The Metal Cation Chelating Capacity of Astaxanthin. Does This Have Any Influence on Antiradical Activity?

Abstract
:1. Introduction
2. Results and Discussion



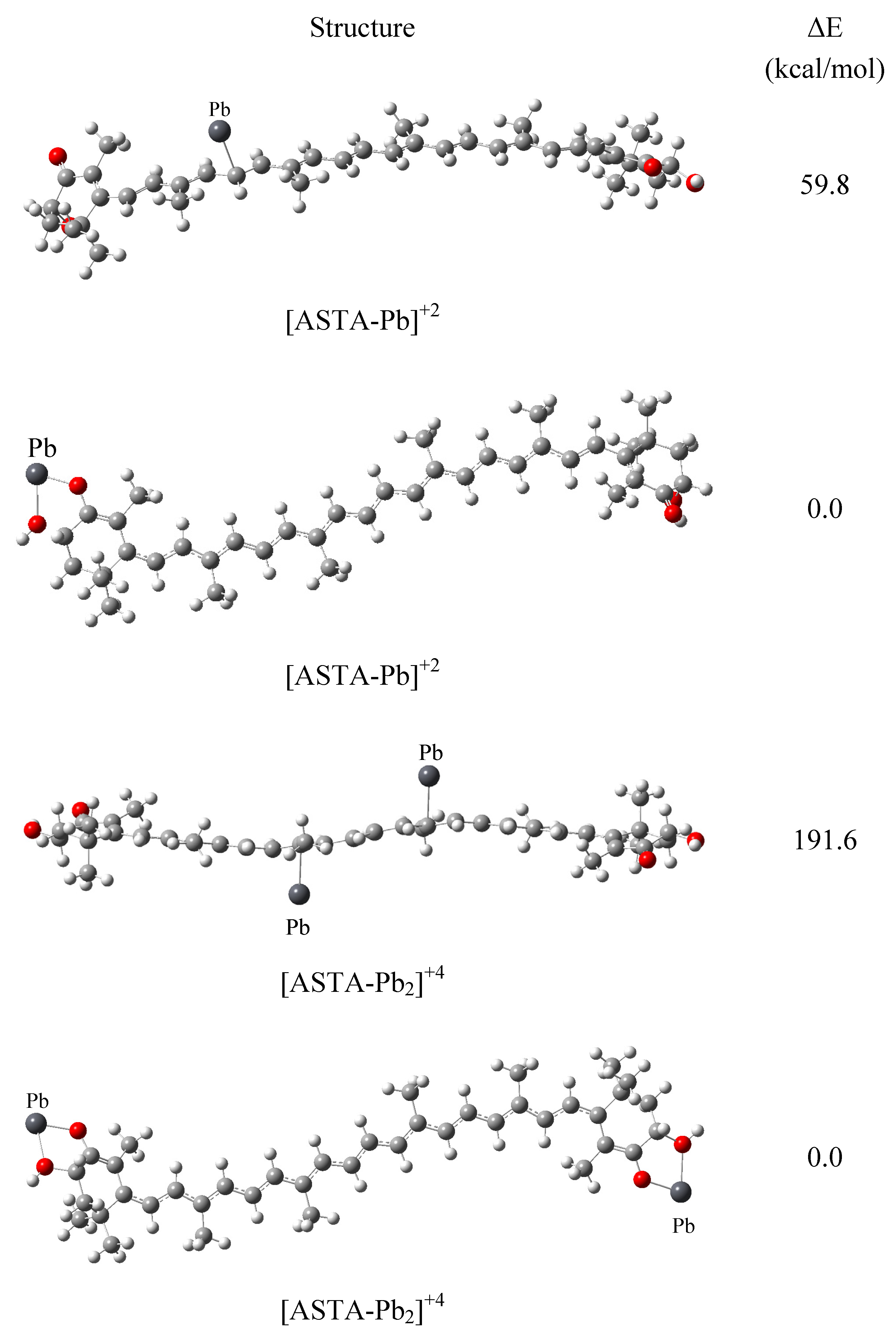{kind=link}
{kind=link}
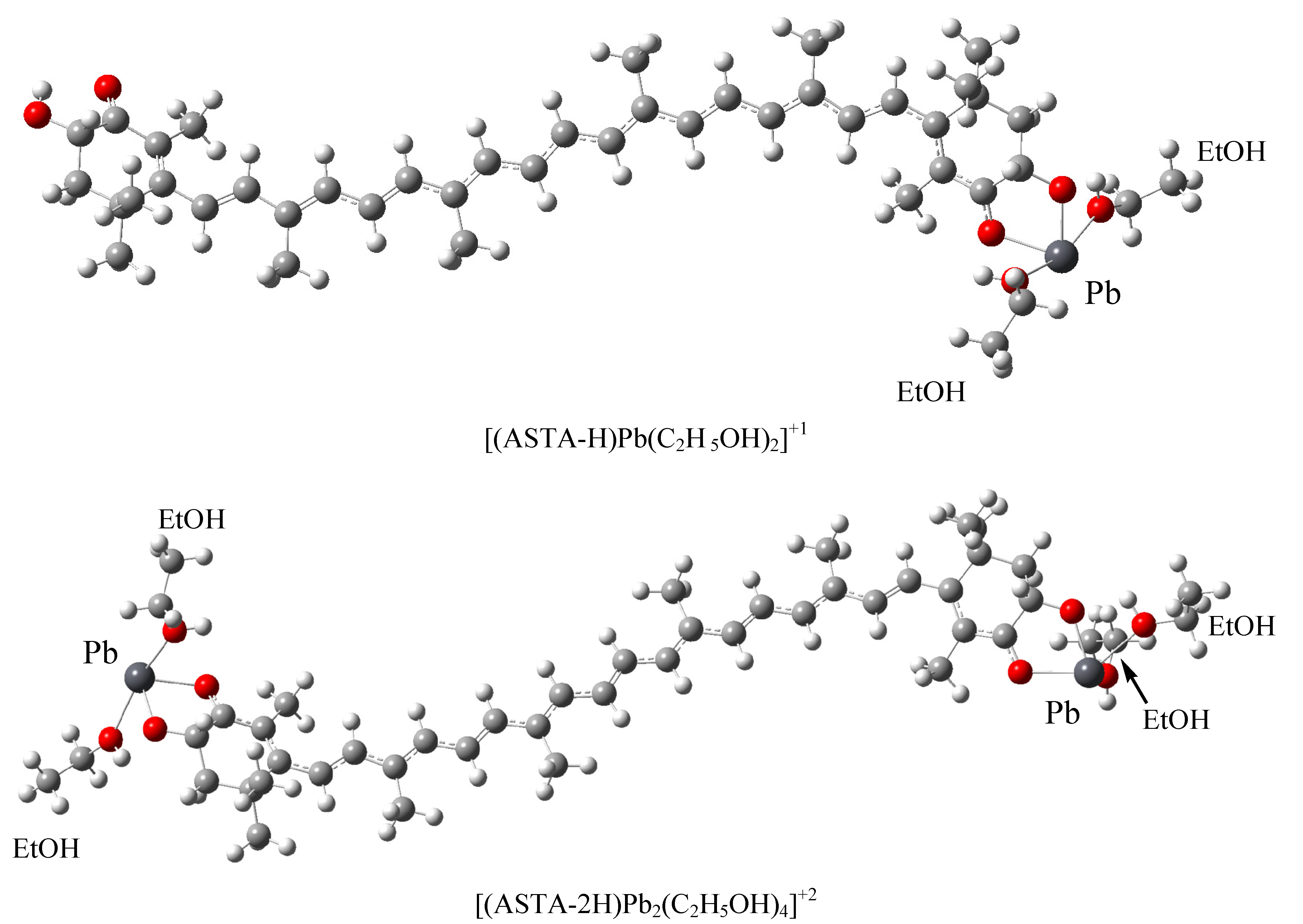{kind=link}
| Chemical Formula | λmax (f)(gas) | EE(gas) | λmax (f)(ethanol) | EE(ethanol) | Exp λmax(ethanol) [42] | Exp EE (ethanol) [42] |
|---|---|---|---|---|---|---|
| ASTA | 447 (4) | 2.77 | 471 (4) | 2.63 | 480 | 2.58 |
| [ASTA-Ca]+2 | 651 (4) | 1.90 | 712 (4) | 1.74 | 492 * | 2.52 |
| [ASTA-Ca2]+4 | 603 (5) | 2.06 | 516 (4) | 2.40 | ||
| [ASTA-Zn(H 2O)2]+2 | 642 (4) | 1.93 | 680 (4) | 1.82 | 492 * | 2.52 |
| [ASTA-Zn2(H 2O)4]+4 | 615 (5) | 2.02 | 551 (5) | 2.25 | ||
| [ASTA-Pb]+2 | 605 (4) | 2.05 | 634 (4) | 1.96 | ||
| [ASTA-Pb2]+4 | 673 (5) | 1.84 | 680 (5) | 1.82 | ||
| [ASTA-Cu(H 2O)2]+2 | 817 (4) | 1.52 | 910 (4) | 1.36 | ||
| [ASTA-Cu2(H 2O)4]+4 | 564 (4) | 2.20 | 587 (4) | 2.11 | ||
| [ASTA-Cd(H 2O)2]+2 | 671 (4) | 1.85 | 737 (4) | 1.68 | ||
| [ASTA-Cd2(H 2O)4]+4 | 595 (5) | 2.08 | 515 (5) | 2.41 | ||
| [ASTA-Hg(H 2O)2]+2 | 642 (4) | 1.93 | 682 (4) | 1.82 | ||
| [ASTA-Hg2(H 2O)4]+4 | 601 (5) | 2.06 | 516 (5) | 2.40 |

| Chemical formula | λmax (f)(ethanol) | EE(ethanol) | Exp λmax(ethanol) [42] | Exp EE |
|---|---|---|---|---|
| ASTA | 471 (4) | 2.63 | 480 | 2.58 |
| [(ASTA-H) Ca (C2H 5OH)2]+1 | 514 (4) | 2.41 | 492 * | 2.52 |
| [(ASTA-2H) Ca2 (C2H5OH)4 ]+2 | 500 (5) | 2.48 | ||
| [(ASTA-H) Zn (C2H 5OH)2]+1 | 547 (4) | 2.27 | 492 * | 2.52 |
| [(ASTA-2H) Zn2 (C2H5OH)4]+2 | 531 (5) | 2.33 | ||
| [(ASTA-H) Pb (C2H 5OH)2]+1 | 562 (4) | 2.09 | ||
| [(ASTA-2H) Pb2 (C2H5OH)4]+2 | 548(5) | 2.26 | ||
| [(ASTA-H) Cu (C2H 5OH)2]+1 | 583 (2) | 2.13 | ||
| [(ASTA-2H) Cu2 (C2H5OH)4]+2 | 851 (3) | 1.46 | ||
| [(ASTA-H) Cd (C2H 5OH)2]+1 | 530 (4) | 2.34 | ||
| [(ASTA-2H) Cd2 (C2H5OH)4]+2 | 513 (5) | 2.42 | ||
| [(ASTA-H) Hg (C2H 5OH)2]+1 | 528 (4) | 2.35 | ||
| [(ASTA-2H) Hg2 (C2H5OH)4]+2 | 509 (5) | 2.44 |
| (ASTA-H)−1 + M+2 + 2 C2H 5OH → [(ASTA-H)M(C2H 5OH)2]+1 | (ASTA-H)−2 + 2M+2 + 4 C2H 5OH→[(ASTA-2H)M2(C2H5OH)4]+2 | ||
|---|---|---|---|
| Ca2 + | −73.2 | Ca2 + | −139.0 |
| Pb2 + | −166.2 | Pb2 + | −336.2 |
| Cu2 + | −239.9 | Cu2 + | −404.9 |
| Zn2 + | −112.4 | Zn2 + | −223.0 |
| Cd2 + | −74.3 | Cd2 + | −144.7 |
| Hg2 + | −47.5 | Hg2 + | −93.8 |
| Compound | VIE (eV) | VEA (eV) |
|---|---|---|
| ASTA | 4.96 | 3.00 |
