Hybridization vs. Bond Stretching Isomerism in Ru(II) Cyclometalated Complexes of 2-Phenylpyridine

Abstract
:1. Introduction

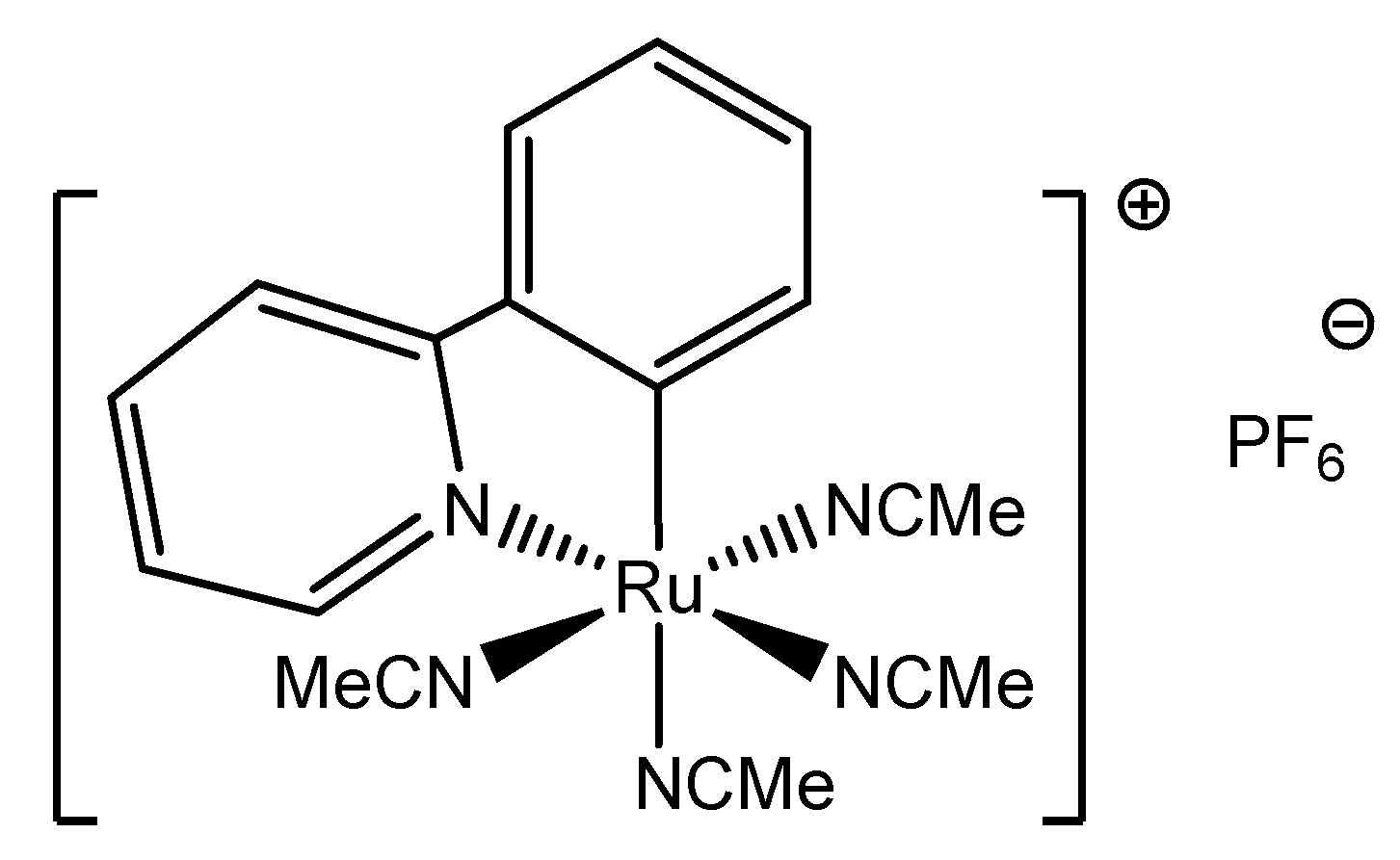
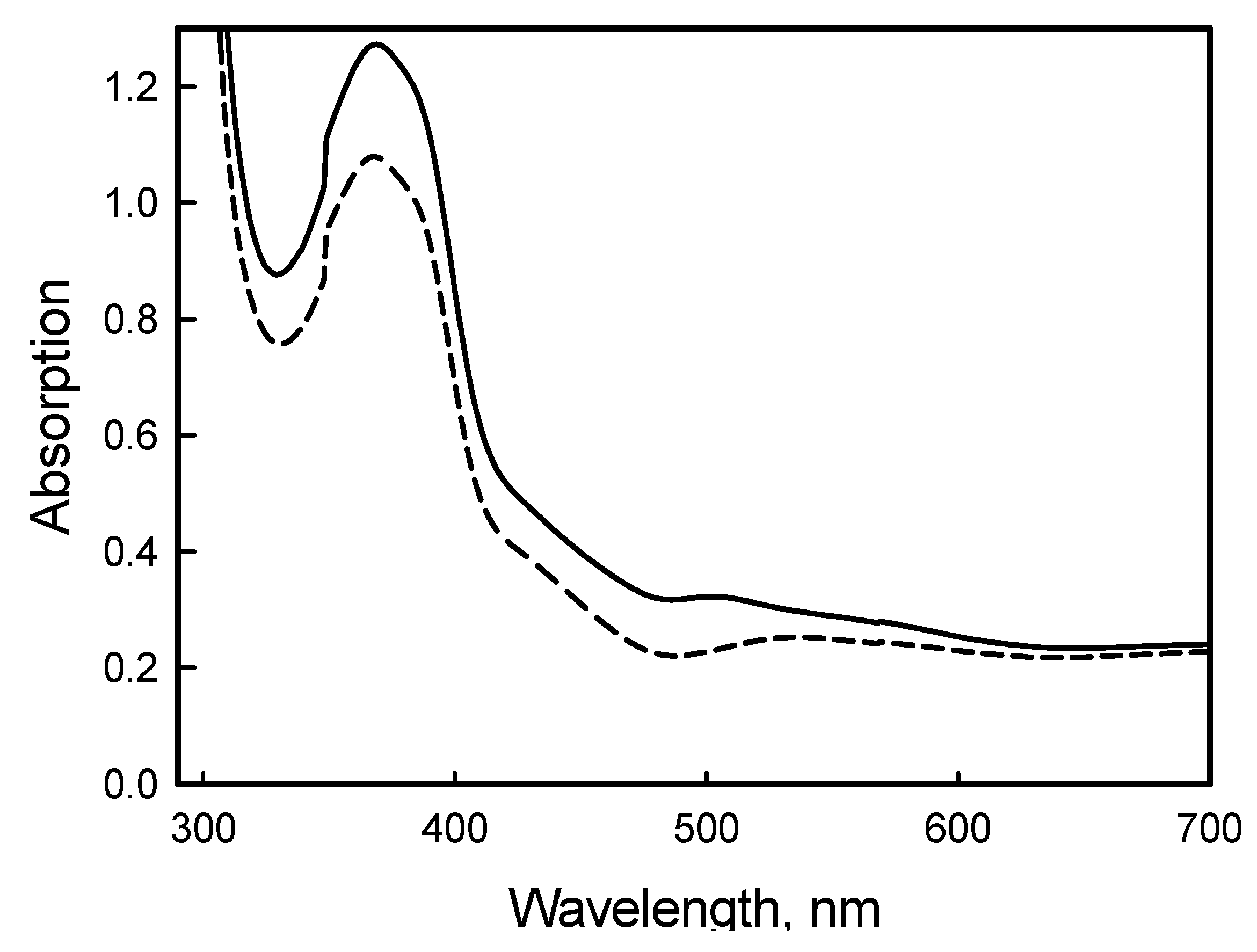
2. Results and Discussion
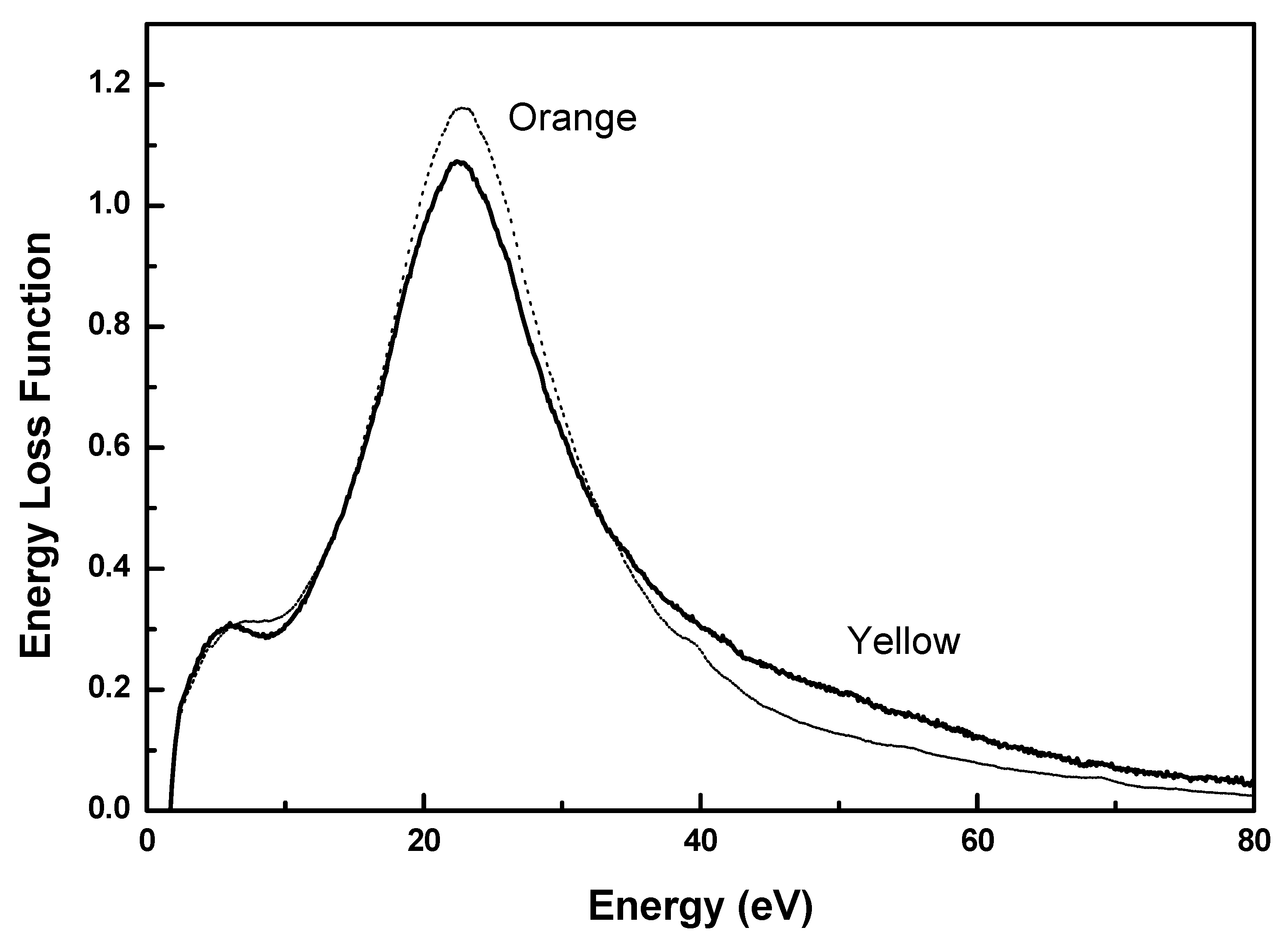
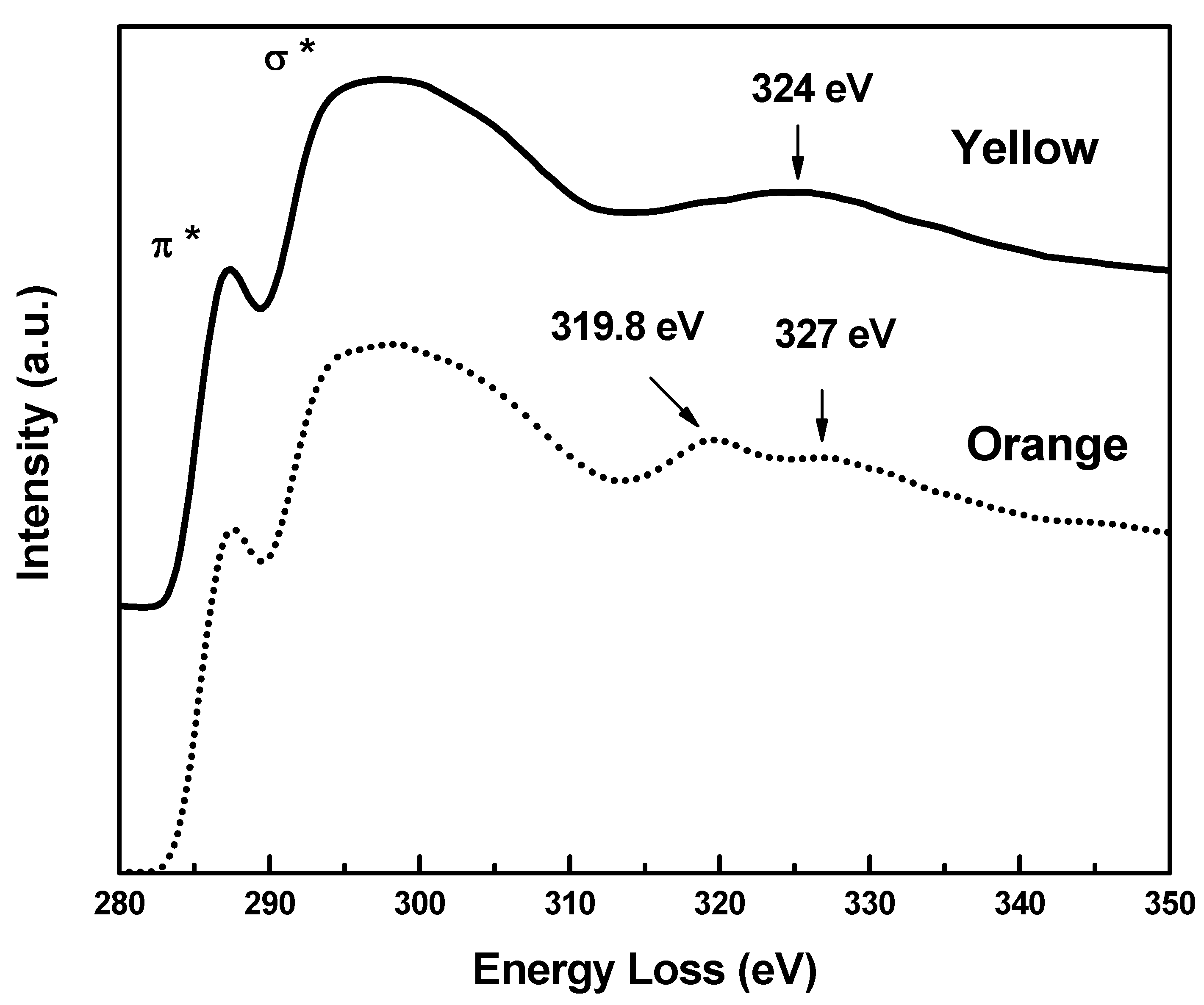
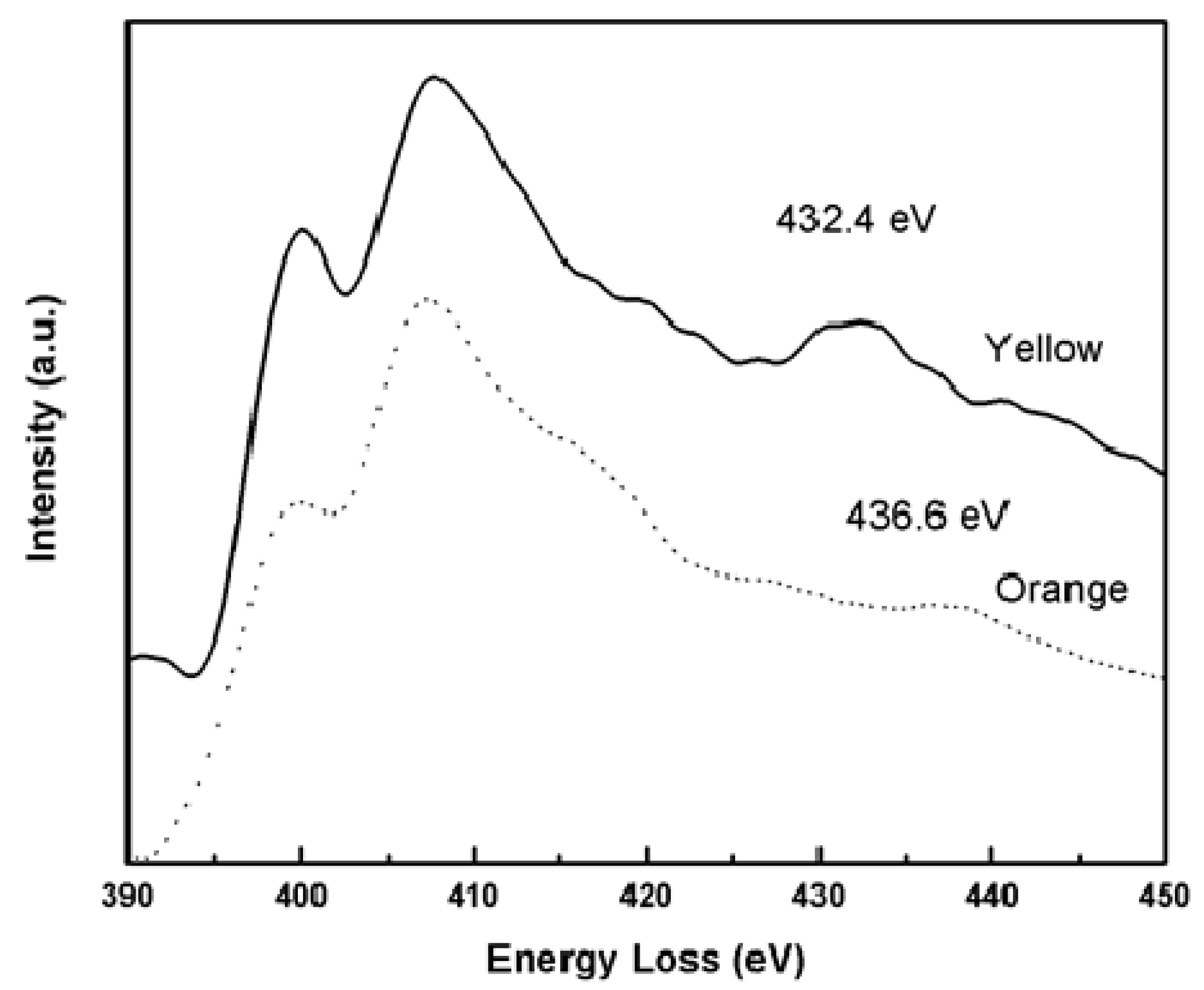
2.1. EELS Results








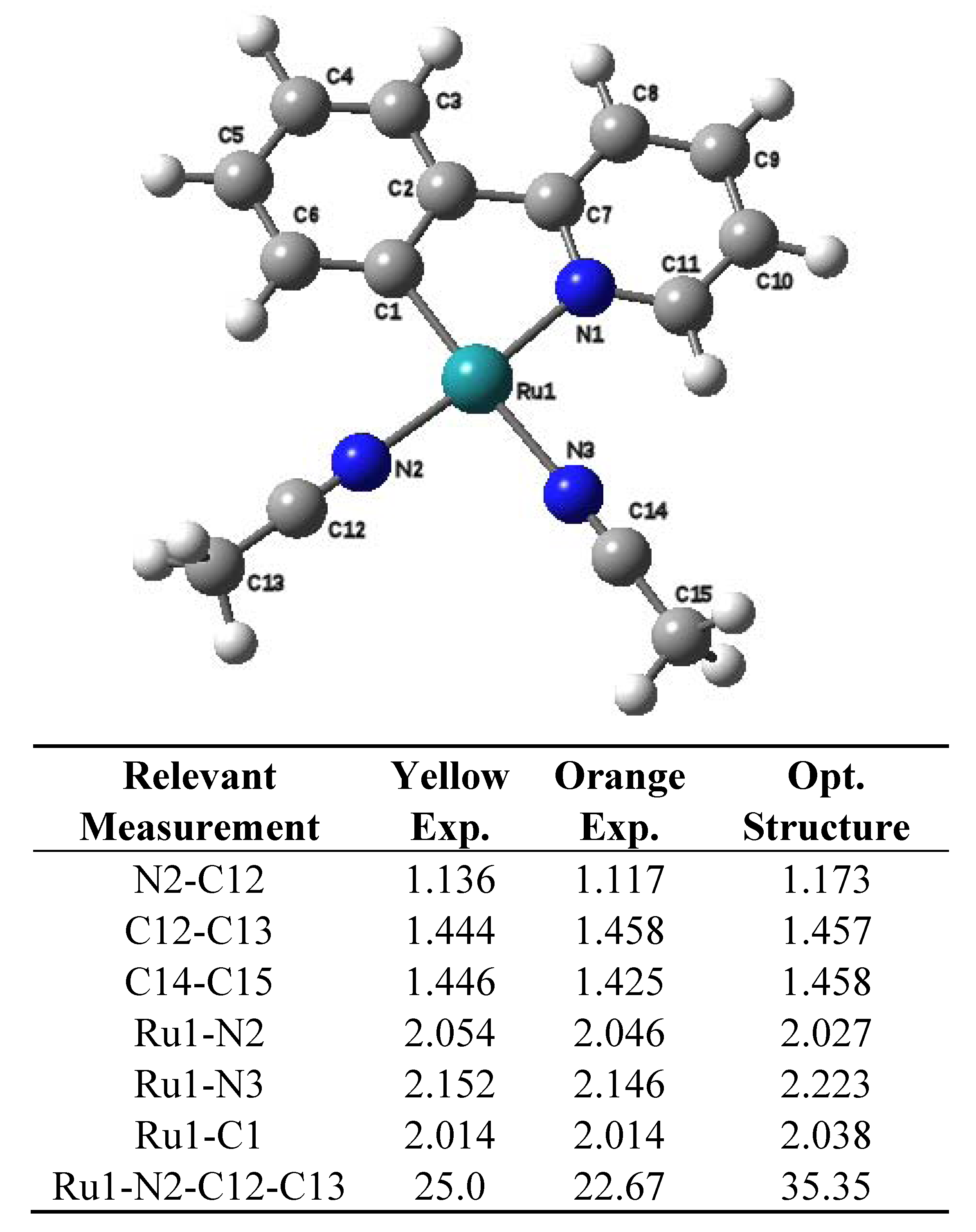
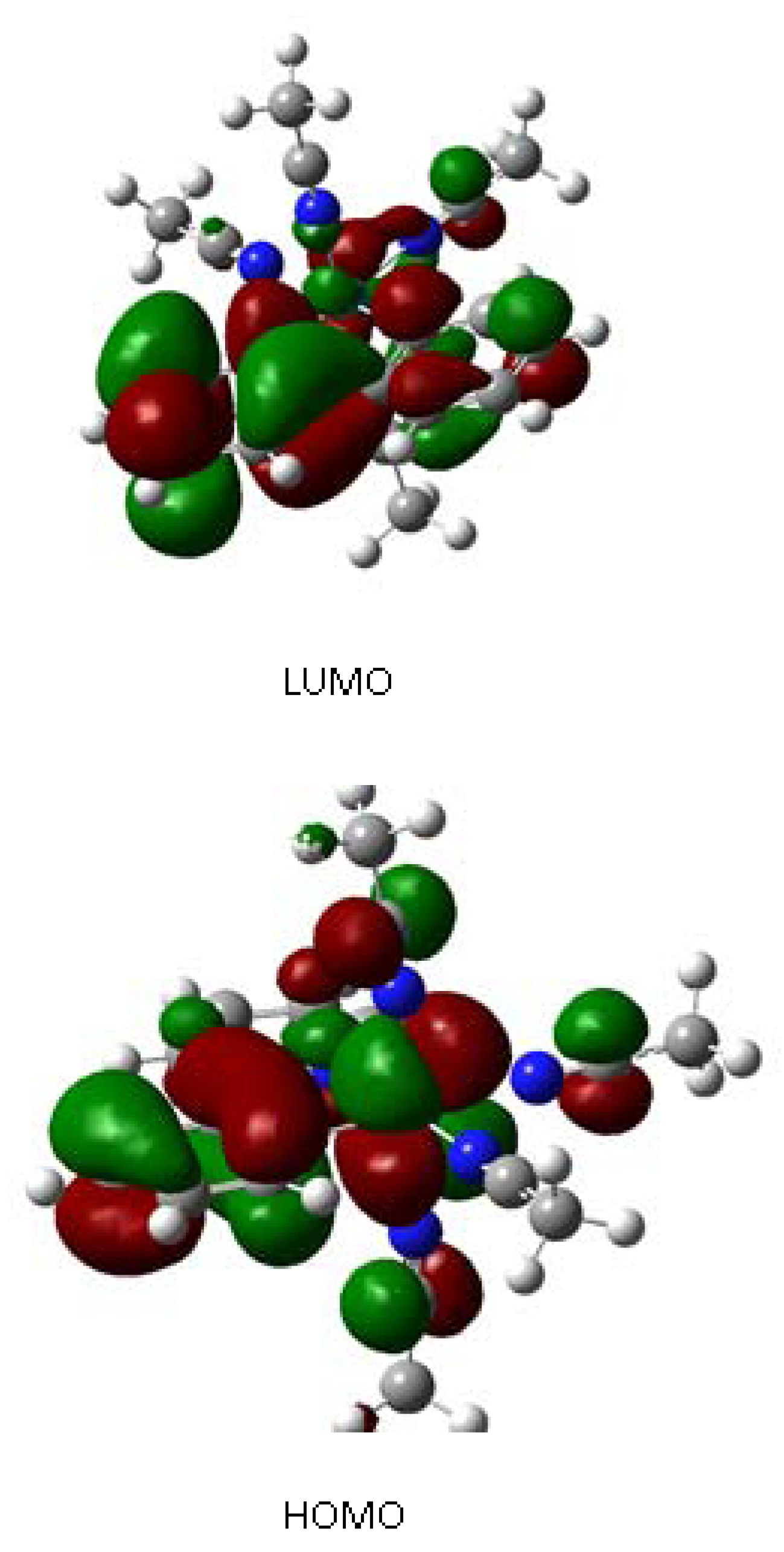
2.2. Computational Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yellow compound | Orange compound | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| MC | %P | EE | λ | f | MC | %P | EE | λ | f |
| S92→93 | 69.29 | 2.56 | 484 | 0.0016 | S92→93 | 69.04 | 2.57 | 482 | 0.0028 |
| S91→93 | 70.32 | 2.67 | 464 | 0.0002 | S91→93 | 70.28 | 2.68 | 463 | 0.0003 |
| S92→94 | 65.77 | 2.96 | 418 | 0.0023 | S92→94 | 65.34 | 2.99 | 415 | 0.0021 |
| S91→94 | 70.44 | 3.07 | 404 | 0.0002 | S91→94 | 70.23 | 3.10 | 400 | 0.0005 |
| S90→93 | 60.34 | 3.21 | 387 | 0.1327 | S90→93 | 59.56 | 3.19 | 389 | 0.1140 |
| S90→94 | 65.96 | 3.40 | 365 | 0.0219 | S90→94 | 65.36 | 3.45 | 359 | 0.0362 |
