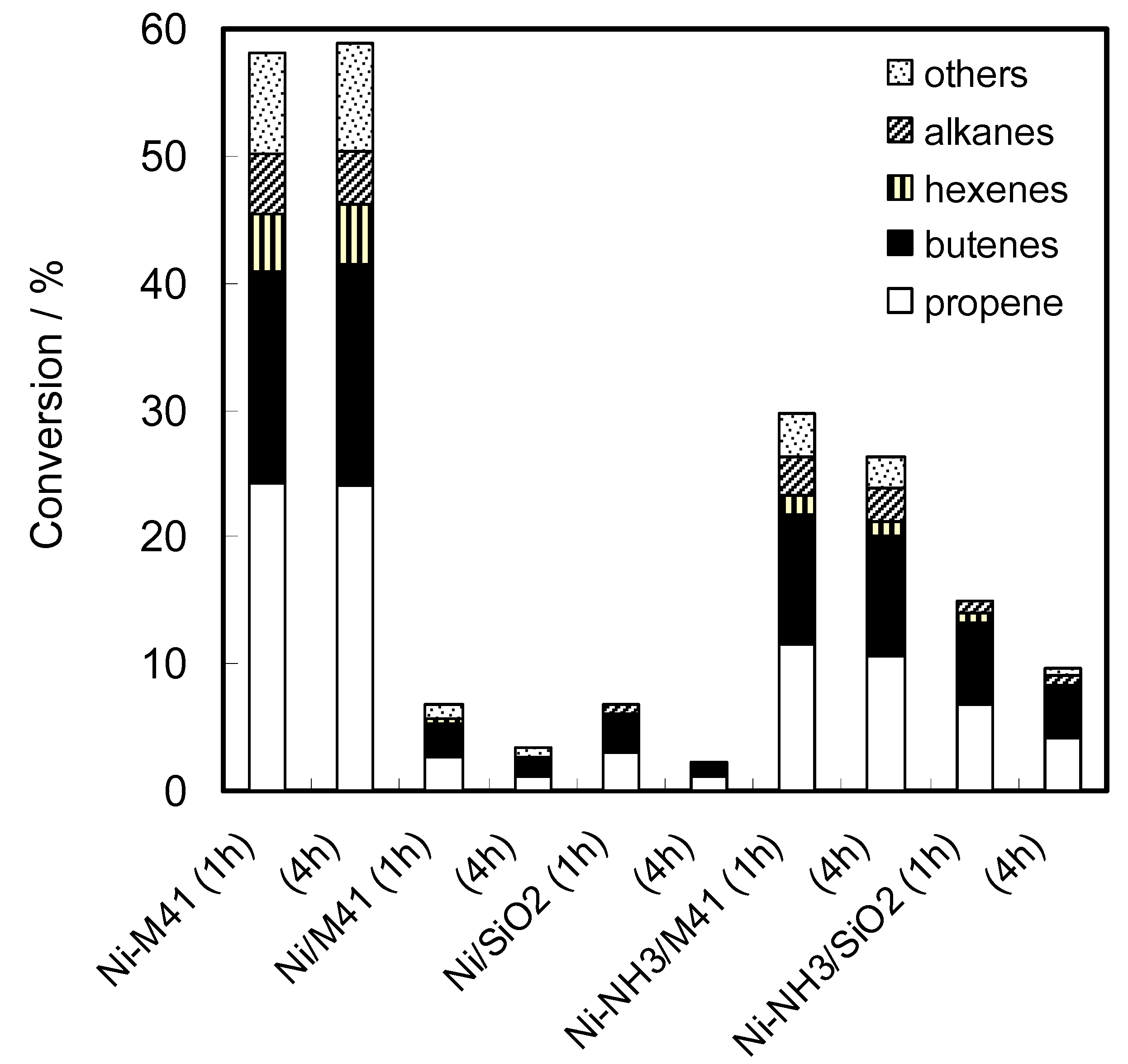
2.2. Reaction of Ethanol on Ni-MCM-41















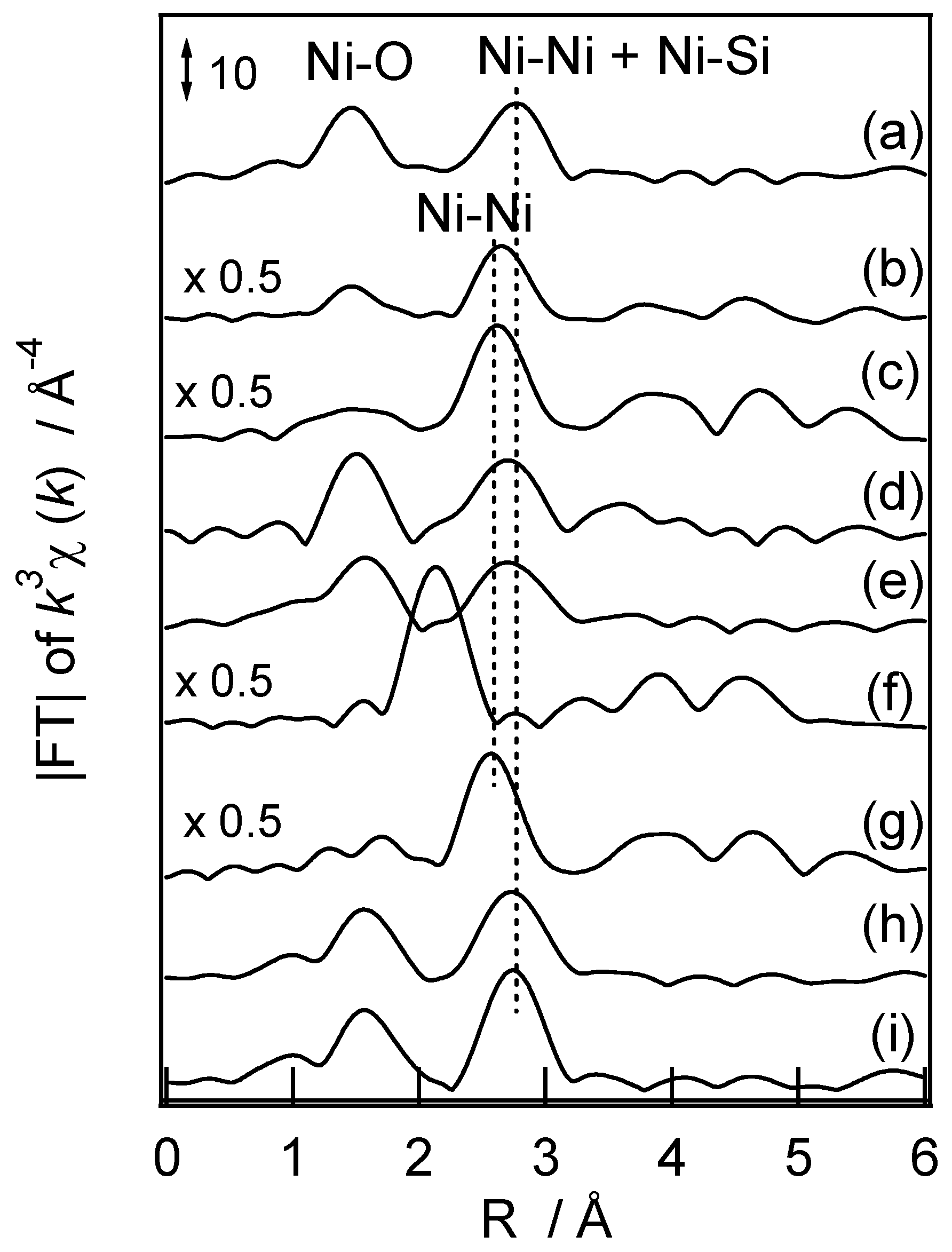
2.3. Characterization of Nickel Species Loaded on the Mesoporous Silica


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Shell | C. N. a | D/nm b | Δσ2/nm2 c | R/% d |
|---|---|---|---|---|---|
| Ni-M41 | Ni-O | 6.9 | 0.208 | 6.08 × 10−5 | 16.2 |
| Ni-Ni | 5.1 | 0.305 | 4.90 × 10−5 | 4.4 | |
| Ni-Si | 2.0 | 0.336 | 1.02 × 10−5 | ||
| Ni/M41 | Ni-O | f | |||
| Ni-Ni | 10.3 | 0.300 | 5.04 × 10−5 | 3.0 | |
| Ni/SiO2 | Ni-O | f | |||
| Ni-Ni | 11.6 | 0.296 | 3.84 × 10−5 | 1.3 | |
| Ni-NH3/M41 | Ni-O | f | |||
| Ni-Ni | 3.8 | 0.305 | 3.25 × 10−5 | 6.1 | |
| Ni-Si | 2.4 | 0.337 | 0.6 × 10−5 | ||
| NiO | Ni-O | 6 | 0.208 | ||
| Ni-Ni | 12 | 0.295 | |||
| Ni-talcite e | Ni-Ni | 6.0 | 0.305 | ||
| Ni-Si | 5.0 | 0.327 | |||
| Nepouite e | Ni-Ni | 6.0 | 0.309 | ||
| Ni-Si | 2.4 | 0.327 |

3. Experimental Section
4. Conclusions
Acknowledgments
- Sample Availability: No sample of the compounds is available from the author.
