Inhibition of Sevoflurane Postconditioning Against Cerebral Ischemia Reperfusion-Induced Oxidative Injury in Rats

Abstract
:1. Introduction
2. Results and Discussion
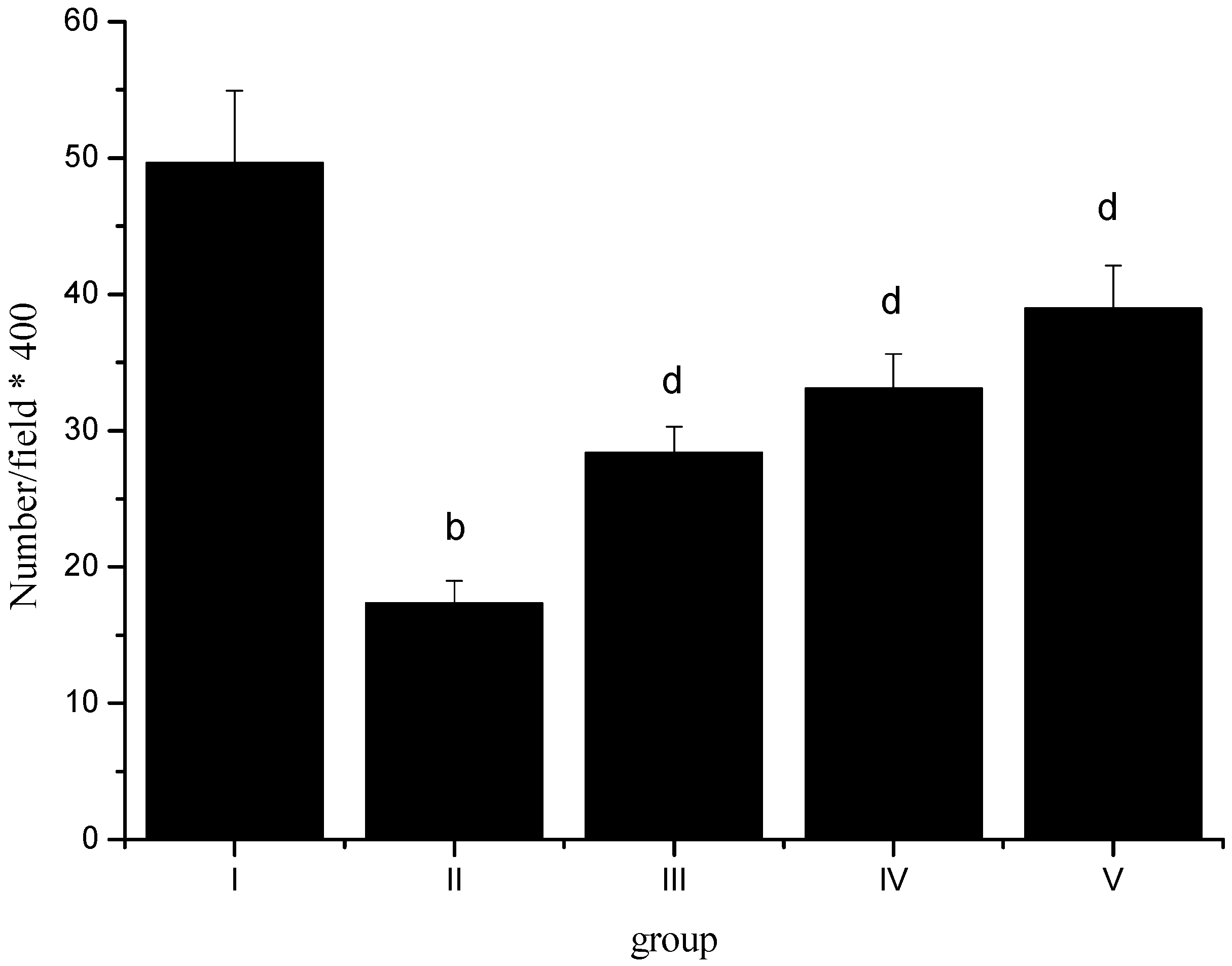
2.1. Effects of Sevoflurane Postconditioning on Serum TNF-α, IL-10 and IL-1β Concentrations

{kind=link}
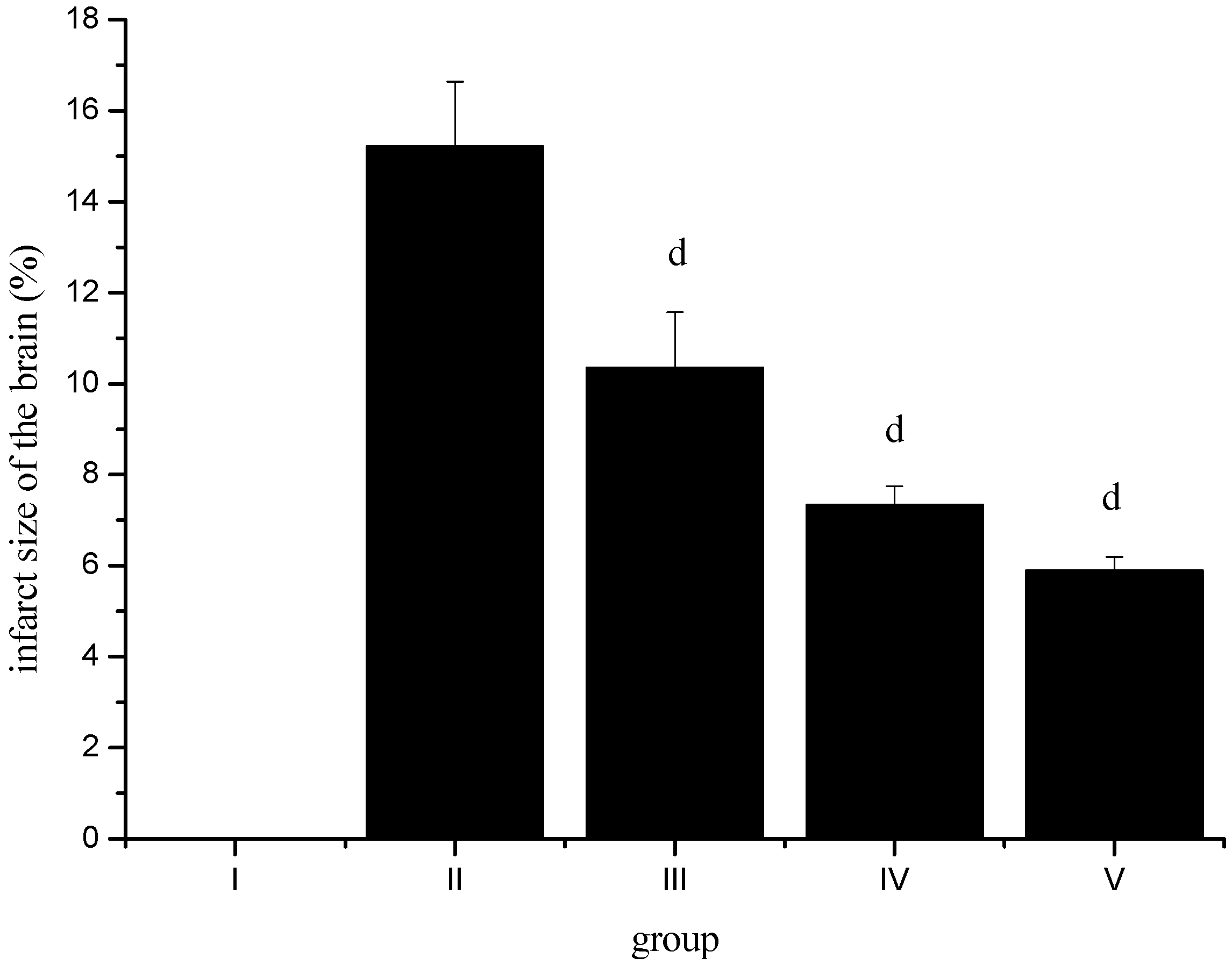{kind=link}
| Group | TNF-α (ng/mL) | IL-1β (ng/L) | IL-10 (ng/L) |
|---|---|---|---|
| I | 1.95 ± 0.11 | 10.65 ± 1.07 | 42.06 ± 3.08 |
| II | 4.65 ± 0.27 b | 19.66 ± 1.43 b | 21.52 ± 1.37 b |
| III | 3.06 ± 0.24 d | 17.03 ± 1.49 c | 29.29 ± 1.33 d |
| IV | 2.66 ± 0.29 d | 14.72 ± 1.35 d | 33.03 ± 1.97 d |
| V | 1.99 ± 0.2 d | 12.18 ± 1.22 d | 38.58 ± 1.85 d |
2.2. Effects of Sevoflurane Postconditioning on Serum NO and NOS Concentrations
| Group | NO (μmol/L) | NOS (U/mL) |
|---|---|---|
| I | 21.57 ± 2.07 | 22.18 ± 1.43 |
| II | 40.62 ± 2.31 b | 51.43 ± 0.96 b |
| III | 35.15 ± 1.99 c | 39.09 ± 1.37 d |
| IV | 29.07 ± 1.26 d | 29.04 ± 1.54 d |
| V | 21.36 ± 1.65 d | 20.13 ± 1.22 d |
2.3. Effects of Sevoflurane Postconditioning on Serum Lipids Levels
| Group | TC (mmol/L) | TG (mmol/L) | HDL-c (mmol/L) | LDL-c (mmol/L) | LDL/HDL |
|---|---|---|---|---|---|
| I | 2.92 ± 0.2 | 0.74 ± 0.05 | 1.63 ± 0.12 | 1.02 ± 0.08 | 0.64 ± 0.04 |
| II | 5.84 ± 0.26 b | 2.03 ± 0.13 b | 0.72 ± 0.08 b | 5.21 ± 0.21 b | 7.09 ± 0.32 b |
| III | 5.17 ± 0.31 c | 1.75 ± 0.11 c | 1.03 ± 0.05 d | 4.52 ± 0.16 c | 4.37 ± 0.18 d |
| IV | 4.53 ± 0.19 d | 1.33 ± 0.09 d | 1.46 ± 0.08 d | 3.62 ± 0.18 d | 2.47 ± 0.13 d |
| V | 3.27 ± 0.22 d | 0.94 ± 0.07 d | 1.55 ± 0.09 d | 2.37 ± 0.11 d | 1.53 ± 0.09 d |
2.4. Effects of Sevoflurane Postconditioning on Serum and Brain MDA, Reduced Glutathione (GSH) Concentrations
| Group | MDA | GSH | ||
|---|---|---|---|---|
| Blood (nmol/mL) | Brain (nmol/g prot) | Blood (nmol/mL) | Brain (nmol/mg protein) | |
| I | 4.21 ± 0.21 | 3.15 ± 0.15 | 43.98 ± 3.08 | 58.32 ± 2.31 |
| II | 8.53 ± 0.37 b | 7.04 ± 0.43 b | 22.17 ± 1.43 b | 20.61 ± 1.26 b |
| III | 7.02 ± 0.42 c | 5.82 ± 0.22 d | 29.51 ± 1.24 d | 32.51 ± 1.32 d |
| IV | 5.97 ± 0.22 d | 5.01 ± 0.27 d | 35.66 ± 1.75 d | 41.28 ± 1.85 d |
| V | 4.88 ± 0.28 d | 4.22 ± 0.17 d | 40.71 ± 1.95 d | 59.03 ± 2.08 d |
2.5. Effects of Sevoflurane Postconditioning on Serum and Brain SOD, CAT, GSH-Px and GR Activities
| Group | SOD | CAT | ||
| Serum (U/mL) | Brain(U/mg) | Serum (U/mL) | Brain(U/mg) | |