| S89→93 | 59.50 | 3.90 | 318 | 0.0678 | S89→93 | 53.64 | 3.99 | 311 | 0.0690 |
| S92→95 | 62.05 | 3.94 | 315 | 0.0013 | S92→95 | 51.16 | 4.01 | 309 | 0.0166 |
| S92→96 | 48.26 | 4.08 | 304 | 0.0227 | S92→96 | 52.78 | 4.12 | 301 | 0.0327 |
| S88→93 | 53.81 | 4.09 | 303 | 0.0104 | S88→93 | 62.60 | 4.13 | 300 | 0.0037 |


3. Experimental
3.1. Synthesis of the Ruthenium(II) Complexes
3.2. EELS Spectroscopy
3.3. Molecular Modelling Details
4. Conclusions
Acknowledgements
References and Notes
- Stohrer, W.D.; Hoffmann, R. The electronic Structure and reactivity of strained tricyclic hydrocarbons. J. Am. Chem. Soc. 1972, 94, 779–786. [Google Scholar] [CrossRef]
- Parkin, G. Bond-stretch isomerism in transition metal complexes: A reevaluation of crystallographic data. Chem. Rev. 1993, 93, 887–911. [Google Scholar]
- Yoon, K.; Parkin, G.; Rheingold, A.L. Bond-stretch isomerism in the chlorooxomolybdenum complexes cis-mer-Moocl2(PR3)3: A reinvestigation. J. Am. Chem. Soc. 1992, 114, 2210–2218. [Google Scholar]
- Rohmer, M.M.; Bénard, M. Bond-stretch isomerism in strained inorganic molecules and in transition metal complexes: A revival? Chem. Soc. Rev. 2001, 30, 340–354. [Google Scholar]