References
- Arpe, H.J. Industrial Organic Chemistry, 4th ed; Wiley-VCH Verlag BmbH & Co.: Weinheim, Germany, 2003. [Google Scholar]
- Olah, G.A.; Molnar, A. Hydrocarbon Chemistry, 2nd ed; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003. [Google Scholar]
- Banks, R.L.; Kukes, S.G. New developments and concepts in enhancing activities of heterogeneous metathesis catalysts. J. Mol. Catal. 1985, 28, 117–131. [Google Scholar] [CrossRef]
- O’Nill, P.P.; Rooney, J.J. Direct transformation of ethylene to propylene on an olefin metathesis catalyst. J. Am. Chem. Soc. 1972, 94, 4383–4384. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Tanaka, Y.; Tanabe, K. Isomerization and disproportionation of olefins over tungsten oxides supported on various oxides. J. Catal. 1980, 65, 442–447. [Google Scholar]
- Lu, J.; Zhao, Z.; Xu, C.; Duan, A.; Zhang, P. CrHZSM-5 Zeolites - Highly Efficient Catalysts for Catalytic Cracking of Isobutane to Produce Light Olefins. Catal. Lett. 2006, 109, 65–70, and the references therein. [Google Scholar] [CrossRef]
- Oikawa, H.; Shibata, Y.; Baba, T. Highly selective conversion of ethene to propene over SAPO-34 as a solid acid catalyst. Appl. Catal. A Gen. 2006, 312, 181–185. [Google Scholar] [CrossRef]
- Speiser, F.; Braunstein, P.; Saussine, L. Catalytic Ethylene Dimerization and Oligomerization: Recent Developments with Nickel Complexes Containing P,N-Chelating Ligands. Acc. Chem. Res. 2005, 38, 784–793, and the references therein. [Google Scholar] [CrossRef]
- Shiba, T.; Ozaki, A. A catalyst consisting of nickel oxide and silica. I. The catalytic activity of ethylene polymerization caused by vacuum heating. Nippon Kagaku Zasshi 1953, 74, 295–297. [Google Scholar] [CrossRef]
- Ozaki, A. Mixed catalyst composed of nickel oxide and silica. V. Relation between the composition and the activity for the polymerization of ethylene. Nippon Kagaku Zasshi 1956, 77, 888–892. [Google Scholar] [CrossRef]
- Kimura, K.; Ai, H.; Ozaki, A. Tracer study of ethylene dimerization over nickel oxide-silica catalyst. J. Catal. 1970, 18, 271–280. [Google Scholar]
- Sohn, J.R.; Ozaki, A. Acidity of nickel silicate and its bearing on the catalytic activity for ethylene dimerization and butene isomerization. J. Catal. 1980, 61, 29–38. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Kevan, L. Electron spin resonance studies of ethylene dimerization catalyzed by nickel species on Y zeolites. J. Phys. Chem. 1990, 94, 3117–3121. [Google Scholar] [CrossRef]
- Zheng, L.; Wang, G.; Bai, X. Interaction of nickel ions with ethylene molecules in ethylene dimerization over Ni-X zeolites. Stud. Surf. Sci. Catal. 1986, 28, 965–972. [Google Scholar] [CrossRef]
- Hertmann, M.; Poppl, A.; Kevan, L. Ethylene Dimerization and Butene Isomerization in Nickel-Containing MCM-41 and AlMCM-41 Mesoporous Molecular Sieves: An Electron Spin Resonance and Gas Chromatography Study. J. Phys. Chem. 1996, 100, 9906–9910. [Google Scholar] [CrossRef]
- Iwamoto, M.; Tanaka, Y.; Sawamura, N.; Namba, S. Remarkable Effect of Pore Size on the Catalytic Activity of Mesoporous Silica for the Acetalization of Cyclohexanone with Methanol. J. Am. Chem. Soc. 2003, 125, 13032–13033. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sawamura, N.; Iwamoto, M. Highly effective acetalization of aldehydes and ketones with methanol on siliceous mesoporous material. Tetrahedron Lett. 1998, 39, 9457–9460. [Google Scholar] [CrossRef]
- Ishitani, H.; Iwamoto, M. Selective aldol reactions of acetals on mesoporous silica catalyst. Tetrahedron Lett. 2003, 44, 299–301. [Google Scholar] [CrossRef]