| I | 276.5 ± 16.3 | 303.1 ± 22.9 | 39.11 ± 1.54 | 33.12 ± 1.84 |
| II | 132.1 ± 9.6 b | 137.4 ± 10.7 b | 15.03 ± 1.11 b | 12.84 ± 1.02 b |
| III | 176.4 ± 8.6 d | 199.2 ± 12.4 d | 19.99 ± 2.09 d | 21.87 ± 1.15 d |
| IV | 220.6 ± 11.6 d | 274.8 ± 17.3 d | 24.08 ± 1.57 d | 28.44 ± 1.36 d |
| V | 281.5 ± 20.5 d | 314.1 ± 19.2 d | 34.17 ± 1.72 d | 32.17 ± 1.83 d |
| Group | GSH-Px | GR | ||
| Serum (U/mL) | Brain(U/mg) | Serum (U/mL) | Brain(U/mg) | |
| I | 52.19 ± 2.54 | 63.19 ± 2.98 | 29.07 ± 1.54 | 32.18 ± 1.76 |
| II | 22.15 ± 1.54 b | 27.51 ± 1.06 b | 12.16 ± 1.06 b | 15.27 ± 1.13 b |
| III | 32.97 ± 1.97 d | 37.82 ± 1.44 d | 19.03 ± 1.17 d | 21.54 ± 1.08 d |
| IV | 40.63 ± 2.61 d | 47.18 ± 2.31 d | 24.01 ± 1.32 d | 29.41 ± 1.57 d |
| V | 49.76 ± 2.88 d | 58.29 ± 2.28 d | 32.18 ± 1.69 d | 34.29 ± 1.39 d |
2.6. Normal Pyramidal Neurons Density

2.7. Histopathological Study
2.8. Effect of Sevoflurane Postconditioning on Infarct Volume after Ischemia-Reperfusion

3. Experimental
3.1. Animals
3.2. Experimental Protocol
3.3. Middle Cerebral Arterial Occlusion
3.4. Biochemical Analysis
3.5. Histological Study
3.6. Measurement of Infarct Volume
3.7. Statistical Analysis
4. Conclusions
Conflict of Interest
Reference and Notes
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar]
- Upham, B.L.; Wagner, J.G. Toxicant-induced oxidative stress in cancer. Toxicol. Sci. 2001, 64, 1–3. [Google Scholar] [CrossRef]
- Dusting, G.J.; Triggle, C. Are we over oxidized? Oxidative stress, cardiovascular disease, and the future of intervention studies with antioxidants. Vasc. Health Risk Manag. 2005, 1, 93–97. [Google Scholar] [CrossRef]
- Harrison, D.G.; Gongora, M.C. Oxidative stress and hypertension. Med. Clin. N. Am. 2009, 93, 621–635. [Google Scholar] [CrossRef]
- Ozsoy, N.; Can, A.; Yanardag, R.; Akev, N. Antioxidant activity of Smilax excelsa L. leaf extracts. Food Chem. 2008, 110, 571–583. [Google Scholar] [CrossRef]
- Mies, G.; Kohno, K.; Hossman, K. MK-801, a glutamate antagonist, lowers flow threshold for inhibition of protein synthesis after middle cerebral artery occlusion of rat. Neurosci. Lett. 1993, 155, 65–68. [Google Scholar] [CrossRef]
- Hossman, K. Viability thresholds and the penumbra of focal ischemia. Ann. Neurol. 1994, 36, 557–565. [Google Scholar] [CrossRef]
- Chagnac-Amitai, Y.; Connors, B.W. Horizontal spread of synchronized activity inneocortex by GABA-mediated inhibition. J. Neurophysiol. 1989, 61, 747–758. [Google Scholar]