- Niecke, E.; Fuchs, A.; Nieger, M. Valence isomerization of a 1,3-diphosphacyclobutane-2,4-diyl: Photochemical ring closure to 2,4-diphosphabicyclo[1.1.0]butane and its thermal ring opening to gauche-1,4-diphosphabutadiene. Angew. Chem. Int. Ed. 1999, 38, 3028–3031. [Google Scholar] [CrossRef]
- Rodriguez, A.; Olsen, R.A.; Ghaderi, N.; Scheschkewitz, D.; Tham, F.S.; Mueller, L.J.; Bertrand, G. Evidence for the coexistence of two bond-stretch isomers in solution. Angew. Chem. Int. Ed. 2004, 43, 4880–4883. [Google Scholar]
- Soleilhavoup, M.; Bertrand, G. Let’s play with valence isomers: The influence of different main group elements on their relative stability. Bull. Chem. Soc. Jpn. 2007, 80, 1241–1252. [Google Scholar] [CrossRef]
- Breher, F. Stretching bonds in main group element compounds—Borderlines between biradicals and closed-shell species. Coord. Chem. Rev. 2007, 251, 1007–1043. [Google Scholar]
- Grützmacher, H.; Breher, F. Odd-electrons bonds and biradicals in main group element chemistry. Angew. Chem. Int. Ed. 2002, 41, 4006–4011. [Google Scholar] [CrossRef]