- Murata, H.; Ishitani, H.; Iwamoto, M. Synthesis of Biginelli dihydropyrimidinone derivatives with various substituents on aluminium-planted mesoporous silica catalyst. Org. Biomol. Chem. 2010, 8, 1202–1211. [Google Scholar] [CrossRef]
- Murata, H.; Ishitani, H.; Iwamoto, M. Highly ordered aluminum-planted mesoporous silica as active catalyst for Biginelli reaction and formyl C-H insertion reaction with diazo ester. Phys. Chem. Chem. Phys. 2010, 12, 14452–14455. [Google Scholar]
- Trong, D.; Joshi, P.N.; Lemay, G.; Kaliaguine, S. Acidity and structural state of boron in mesoporous boron silicate MCM-41. Stud. Surf. Sci. Catal. 1995, 97, 543–549. [Google Scholar] [CrossRef]
- Yamamoto, T.; Tanaka, T.; Funabiki, T.; Yoshida, S. Acidic Property of FSM-16. J. Phys. Chem. B 1998, 102, 5830–5839. [Google Scholar] [CrossRef]
- Ito, A.; Kodama, T.; Maeda, S.; Masaki, Y. Selective acceleration for deprotection of benzyl ethers with Ti-HMS. Tetrahedron Lett. 1998, 39, 9461–9464. [Google Scholar] [CrossRef]
- Phillips, C.B.; Datta, R. Production of Ethylene from Hydrous Ethanol on H-ZSM-5 under Mild Conditions. Ind. Eng. Chem. Res. 1997, 36, 4466–4475. [Google Scholar] [CrossRef]
- Brando, P.; Philippou, A.; Rocha, J.; Anderson, M.W. Dehydration of Alcohols by Microporous Niobium Silicate AM-11. Catal. Lett. 2002, 80, 99–102. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Gayubo, A.G.; Atutxa, A.; Olazar, M.; Bilbao, J. Catalyst Deactivation by Coke in the Transformation of Aqueous Ethanol into Hydrocarbons. Kinetic Modeling and Acidity Deterioration of the Catalyst. Ind. Eng. Chem. Res. 2002, 41, 4216–4224. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Gayubo, A.G.; Tarro, A.M.; Atutxa, A.; Bilbao, J. Study of operating variables in the transformation of aqueous ethanol into hydrocarbons on an HZSM-5 zeolite. J. Chem. Technol. Biotechnol. 2002, 77, 211–216. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Alonso, A.; Valle, B.; Aguayo, A.T.; Bilbao, J. Kinetic Model for the Transformation of Bioethanol into Olefins over a HZSM-5 Zeolite Treated with Alkali. Ind. Eng. Chem. Res. 2010, 49, 10836–10844. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Alonso, A.; Valle, B.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Kinetic modelling for the transformation of bioethanol into olefins on a hydrothermally stable Ni-HZSM-5 catalyst considering the deactivation by coke. Chem. Eng. J. 2011, 167, 262–277. [Google Scholar] [CrossRef]
- Takahara, I.; Saito, M.; Inaba, M.; Murata, K. Dehydration of Ethanol into Ethylene over Solid Acid Catalysts. Catal. Lett. 2005, 105, 249–252. [Google Scholar] [CrossRef]
- Takahara, I.; Saito, M.; Matsuhasi, H.; Inaba, M.; Murata, K. Increase in the number of acid sites of a H-ZSM 5 zeolite during the dehydration of ethanol. Catal. Lett. 2007, 113, 82–85. [Google Scholar] [CrossRef]
- Inaba, M.; Murata, K.; Saito, M.; Takahara, I. Production of olefins from ethanol by Fe-supported zeolite catalysts. Green Chem. 2007, 9, 638–646. [Google Scholar] [CrossRef]
- Song, Z.; Takahashi, A.; Mimura, N.; Fujitani, T. Production of Propylene from Ethanol Over ZSM-5 Zeolites. Catal. Lett. 2009, 131, 364–369. [Google Scholar] [CrossRef]
- Song, Z.; Takahashi, A.; Nakamura, I.; Fujitani, T. Phosphorus-modified ZSM-5 for conversion of ethanol to propylene. Appl. Catal. A Gen. 2010, 384, 201–205. [Google Scholar] [CrossRef]
- Arias, D.; Colmenares, A.; Cubeiro, M.L.; Goldwasser, J.; Lopez, C.M.; Machado, F.J.; Sazo, V. The transformation of ethanol over AlPO4 and SAPO molecular sieves with AEL and AFI topology. Kinetic and thermodynamic approach. Catal. Lett. 1997, 45, 51–58. [Google Scholar] [CrossRef]