- Traystman, R.J.; Kirsch, J.R.; Koehler, R.C. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. J. Appl. Physiol. 1991, 71, 1185–1195. [Google Scholar]
- Paradis, E.; Clavel, S.; Julien, P.; Murthy, M.R.V.; de Bilbao, F.; Arsenijevic, D.; Giannakopoulos, P.; Vallet, P.; Richard, D. Lipoprotein lipase and endothelial lipase expression in mouse brain: Regional distribution and selective induction following kainic acid-induced lesion and focal cerebral ischemia. Neurobiology 2004, 15, 312–325. [Google Scholar]
- Bolwell, G.P.; Davies, D.R.; Gerrish, C.; Auh, C.K.; Murphy, T.M. Comparative biochemistry of the oxidative burst produced by rose and French bean cells reveals two distinct mechanisms. Plant Physiol. 1998, 116, 1379–1385. [Google Scholar] [CrossRef]
- Bolwell, G.P.; Davies, D.R.; Gerrish, C.; Auh, C.K.; Murphy, T.M. Comparative biochemistry of the oxidative burst produced by rose and French bean cells reveals two distinct mechanisms. Plant Physiol. 1998, 116, 1379–1385. [Google Scholar] [CrossRef]
- Wang, H.Y.; Wang, G.L.; Yu, Y.H.; Wang, Y. The role of phosphoinositide-3-kinase/Akt pathway in propofol-induced postconditioning against focal cerebral ischemia-reperfusion injury in rats. Brain Res. 2009, 1297, 177–184. [Google Scholar]
- Young, Y.; Menon, D.K.; Tisavipat, N.; Matta, B.F.; Jones, J.G. Propofol neuroprotection in a rat model of ischaemia reperfusion injury. Eur. J. Anaesthesiol. 1997, 14, 320–326. [Google Scholar] [CrossRef]
- Maier, C.; Steinberg, G.K.; Sun, G.H.; Zhi, G.T.; Maze, M. Neuroprotection by the alpha 2-adrenoreceptor agonist dexmedetomidine in a focal model of cerebral ischemia. Anesthesiology 1993, 79, 306–312. [Google Scholar] [CrossRef]
- Rajakumaraswamy, N.; Ma, D.; Hossain, M.; Sanders, R.D.; Franks, N.P.; Maze, M. Neuroprotective interaction produced by xenon and dexmedetomidine on in vitro and in vivo neuronal injury models. Neurosci. Lett. 2006, 409, 128–133. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, D.; Ieong, E.; Sanders, R.D.; Yu, B.; Hossain, M.; Maze, M. Xenon and sevoflurane protect against brain injury in a neonatal asphyxia model. Anesthesiology 2008, 109, 782–789. [Google Scholar] [CrossRef]
- Sakai, H.; Sheng, H.; Yates, R.B.; Ishida, K.; Pearlstein, R.D.; Warner, D.S. Isoflurane provides long-term protection against focal cerebral ischemia in the rat. Anesthesiology 2007, 106, 92–99. [Google Scholar] [CrossRef]