| [(ASTA-H)Ca(C2H 5OH)2]+1 | 4.87 | 3.70 |
| [(ASTA-2H)Ca2(C2H5OH)4]+2 | 4.96 | 3.20 |
| [(ASTA-H)Zn(C2H 5OH)2]+1 | 4.86 | 4.06 |
| [(ASTA-2H)Zn2(C2H5OH)4]+2 | 4.97 | 3.36 |
3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgements
References and Notes
- Burton, G.W.; Ingold, K.U. beta-Carotene: An unusual type of lipid antioxidant. Science 1984, 224, 569–573. [Google Scholar]
- Edge, R.; McGarvey, D.J.; Truscott, T.G. The carotenoids as anti-oxidants—A review. J. Photochem. Photobio. B Biol. 1997, 41, 189–200. [Google Scholar] [CrossRef]
- Edge, R.; Land, E.J.; McGarvey, D.J.; Mulroy, L.; Truscott, T.G. Relative one-electron reduction potentials of carotenoid radical cations and the interactions of carotenoids with the vitamin E radical cation. J. Am. Chem. Soc. 1998, 120, 4087–4090. [Google Scholar] [CrossRef]
- Galano, A.; Vargas, R.; Martínez, A. Carotenoids can act as antioxidants by oxidizing the superoxide radical anion. Phys. Chem. Chem. Phys. 2010, 12, 193–200. [Google Scholar]
- Øpstad, C.L.; Sliwka, H.R.; Partali, V. Facile electron uptake by carotenoids under mild, non-radiative conditions: Formation of carotenoid anions. Eur. J. Org. Chem. 2010, 24, 4637–4641. [Google Scholar]
- Martin, H.D.; Jäger, C.; Ruck, C.; Schmidt, M.; Walsh, R.; Paust, J. Anti- and prooxidant properties of carotenoids. J. Prakt. Chem. 1999, 341, 302–308. [Google Scholar] [CrossRef]
- Mairanosvky, V.G.; Engovatov, A.A.; Ioffe, N.T.; Samokhvalov, G.I. Electron-donor and electron-acceptor properties of carotenoids: Electrochemical study of carotenes. J. Electroanal. Chem. 1975, 66, 123–137. [Google Scholar] [CrossRef]
- Polyakov, N.E.; Leshina, T.V.; Konovalova, T.A.; Kispert, L.D. Carotenoids as scavengers of free radicals in a Fenton reaction: Antioxidants or pro-oxidants? Free Radic. Biol. Med. 2001, 31, 398–404. [Google Scholar] [CrossRef]
- Polyakov, N.E.; Kruppa, A.I.; Leshina, T.V.; Konovalova, T.A.; Kispert, L.D. Carotenoids as antioxidants: Spin trapping EPR and optical study. Free Rad. Biol. Med. 2001, 31, 43–52. [Google Scholar] [CrossRef]
- Mortensen, A.; Skibsted, L.H.; Sampson, J.; Rice-Evans, A.C.; Everett, S.A. Comparative mechanisms and rates of free radical scavenging by carotenoid antioxidants. FEBS Lett. 1997, 418, 91–97. [Google Scholar] [CrossRef]