- Golay, S.; Doepper, R.; Renken, A. Reactor performance enhancement under periodic operation for the ethanol dehydration over γ-alumina, a reaction with a stop-effect. Chem. Eng. Sci. 1999, 54, 4469–4474. [Google Scholar] [CrossRef]
- Bakoyannakis, D.N.; Zamboulis, D.; Stalidis, G.A.; Deliyanni, E.A. The effect of preparation method on the catalytic activity of amorphous aluminas in ethanol dehydration. J. Chem. Technol. Biotechnol. 2001, 76, 1159–1164. [Google Scholar] [CrossRef]
- Doheim, M.M.; El-Shobaky, H.G. Catalytic conversion of ethanol and iso-propanol over ZnO-treated Co3O4/Al2O3 solids. Colloids Surf. A 2002, 204, 169–174. [Google Scholar] [CrossRef]
- Zaki, T. Catalytic dehydration of ethanol using transition metal oxide catalysts. J. Colloid Interface Sci. 2005, 284, 606–613. [Google Scholar] [CrossRef]
- Varisli, D.; Dogu, T.; Dogu, G. Ethylene and diethyl-ether production by dehydration reaction of ethanol over different heteropolyacid catalysts. Chem. Eng. Sci. 2007, 62, 5349–5352. [Google Scholar] [CrossRef]
- Varisli, D.; Dogu, T.; Dogu, G. Silicotungstic Acid Impregnated MCM-41-like Mesoporous Solid Acid Catalysts for Dehydration of Ethanol. Ind. Eng. Chem. Res. 2008, 47, 4071–4076. [Google Scholar] [CrossRef]
- Varisli, D.; Dogu, T.; Dogu, G. Novel Mesoporous Nanocomposite WOx-Silicate Acidic Catalysts: Ethylene and Diethylether from Ethanol. Ind. Eng. Chem. Res. 2009, 48, 9394–9401. [Google Scholar] [CrossRef]
- Carrasco-Marn, F.; Mueden, A.; Moreno-Castilla, C. Surface-Treated Activated Carbons as Catalysts for the Dehydration and Dehydrogenation Reactions of Ethanol. J. Phys. Chem. B 1998, 102, 9239–9244. [Google Scholar] [CrossRef]
- Kamiguchi, S.; Chihara, T. Catalytic Dehydration of Alcohol to Olefin and Ether by Halide Clusters of Nb, Mo, Ta and W Possessing an Octahedral Metal Core. Catal. Lett. 2003, 85, 97–100. [Google Scholar] [CrossRef]
- Kamiguchi, S.; Nagashima, S.; Komori, K.; Kodomari, M.; Chihara, T. Thermal Activation of Molecular Tungsten Halide Clusters with the Retention of an Octahedral Metal Framework and the Catalytic Dehydration of Alcohols to Olefins as a Solid Acid Catalyst. J. Cluster Sci. 2007, 18, 414–430. [Google Scholar] [CrossRef]
- Kamimura, Y.; Sato, S.; Takahashi, R.; Sodesawa, T.; Akashi, T. Synthesis of 3-pentanone from 1-propanol over CeO2-Fe2O3 catalysts. Appl. Catal. A Gen. 2003, 252, 399–410. [Google Scholar] [CrossRef]
- Nagashima, O.; Sato, S.; Takahashi, R.; Sodesawa, T. Ketonization of carboxylic acids over CeO2-based composite oxides. J. Mol. Catal. A Chem. 2005, 227, 231–239. [Google Scholar] [CrossRef]
- Tsuchida, T.; Kubo, J.; Yoshioka, T.; Sakuma, S.; Takeguchi, T.; Ueda, W. Reaction of ethanol over hydroxyapatite affected by Ca/P ratio of catalyst. J. Catal. 2008, 259, 183–189. [Google Scholar]
- Iwamoto, M.; Kosugi, Y. Highly Selective Conversion of Ethene to Propene and Butenes on Nickel Ion-Loaded Mesoporous Silica Catalysts. J. Phys. Chem. C 2007, 111, 13–15. [Google Scholar] [CrossRef]
- Ikeda, K.; Kawamura, Y.; Yamamoto, T.; Iwamoto, M. Effectiveness of the template-ion exchange method for appearance of catalytic activity of Ni-MCM-41 for the ethene to propene reaction. Catal. Commun. 2008, 9, 106–110. [Google Scholar] [CrossRef]
- Kasai, K.; Haishi, T.; Iwamoto, M. Selective conversion of bio-ethanol to lower olefins on nickel ion-loaded mesoporous silica catalysts. Shokubai 2007, 49, 126–128. [Google Scholar]
- Iwamoto, M.; Kasai, K. Preparation of olefins from alcohols by use of ordered mesoporous catalysts in high yield. Jpn. Tokkyo Koho 2008. JP 2008255104 A 20081023.