- Yu, L.-N.; Yu, J.; Zhang, F.-J.; Yang, M.-J.; Ding, T.-T.; Wang, J.-K.; He, W.; Fang, T.; Chen, G.; Yan, M. Sevoflurane postconditioning reduces myocardial reperfusion injury in rat isolated hearts via activation of PI3K/Akt signaling and modulation of Bcl-2 family proteins. J. Zhejiang Univ. Sci. B 2010, 11, 661–672. [Google Scholar] [CrossRef]
- Payne, R.S.; Akca, O.; Roewer, N.; Schurr, A.; Kehl, F. Sevoflurane-induced preconditioning protects against cerebral ischemic neuronal damage in rats. Brain Res. 2005, 1034, 147–152. [Google Scholar]
- Yao, Y.-T.; Fang, N.-X.; Shi, C.-X.; Li, L.-H. Sevoflurane postconditioning protects isolated rat hearts against ischemia-reperfusion injury. Chin. Med. J. 2010, 123, 1320–1328. [Google Scholar]
- Zhang, M.-K.; Shen, S.-Q.; Liu, R.-Z.; Xu, F.-H.; Chen, X.-Q.; Li, Y.-S.; Chen, T-T. Effects of naoshuning on the plasma TNF-α, IL-1β and ICAM-1 levels in a rat brain ischemia-reperfusion injury model. Sci. Res. Essays 2011, 6, 923–927. [Google Scholar]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Kinoshita, A.; Ikemoto, A.; Nakamura, S.; Kimura, J. Glial expression of cytokines in the brains of cerebrovascular disease patients. Acta Neuropathol. (Berlin) 1996, 92, 281–287. [Google Scholar] [CrossRef]
- Sairanen, T.; Carpén, O.; Karjalainen-Lindsberg, M.L.; Paetau, A.; Turpeinen, U.; Kaste, M.; Lindsberg, P.J. Evolution of cerebral tumor necrosis factor-alpha production during human ischemic stroke. Stroke 2001, 32, 1750–1758. [Google Scholar] [CrossRef]
- Carlstedt, F.; Lind, L.; Lindahl, B. Proinflammatory cytokines, measured in a mixed population on arrival in the emergency department, are related to mortality and severity of disease. J. Int. Med. 1997, 242, 361–365. [Google Scholar]
- Vila, N.; Castillo, J.; Dávalos, A.; Chamorro, A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef]
- Zaremba, J.; Skrobanski, P.; Losy, J. Tumor necrosis factor-alpha is increased in the cerebrospinal fluid and serum of ischaemic stroke patients and correlates with the volume of evolving brain infarct. Biomed. Pharmacother. 2001, 55, 258–263. [Google Scholar] [CrossRef]
- Sairanen, T.R.; Lindsberg, P.J.; Brenner, M.; Carpén, O.; Sirén, A.L. Differential cellular expression of tumor necrosis factor-α and type I tumor necrosis factor receptor after transient global forebrain ischemia. J. Neurol. Sci. 2001, 186, 87–99. [Google Scholar] [CrossRef]