- Jeevarajan, A.S.; Khaleb, M.; Kispert, L.D. Simultaneous Electrochemical and Electron Paramagnetic Resonance Studies of Carotenoids: Effect of Electron Donating and Accepting Substituents. J. Phys. Chem. 1994, 98, 7777–7781. [Google Scholar] [CrossRef]
- Halliwell, B. Vitamin C: Antioxidant or pro-oxidant in vivo. Free Radic. Res. 1996, 25, 439–454. [Google Scholar] [CrossRef]
- Prior, R.L.; Cao, G. In vivo total antioxidant capacity: Comparison of different analytical methods. Free Radic. Biol. Med. 1999, 27, 1173–1181. [Google Scholar] [CrossRef]
- Mortensen, A.; Skibsted, L.H. Importance of carotenoid structure in radical scavenging reactions. J. Agric. Food Chem. 1997, 45, 2970–2977. [Google Scholar] [CrossRef]
- Woodall, A.A.; Lee, W.M.S.; Weesie, R.J.; Jackson, M.J.; Britton, G. Oxidation of carotenoids by free radicals: Relationship between structure and reactivity. Biochim. et Biophys. Acta 1997, 1336, 33–42. [Google Scholar] [CrossRef]
- Woodall, A.A.; Britton, G.; Jackson, M.J. Carotenoids and protection of phospholipids in solution or in liposomes against oxidation by peroxyl radicals: Relationship between carotenoid structure and protective ability. Biochim. Biophys. Acta 1997, 1336, 575–586. [Google Scholar] [CrossRef]
- Guo, J.D.; Luo, Y.; Himo, F. Density functional theory study of the canthaxanthin and other carotenoid radical cations. Chem. Phys. Lett. 2002, 366, 73–81. [Google Scholar] [CrossRef]
- Dreuw, A. Influence of Geometry Relaxation on the Energies of the S1 and S2 States of Violaxanthin, Zeaxanthin and Lutein. J. Phys. Chem. A 2006, 110, 4592–4599. [Google Scholar] [CrossRef]
- Naguib, Y.M.A. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef]
- Böhm, F.; Edge, R.; Land, E.J.; McGarvey, D.J.; Truscott, T.G. Carotenoids enhance vitamin E antioxidant efficiency. J. Am. Chem. Soc. 1997, 119, 621–622. [Google Scholar]
- Krinsky, N.I. Carotenoids as antioxidants. Nutrition 2001, 10, 815–817. [Google Scholar] [CrossRef]
- El-Tinay, A.H.; Chichester, C.O. Oxidation of beta-carotene. Site of initial attack. J. Org. Chem. 1970, 35, 2290–2293. [Google Scholar] [CrossRef]
- Garavelli, M.; Bernardi, F.; Olivucci, M.; Robb, M.A. DFT Study of the Reactions between Singlet-Oxygen and a Carotenoid Model. J. Am. Chem. Soc. 1998, 120, 10210–10222. [Google Scholar] [CrossRef]
- Gao, Y.; Focsan, A.L.; Kispert, L.D.; Dixon, A.D. Density functional theory study of the β-carotene radical cation and deprotonated radicals. J. Phys. Chem. B 2006, 110, 24750–24756. [Google Scholar] [CrossRef]