- McGrady, J.E. [(Cp*RuCl)2(μ-Cl)2]: Bond-stretch or spin-state isomerism? Angew. Chem. Int. Ed. 2000, 39, 3077–3079. [Google Scholar] [CrossRef]
- Feng, Q.; Rauchfuss, T.B.; Wilson, S. Geometric isomerism based on metal-metal bonds. J. Am. Chem. Soc. 1995, 117, 4702–4703. [Google Scholar] [CrossRef]
- Pantazis, D.A.; McGrady, J.E. A three-state model for the polymorphism in linear tricobalt compounds. J. Am. Chem. Soc. 2006, 128, 4128–4135. [Google Scholar] [CrossRef]
- Clérac, R.; Cotton, F.A.; Daniels, L.M.; Dunbar, K.R.; Murillo, C.A.; Wang, X. Tuning the metal metal bonds in the linear tricobalt compound Co3(Dpa)4Cl2: Bond-stretch and spin-state isomers. Inorg. Chem. 2001, 40, 1256–1264. [Google Scholar] [CrossRef]
- Rohmer, M.M.; Strich, A.; Bénard, M.; Malrieu, J.P. Metal-metal bond length variability in Co(3)(Dipyridylamide)(4)Cl(2): Bond-stretch isomerism, crystal field effects, or spin transition process? A DFT study. J. Am. Chem. Soc. 2001, 123, 9126–9134. [Google Scholar] [CrossRef]
- Berry, J.F.; Cotton, F.A.; Lu, T.; Murillo, C.A.; Roberts, B.K.; Wang, X. Molecular and electronic structures by design: Tuning symmetrical and unsymmetrical linear trichromium chains. J. Am. Chem. Soc. 2004, 126, 7082–7096. [Google Scholar]