- Saito, K.; Suyama, K.; Nishida, K.; Sei, Y.; Basile, A.S. Early increases in TNF-α, IL-6, and IL-1β levels following transient cerebral ischemia in gerbil brain. Neurosci. Lett. 1996, 206, 149–152. [Google Scholar] [CrossRef]
- Liu, Y.; Jacobowitz, D.M.; Barone, F.; McCarron, R.; Spatz, M.; Feuerstein, G.; Hallenbeck, J.M.; Siren, A.L. Quantitation of perivascular monocytes and macrophages around cerebral blood vessels of hypertensive and aged rats. J. Cereb. Blood Flow Metab. 1994, 14, 348–352. [Google Scholar] [CrossRef]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef]
- Toda, N.; Ayajiki, K.; Okamura, T. Cerebral blood flow regulation by nitric oxide: Recent advances. Pharmacol. Rev. 2009, 61, 62–97. [Google Scholar] [CrossRef]
- Vestergaard, S.; Loft, S.; Møller, P. Role of inducible nitrogen oxide synthase in benzene-induced oxidative DNA damage in the bone marrow of mice. Free Radic. Biol. Med. 2002, 32, 481–484. [Google Scholar] [CrossRef]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar]
- Traystman, R.J.; Kirsch, J.R.; Koehler, R.C. Oxygen radical mechanism of brain injury following ischemia and reperfusion. J. Appl. Physiol. 1991, 71, 1185–1195. [Google Scholar]
- Wang, Y.-C.; Zhang, S.; Du, T.-Y.; Wang, B.; Sun, X.-Q. Hyperbaric oxygen preconditioning reduces ischemia-reperfusion injury by stimulating autophagy in neurocyte. Brain Res. 2010, 1323, 149–151. [Google Scholar]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Oh, S.; Betz, A.L. Interaction between free radicals and excitatory amino acids in the formation of ischemic brain edema in rats. Stroke 1991, 22, 915–921. [Google Scholar] [CrossRef]
- Watson, B.D.; Busto, R.; Goldberg, W.J.; Santiso, M.; Yoshida, S.; Ginsberg, M.D. Lipid peroxidation in vivo induced reversible global ischemia in rat brain. J. Neurochem. 1984, 42, 268–274. [Google Scholar] [CrossRef]
- Evans, P.H. Free radicals in brain metabolism and pathology. Br. Med. Bull. 1993, 49, 577–587. [Google Scholar]
- Candelario-Jalil, E.; Alvarez, D.; Merino, N.; León, O.S. Delayed treatment with nimesulide reduces measures of oxidative stress following global ischemic brain injury in gerbils. Neurosci. Res. 2003, 47, 245–253. [Google Scholar] [CrossRef]