- Himo, F. Density functional theory study of the beta-carotene radical cation. J. Phys. Chem. A 2001, 105, 7933–7937. [Google Scholar] [CrossRef]
- Hashimoto, H.; Yoda, T.; Kobayashi, T.; Young, A.J. Molecular structures of carotenoids as predicted by MNDO-AM1 molecular orbital calculations. J. Mol. Struct. 2002, 604, 125–146. [Google Scholar] [CrossRef]
- Mathews-Roth, M.M.; Krinsky, N.I. Carotenoids affect development of UV-B-induced skin cancer. Photochem. Photobiol. 1987, 46, 507–509. [Google Scholar]
- Rice-Evans, C.A.; Sampson, J.; Bramley, P.M.; Holloway, D.E. Why do we expect carotenoids to be antioxidants in vivo? Free Radic. Res. 1997, 26, 381–398. [Google Scholar]
- Hill, G.E. Plumage coloration is a sexually selected indicator of male quality. Nature 1991, 350, 337–339. [Google Scholar] [CrossRef]
- McGraw, K.J. Antioxidant function of many animal pigments: Consistent health benefits of sexually selected colorants? Anim. Behav. 2005, 69, 757–764. [Google Scholar] [CrossRef]
- Hill, G.E.; McGraw, K.J. Mechanisms and measurements. In Bird Coloration; Harvard University Press: Cambdrige, MA, USA, 2006; Volume 1. [Google Scholar]
- Hill, G.E. The function and evolution of colorful plumage in the house finch. In A Red Bird in a Brown Bag; Oxford University Press: New York, NY, USA, 2002. [Google Scholar]
- Galano, A.; Francisco-Márquez, M. Reactions of OOH radical with β-carotene, lycopene and torulene: Hydrogen atom transfer and adduct formation mechanisms. J. Phys. Chem. B 2009, 113, 11338–11345. [Google Scholar] [CrossRef]
- Mougeot, F.; Pérez-Rodríguez, L.; Martínez-Padilla, J.; Leckie, F.; Redpath, S.M. Parasites, testosterone and honest carotenoid based signalling of health. Func. Ecol. 2007, 21, 886–898. [Google Scholar] [CrossRef]
- Navarro, C.; Pérez-Contreras, T.; Avilés, J.M.; McGraw, K.J.; Soler, J.J. Beak colour reflects circulating carotenoid and vitamin A levels in spotless starlings (Sturnus unicolor). Behav. Ecol. Sociobiol. 2010, 64, 1057–1067. [Google Scholar] [CrossRef]
- Martínez, A.; Rodríguez-Gironés, M.A.; Barbosa, A.; Costas, M. Donator acceptor map for carotenoids, melatonin and vitamins. J. Phys. Chem. A 2008, 112, 9037–9042. [Google Scholar]
- Martínez, A.; Vargas, R.; Galano, A. What is important to prevent oxidative stress? A theoretical study on electron-transfer reactions between carotenoids and free radicals. J. Phys. Chem. B 2009, 113, 12113–12120. [Google Scholar]