- Hammann, B.; Chen, C.; Flörke, U.; Hauptmann, R.; Bill, E.; Sinnecker, S.; Henkel, G. Alpha- and beta-[Fe(2)(Mu-Stbu)(2)(Stbu)(4)](2-): Coexistence of two bond-stretch isomers of a classical bitetrahedral metal chalcogenolate compound. Angew. Chem. Int. Ed. 2006, 45, 8245–8249. [Google Scholar] [CrossRef]
- Cavigliasso, G.; Stranger, R. Electronic structure and metal-metal interactions in trinuclear face-shared [M3X12]3- (M = Mo, W; X = F, Cl, Br, I) systems. Inorg. Chem. 2008, 47, 3072–3083. [Google Scholar] [CrossRef]
- Born, K.; Comba, P.; Kerscher, M.; Linti, G.; Pritzkow, H.; Rohwer, H. Distortional isomerism with Copper(I) complexes of 3,7-diazabicyclo[3.3.1]nonane derivatives. Dalton Trans. 2009, 362–367. [Google Scholar]
- Parkin, G.; Hoffmann, R. Spin-state isomers and bond stretch isomers: A comment on the article “bond-stretch isomers: Fact not fictio”. Angew. Chem. Int. Ed. 1994, 33, 1462. [Google Scholar] [CrossRef]
- Collman, J.P.; Farnham, P.; Dolcetti, G. Intramolecular redox equilibriums of cobalt-nitrosyl complexes. J. Am. Chem. Soc. 1971, 93, 1788–1790. [Google Scholar] [CrossRef]