- Ozkul, A.; Akyol, A.; Yenisey, C.; Arpaci, E.; Kiylioglu, N.; Tataroglu, C. Oxidative stress in acute ischemic stroke. J. Clin. Neurosci. 2007, 14, 1062–1066. [Google Scholar]
- Ross, D. Glutathione, free radicals and chemotherapeutic agents. Pharmacol. Ther. 1988, 37, 231–249. [Google Scholar] [CrossRef]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar]
- Cuevas, P.; Carceller-Benito, F.; Reimers, D. Administration of bovine superoxide dismutase prevents sequelae of spinal cord ischemia in the rabbit. Anat. Embryol. 1989, 179, 251–253. [Google Scholar] [CrossRef]
- Cuevas, P.; Reimers, D.; Carceller, F.; Iimenez, A. Ischemic reperfusion injury in rabbit spinal cord. Protective effect of superoxide dismutase on neurological recovery and spinal infarction. Acta Anat. 1990, 137, 303–310. [Google Scholar] [CrossRef]
- Lim, K.H.; Connolly, M.; Rose, D.; Siegman, F.; Jacobowitz, I.; Acinapura, A.; Cunningham, J.N., Jr. Ischemic reperfusion injury in rabbit spinal cord. Protective effect of superoxide dismutase on neurological recovery and spinal infarction. Ann. Thorac. Surg. 1986, 42, 282–286. [Google Scholar] [CrossRef]
- Taoka, Y.; Naruo, M.; Koyanagi, E.; Urakado, M.; Inoue, M. Superoxide radicals play important roles in the pathogenesis of spinal cord injury. Paraplegia 1995, 33, 450–453. [Google Scholar] [CrossRef]
- Shah, Z.A.; Gilani, R.A.; Sharma, P.; Vohora, S.B. Cerebroprotective effect of Korean ginseng tea against global and focal models of ischemia in rats. J. Ethno. Pharmacol. 2005, 101, 299–307. [Google Scholar]
- Li, J.; Han, C.F.; Li, X.X.; Du, Y.; Yang, W.Q. The effect of sevoflurane postconditioning on antioxidation of rat hearts in vivo. Chin. Med. Herald 2011, 8, 15–16. [Google Scholar]
- Kilian, J.G.; Nakhla, S.; Griffith, K.; Harmer, J.; Skilton, M.; Celermajer, D.S. Reperfusion injury in the human forearm is mild and not attenuated by short-term ischaemic preconditioning. Clin. Exp. Pharmacol. Physiol. 2005, 32, 86–90. [Google Scholar] [CrossRef]
- Badhwar, A.; Bihari, A.; Dungey, A.A.; Scott, J.R.; Albion, C.D.; Forbes, T.L.; Harris, K.A.; Potter, R.F. Protective mechanisms during ischemic tolerance in skeletal muscle. Free Radic. Biol. Med. 2004, 36, 371–379. [Google Scholar] [CrossRef]
- Bedirli, A.; Kerem, M.; Pasaoglu, H.; Erdem, O.; Ofluoglu, E.; Sakrak, O. Effects of ischemic preconditioning on regenerative capacity of hepatocyte in the ischemically damaged rat livers. J. Surg. Res. 2005, 125, 42–48. [Google Scholar] [CrossRef]