- Haishi, T.; Kasai, K.; Iwamoto, M. Fast and Quantitative Dehydration of Lower Alcohols to Corresponding Olefins on Mesoporous Silica Catalyst. Chem. Lett. 2011, 40, 614–616. [Google Scholar] [CrossRef]
- Iwamoto, M.; Kasai, K.; Haishi, T. Conversion of Ethanol into Polyolefin Building Blocks: Reaction Pathways on Nickel Ion-loaded Mesoporous Silica. ChemSusChem 2011, 4, 1055–1058. [Google Scholar] [CrossRef]
- Sugiyama, S.; Kato, Y.; Wada, T.; Ogawa, S.; Nakagawa, K.; Sotowa, K. Ethanol Conversion on MCM-41 and FSM-16, and on Ni-Doped MCM-41 and FSM-16 Prepared without Hydrothermal Conditions. Top. Catal. 2010, 53, 550–554. [Google Scholar] [CrossRef]
- Liu, B.; Nakatani, H.; Terano, M. Mechanistic implications of the unprecedented transformations of ethene into propene and butene over Phillips CrOx/SiO2 catalyst during induction period. J. Mol. Catal. A 2003, 201, 189–197. [Google Scholar] [CrossRef]
- Negishi, E.; Takahashi, T. Patterns of Stoichiometric and Catalytic Reactions of Organozirconium and Related Complexes of Synthetic Interest. Acc. Chem. Res. 1994, 27, 124–130. [Google Scholar] [CrossRef]
- Grubbs, R.H., Ed. Handbook of Metathesis; Wiley-VCH Verlag BmbH & Co.: Weinheim, Germany, 2003. [Google Scholar]
- Grubbs, R.H. Olefin metathesis. Tetrahedron 2004, 60, 7117–7140. [Google Scholar] [CrossRef]
- Sato, Y.; Saito, N.; Mori, M. Asymmetric Cyclization of ω-Formyl-1,3-dienes Catalyzed by a Zerovalent Nickel Complex in the Presence of Silanes. J. Org. Chem. 2002, 67, 9310–9317. [Google Scholar] [CrossRef]
- Baker, M.V.; Brown, D.H.; Skelton, B.W.; White, A.H. An investigation into alkenyl-functionalized 1,4,7-triazacyclononanes: Synthesis, metal complexation, and attempted olefin metathesis. Aust. J. Chem. 2002, 55, 655–600. [Google Scholar] [CrossRef]
- Xiao, S.; Meng, Z. X-ray photoelectron spectroscopy characterization of the reduction and oxidation behavior of Ni-containing HZSM-5 zeolites. J. Chem. Soc. Faraday Trans. 1994, 90, 2591–2595. [Google Scholar] [CrossRef]
- Schoonheydt, R.A.; Roodhooft, D. Spectroscopy of the thermal reduction of nickel(II) in the presence of hydrogen in zeolites X and Y. J. Phys. Chem. 1986, 90, 6319–6323. [Google Scholar] [CrossRef]
- Elev, I.V.; Shelimov, B.N.; Kazanskii, V.B. The role of nickel(1+) ions in the activity of NiCaY zeolite catalysts for ethylene dimerization. J. Catal. 1984, 89, 470–477. [Google Scholar] [CrossRef]
- Prakash, A.M.; Wasowicz, T.; Kevan, L. Reducibility, Location, and Adsorbate Interactions of Ni(I) Ions in Ni(II)-Exchanged Silicoaluminophosphate Type 41 Studied by Electron Spin Resonance and Electron Spin Echo Modulation Spectroscopies. J. Phys. Chem. 1996, 100, 15947–15953. [Google Scholar] [CrossRef]
- Hartmann, M.; Poppl, A.; Kevan, L. Formation and Stability of Ni(I) Ions in MCM-41 Mesoporous Molecular Sieves. J. Phys. Chem. 1995, 99, 17494–17496. [Google Scholar] [CrossRef]
- Taoufik, M.; Le Roux, E.; Thivolle-Cazat, J.; Basset, J.M. Direct transformation of ethylene into propylene catalyzed by a tungsten hydride supported on alumina: trifunctional single-site catalysis. Angew. Chem. Int. Ed. 2007, 46, 7202–7205. [Google Scholar] [CrossRef]