- Brown, J.E.; Khodr, H.; Hider, R.C.; Rice-Evans, C.A. Structural dependence of flavonoid interactions with Cu2+ ions: Implications for their antioxidant properties. Biochem. J. 1998, 330, 1173–1178. [Google Scholar]
- Morel, I.; Lescoat, G.; Cillard, P. Role of flavonoids and iron chelation in antioxidant action. Methods Enzymol. 1994, 234, 437–443. [Google Scholar]
- Paganga, G.; Al-Hashim, A.; Khodr, H.; Scott, B.C.; Aruoma, O.I.; Hider, R.C.; Halliwell, B.; Rice-Evans, C.A. Mechanisms of antioxidant activities of quercetin and catechin. Redox Rep. 1996, 3, 359–364. [Google Scholar]
- Gao, Y.; Konovalova, T.A.; Lawrence, J.N.; Smitha, M.A.; Nunley, J.; Schad, R.; Kispert, L.D. Interaction of carotenoids and Cu2+ in Cu-MCM-41: Distance-dependent reversible electron transfer. J. Phys. Chem. B 2003, 107, 2459–2465. [Google Scholar] [CrossRef]
- Polyakov, N.E.; Focsan, A.L.; Bowman, M.K.; Kispert, L.D. Free radical formation in novel carotenoid metal ion complexes of astaxanthin. J. Phys. Chem. B 2010, 114, 16968–16977. [Google Scholar] [CrossRef]
- Hsu, P.C.; Guo, Y.L. Antioxidant nutrients and lead toxicity. Toxicology 2002, 180, 33–44. [Google Scholar] [CrossRef]
- Mateo, A.J.; Green, A.J.; LeFranc, H.; Baos, R.; Figuerola, J. Lead poisoning in wild birds from southern Spain: A comparative study of wetland areas and species affected, and trends over time. Ecotox. Environ. Saf. 2007, 66, 119–126. [Google Scholar] [CrossRef]
- Geyer, B.P.; McP Smith, G. Preparation and properties of some phthiocol inner complexes. J. Am. Chem. Soc. 1941, 63, 3071–3075. [Google Scholar] [CrossRef]
- Salunke-Gawali, S.; Rane, S.Y.; Puranik, V.G.; Guyard-Duhayon, C.; Varret, F. Three dimensional hydrogen-bonding network in a copper complex of 2-hydroxy-1,4-naphthoquinone: Structural, spectroscopic and magnetic properties. Polyhedron 2004, 23, 2541–2551. [Google Scholar] [CrossRef]
- Kataoka, K.; Takagi, T.; Sasaki, Y. Solvation Effect on the complex formation between o-quinones and metal chlorides in nonaqueous media. Bull. Chem. Soc. Jpn. 1982, 55, 1344–1347. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. Extensive TD-DFT investigation of the first electronic transition in substituted azobenzenes. Chem. Phys. Lett. 2008, 465, 226–229. [Google Scholar] [CrossRef]