- Alam, M.S.; Xi, H.-W.; Lim, K.H. Theoretical studies of germaallenic structure, bonding and hyperconjugation. Organometallics 2009, 28, 3678–3685. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Sukharev, V.S.; Alexandrova, L.; Le Lagadec, R.; Pfeffer, M. New synthesis and new bio-application of cyclometalated Ruthenium(II) complexes for fast mediated electron transfer with peroxidase and glucose oxidase. Inorg. Chem. 2001, 40, 6529–6532. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Le Lagadec, R.; Estevez, H.; Toscano, R.A.; Hernández, S.; Alexandrova, L.; Kurova, V.S.; Fischer, A.; Sirlin, C.; Pfeffer, M. Synthesis, characterization, and electrochemistry of biorelevant photosensitive low-potential orthometalated ruthenium complexes. Inorg. Chem. 2005, 44, 1626–1634. [Google Scholar]
- Ryabov, A.D.; Estevez, H.; Alexandrova, L.; Pfeffer, M.; Le Lagadec, R. Unusual phenomenon in the chemistry of orthometalated Ruthenium(II) complexes. Inorg. Chim. Acta 2006, 359, 883–887. [Google Scholar] [CrossRef]
- Ahn, C.C. Transmission Electron Energy Loss Spectrometry in Materials Science and the EELS Atlas; WILEY-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2004. [Google Scholar]