- Lin, B.; Zhang, Q.; Wang, Y. Catalytic Conversion of Ethylene to Propylene and Butenes over H-ZSM-5. Ind. Eng. Chem. Res. 2009, 48, 10788–10795. [Google Scholar] [CrossRef]
- Tsuji, K.; Uchida, H.; Nakajou, T.; Sano, K. Development of Novel Production Processes for Ethyl Acetate and Acetic Acid Catalyzed by Solid Heteropolyacids. Chem. Chem. Ind. 2008, 61, 206–208. [Google Scholar]
- Clause, O.; Kermarec, M.; Bonneviot, L.; Villain, F.; Che, M. Nickel(II) ion-support interactions as a function of preparation method of silica-supported nickel materials. J. Am. Chem. Soc. 1992, 114, 4709–4717. [Google Scholar] [CrossRef]
- Carriat, J.Y.; Che, M.; Kermarec, M.; Verdaguer, M.; Michalowicz, A. Control of Dispersion of Ni2+ Ions via Chelate Ligands in the Preparation of Ni/SiO2 Materials. A XAFS Study. J. Am. Chem. Soc. 1998, 120, 2059–2070. [Google Scholar]
- Yang, Y.; Lim, S.; Du, G.; Chen, Y.; Ciuparu, D.; Haller, G.L. Synthesis and Characterization of Highly Ordered Ni-MCM-41 Mesoporous Molecular Sieves. J. Phys. Chem. B 2005, 109, 13237–13246. [Google Scholar]
- Martin, G.A.; Renoupre, A.; Dalmaiim, G.; Imelik, B. Synthesis of nickel talc and antigorite. Study of their thermal decomposition and reduction to obtain nickel catalysts on silica. J. Chim. Phys. 1970, 67, 1149–1160. [Google Scholar]
- Wells, A.F. Structural Inorganic Chemistry; Clarendon Press: Oxford, UK, 1984. [Google Scholar]
- Farges, F.; Munoz, M.; Siewert, R.; Malavergne, V.; Brown, G.E.; Behrens, H.; Nowak, M.; Petit, P.E. Transition elements in water-bearing silicate glasses/melts. Part II. Ni in water-bearing glasses. Geochim. Cosmochim. Acta 2001, 65, 1679–1693. [Google Scholar] [CrossRef]
- Burattin, P.; Che, M.; Louis, C. Characterization of the Ni(II) Phase Formed on Silica Upon Deposition-Precipitation. J. Phys. Chem. B 1997, 101, 7060–7074. [Google Scholar] [CrossRef]
- Yang, J.C.; Shul, Y.G.; Louis, C.; Che, M. In situ EXAFS study of the nucleation and crystal growth of Ni particles on SiO2 support. Catal. Today 1998, 44, 315–325. [Google Scholar] [CrossRef]
- Bonneviot, L.; Clause, O.; Che, M.; Manceau, A.; Decarreau, A.; Villian, F.; Bazin, D.; Dexpert, H. Investigation by EXAFS of the effect of pH on the structure of nickel(2+) ions impregnated on silica. Phys. B 1989, 158, 43–44. [Google Scholar] [CrossRef]
- Clause, O.; Bonneviot, L.; Che, M.; Dexpert, H. EXAFS characterization of the adsorbed state of nickel(II) ions in nickel/silica materials prepared by deposition-precipitation. J. Catal. 1991, 130, 21–28. [Google Scholar]
- Espinos, J.P.; Gonzalez-Elipe, A.R.; Munuera, G.; Garcia, J.; Conesa, J.C.; Burattini, E. EXAFS study of catalyst preparation procedure in nickel-silica and nickel-titania. Phys. B 1989, 158, 174–175. [Google Scholar] [CrossRef]
- Grauby, O.; Petit, S.; Decarreau, A.; Baronenet, A. The beidellite-saponite series: An experimental approach. Eur. J. Mineral. 1993, 5, 623–635. [Google Scholar]