- Barone, V.; Hod, O.; Perakta, J.E.; Scuseria, G.E. Accurate prediction of the electronic properties of low-dimensional graphene derivatives using a screened hybrid density functional. Acc. Chem. Res. 2011, 44, 269–279. [Google Scholar] [CrossRef]
- Jimenez-Hoyos, C.A.; Janesko, B.G.; Scuseria, G.E. Evaluation of range-separated hybrid and other density functional approaches on test sets relevant for transition metal-based homogeneous catalysts. J. Phys. Chem. A 2009, 113, 11742–11749. [Google Scholar]
- Vydrov, O.A.; Scuseria, G. Assessment ofa long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef]
- Krukav, A.V.; Scuseria, G.; Perdew, J.P.; Savi, A. Hybrid functionals with local range separation. J. Chem. Phys. 2008, 129, 124103. [Google Scholar] [CrossRef]
- Nayyar, I.H.; Batista, E.R.; Tretiak, S.; Saxena, A.; Smith, D.L.; Martin, R.L. Localization of electronic excitations inconjugated polymers studied by DFT. J. Phys. Chem. Lett. 2011, 2, 566–571. [Google Scholar] [CrossRef]
- Sonk, J.A.; Schlegel, H.B. TD-CI simulation of the electronic optical response of molecules in intense fields II: Comparison of DFT functionals and EOM-CCSD. J. Phys. Chem. A 2011, 115, 11832–11840. [Google Scholar] [CrossRef]
- Haunschild, R.; Scuseria, G. Range-separated local hybrids. J. Chem. Phys. 2010, 132, 224106. [Google Scholar] [CrossRef]
- Körzdörfer, T.; Sears, J.S.; Sutton, C.; Brédas, L.C. Long-Range corrected hybrid functionals for π-conjugated systems: Dependence of the range-separation parameter on conjugation length. J. Chem. Phys. 2011, 135, 204107. [Google Scholar]
- Jacquemin, D.; Perpète, E.A.; Ciofini, I.; Adamo, C. On the TD-DFT UV/Vis spectra accuracy: The azoalkanes. Theor. Chem. Acc. 2008, 120, 405–410. [Google Scholar] [CrossRef]
- Autschbach, J. Charge-transfer excitations and time-dependent density functional theory: Problems and some proposed solutions. Chem. Phys. Chem. 2009, 10, 1757–1760. [Google Scholar] [CrossRef]
- Chen, C.-S.; Wu, S.-H.; Wu, Y.-Y.; Fang, J.-M.; Wu, T.-H. Properties of astaxanthin/Ca2+ complex formation in the deceleration of cis/trans isomerization. Org. Lett. 2007, 9, 2985–2988. [Google Scholar] [CrossRef]