- Ponsonnet, L.; Donnet, C.; Varlot, K.; Martin, J.M.; Grill, A.; Patel, V. EELS analysis of hydrogenated diamond-like carbon films. Thin SolidFilms 1998, 319, 97–100. [Google Scholar] [CrossRef]
- Egerton, R.F. Electron Energy Loss Spectroscopy in the Electron Microscope; Plenum Press: New York, NY, USA, 1996. [Google Scholar]
- Kurata, H.; Lefevre, E.; Colliex, C. Electron-energy-loss near-edge structures in the oxygen K-edge spectra of transition-metal oxides. Phys. Rev. B 1992, 47, 13763–13768. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseira, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A., Jr.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian03; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- van Gisbergen, S.J.A.; Snijders, J.G.; Baerends, E.J. A Density functional theory study of frequency-dependent polarizabilities and van der waals dispersion coefficients for polyatomic molecules. J. Chem. Phys. 1995, 103, 9347–9354. [Google Scholar] [CrossRef]
- Jamorski, C.; Casida, M.E.; Salahub, D.R. Dynamic polarizabilities and excitation spectra from a molecular implementation of time–dependent density–functional response theory: N2 as a case study. J. Chem. Phys. 1996, 104, 5134–5147. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compound [Ru(o-C6H4-py)(MeCN)4]+ are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Molina, B.; Alexandrova, L.; LeLagadec, R.; Sansores, L.E.; Rios-Jara, D.; Espinosa-Magaña, F.; Salcedo, R. Hybridization vs. Bond Stretching Isomerism in Ru(II) Cyclometalated Complexes of 2-Phenylpyridine. Molecules 2012, 17, 34-47. https://doi.org/10.3390/molecules17010034
Molina B, Alexandrova L, LeLagadec R, Sansores LE, Rios-Jara D, Espinosa-Magaña F, Salcedo R. Hybridization vs. Bond Stretching Isomerism in Ru(II) Cyclometalated Complexes of 2-Phenylpyridine. Molecules. 2012; 17(1):34-47. https://doi.org/10.3390/molecules17010034
Chicago/Turabian StyleMolina, Bertha, Larissa Alexandrova, Ronan LeLagadec, Luis E. Sansores, David Rios-Jara, Francisco Espinosa-Magaña, and Roberto Salcedo. 2012. "Hybridization vs. Bond Stretching Isomerism in Ru(II) Cyclometalated Complexes of 2-Phenylpyridine" Molecules 17, no. 1: 34-47. https://doi.org/10.3390/molecules17010034