- Decarreau, A. Partitioning of divalent transition elements between octahedral sheets of trioctahedral smectites and water. Geochim. Cosmochim. Acta 1985, 49, 1537–1544. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Mihaylov, M.; Klissurski, D.; Stefanov, P.; Abadjieva, N.; Vassileva, E.; Mintchev, L. Characterization of Ni/SiO2 Catalysts Prepared by Successive Deposition and Reduction of Ni2+ Ions. J. Catal. 1999, 185, 314–323. [Google Scholar] [CrossRef]
- Wojcieszak, R.; Moteverdi, S.; Mercy, M.; Nowak, I.; Ziolek, M.; Bettahar, M.M. Nickel containing MCM-41 and AlMCM-41 mesoporous molecular sieves. Characteristics and activity in the hydrogenation of benzene. Appl. Catal. A Gen. 2004, 268, 241–253. [Google Scholar] [CrossRef]
- Kutseva, L.N. Influence of superstoichiometric oxygen on the catalytic, adsorptive, and electrical properties of nickelous oxide. Dokl. Akad. Nauk. SSSR 1961, 138, 409–411. [Google Scholar]
- Houalla, M.; Delannay, F.; Matsuura, I.; Delmon, B. Physico-chemical characterization of impregnated and ion-exchanged silica-supported nickel oxide. J. Chem. Soc. Faraday. Trans. 1 1980, 76, 2128–2141. [Google Scholar]
- Kermarec, M.; Carriat, J.Y.; Burattin, P.; Che, M.; Decarreau, A. FTIR Identification of the Supported Phases Produced in the Preparation of Silica-Supported Nickel Catalysts. J. Phys. Chem. 1994, 98, 12007–12017. [Google Scholar]
- Lehmann, T.; Wolff, T.; Zahn, V.M.; Veit, P.; Hamel, C.; Seidel-Morgenstern, A. Preparation of Ni-MCM-41 by equilibrium adsorption—Catalytic evaluation for the direct conversion of ethene to propene. Catal. Commun. 2011, 12, 368–374. [Google Scholar] [CrossRef]
- Xu, S.; Wang, X. Highly active and coking resistant Ni/CeO2-ZrO2 catalyst for partial oxidation of methane. Fuel 2005, 84, 563. [Google Scholar] [CrossRef]
- Li, G.; Hu, L.; Hill, J.M. Comparison of reducibility and stability of alumina-supported Ni catalysts prepared by impregnation and co-precipitation. Appl. Catal. A 2006, 301, 16–24. [Google Scholar] [CrossRef]
- Abe, T.; Tachibana, Y.; Uematsu, T.; Iwamoto, M. Preparation and characterization of Fe2O3 nanoparticles in mesoporous silicate. J. Chem. Soc. Chem. Commun. 1995, 1617–1618. [Google Scholar]
- Iwamoto, M.; Tanaka, Y. Preparation of metal ion-planted mesoporous silica by template ion-exchange method and its catalytic activity for asymmetric oxidation of sulfide. Catal. Surv. Jpn. 2001, 5, 25–36. [Google Scholar] [CrossRef]
- Hayashi, F.; Iwamoto, M. Effect of pore structure on the nitridation of mesoporous silica with ammonia. Eur. J. Inorg. Chem. 2010, 15, 2235–2243. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iwamoto, M. One Step Formation of Propene from Ethene or Ethanol through Metathesis on Nickel Ion-loaded Silica. Molecules 2011, 16, 7844-7863. https://doi.org/10.3390/molecules16097844
Iwamoto M. One Step Formation of Propene from Ethene or Ethanol through Metathesis on Nickel Ion-loaded Silica. Molecules. 2011; 16(9):7844-7863. https://doi.org/10.3390/molecules16097844
Chicago/Turabian StyleIwamoto, Masakazu. 2011. "One Step Formation of Propene from Ethene or Ethanol through Metathesis on Nickel Ion-loaded Silica" Molecules 16, no. 9: 7844-7863. https://doi.org/10.3390/molecules16097844