- Galano, A. Relative antioxidant efficiency of a large series of carotenoids in terms of one electron transfer reactions. J. Phys. Chem. B 2007, 111, 12898–12908. [Google Scholar]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J.M.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, O.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian 09, Revision A.02. Gaussian, Inc.:: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Mielich, B.; Savin, A.; Stoll, H.; Peuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Modern Theoretical Chemistry (ed) Schaefer III HF; Plenum: New York, NY, USA, 1976; pp. 1–28. [Google Scholar]
- Zhanpeisov, N.U.; Ju, W.S.; Anpo, M. Local structure of highly dispersed lead containing zeolite. An ab initio and density functional theory study. J. Mol. Struct. (THEOCHEM) 2002, 592, 155–160. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, J.; Inagaki, S. Theoretical design of singlet localized sigma-diradicals: C(MH2)3C (M = Si, Ge, Sn, Pb). Tetrahedron Lett. 2005, 46, 5567–5577. [Google Scholar] [CrossRef]
- Kaenkaew, S.; Sae-Khow, O.; Ruangpornvisuti, V. Cation recognition of thiacalix[2]thianthrene and p-tert-butylthiacalix[2]thianthrene and their conformers and complexes with Zn(II), Cd(II) and Hg(II): A theoretical investigation. J. Mol. Model 2010, 16, 243–253. [Google Scholar] [CrossRef]
- Kandemirli, F.; Köksoy, B.; Arslan, T.; Sagdınc, S.; Berber, H. Synthesis and theoretical study of bis(fluoroisatinato)mercury(II). J. Mol. Struct. 2009, 921, 172–177. [Google Scholar] [CrossRef]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long range-corrected time-dependent Density Functional Theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef]
- Cances, M.T.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3037. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum solvation models: A new approach to the problem of solute’s charge distribution and cavity boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hernández-Marin, E.; Barbosa, A.; Martínez, A. The Metal Cation Chelating Capacity of Astaxanthin. Does This Have Any Influence on Antiradical Activity? Molecules 2012, 17, 1039-1054. https://doi.org/10.3390/molecules17011039
Hernández-Marin E, Barbosa A, Martínez A. The Metal Cation Chelating Capacity of Astaxanthin. Does This Have Any Influence on Antiradical Activity? Molecules. 2012; 17(1):1039-1054. https://doi.org/10.3390/molecules17011039
Chicago/Turabian StyleHernández-Marin, Elizabeth, Andrés Barbosa, and Ana Martínez. 2012. "The Metal Cation Chelating Capacity of Astaxanthin. Does This Have Any Influence on Antiradical Activity?" Molecules 17, no. 1: 1039-1054. https://doi.org/10.3390/molecules17011039