- Glantz, L.; Avramovich, A.; Trembovler, V.; Gurvitz, V.; Kohen, R.; Eidelman, L.A.; Shohami, E. Ischemic preconditioning increases antioxidants in the brain and peripheral organs after cerebral ischemia. Exp. Neurol. 2005, 192, 117–124. [Google Scholar] [CrossRef]
- Sileri, P.; Sica, G.; Gentileschi, P.; Venza, M.; Manzelli, A.; Palmieri, G.; Spagnoli, L.G.; Testa, G.; Benedetti, E.; Gaspari, A.L. Ischemic preconditioning protects intestine from prolonged ischemia. Transplant. Proc. 2004, 36, 283–285. [Google Scholar] [CrossRef]
- Kirino, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982, 239, 57–69. [Google Scholar] [CrossRef]
- Pulsinelli, W.A.; Brierley, J.B.; Plum, F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 1982, 11, 491–498. [Google Scholar] [CrossRef]
- Smith, M.L.; Bendek, G.; Dahlgren, N.; Rosen, I.; Wieloch, T.; Siesjö, B.K. Models for studying long-term recovery following forebrain ischemia in the rat. A 2-vessel occlusion model. Acta Neurol. Scand. 1984, 69, 385–401. [Google Scholar] [CrossRef]
- Kirino, T.; Tamura, A.; Sato, K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol. 1984, 64, 139–147. [Google Scholar] [CrossRef]
- Petito, C.K.; Pusinelli, W.A. Delayed neuronal recovery and neuronal death in rat hippocampus following severe cerebral ischemia: Possible relationship to abnormalities in neuronal processes. J. Cereb. Blood Flow Metab. 1984, 4, 194–205. [Google Scholar] [CrossRef]
- Rafols, J.A.; Daya, A.M.; O’Neil, B.J.; Krause, G.C.; Neumar, R.W.; White, B.C. Global brain ischemia and reperfusion: Golgi apparatus ultrastructure in neurons selectively vulnerable to death. Acta Neuropathol. 1995, 90, 17–30. [Google Scholar] [CrossRef]
- Hu, B.R.; Park, M.; Martone, M.E.; Fischer, W.H.; Ellisman, M.H.; Zivin, J.A. Assembly of proteins to postsynaptic densities after transient cerebral ischemia. Neuroscience 1998, 18, 625–633. [Google Scholar]
- Martone, M.E.; Jones, Y.Z.; Young, S.J.; Ellisman, M.H.; Zivin, J.A.; Hu, B.R. Modification of postsynaptic densities after transient cerebral ischemia: A quantitative and three-dimensional ultrastructural study. Neuroscience 1999, 19, 1988–1997. [Google Scholar]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar]
- Beutler, E.; Duran, O.; Kelly, M.B. Improved method for the determination of blood glutathione. J. Lab. Clin. Med. 1963, 61, 882–886. [Google Scholar]
- Sun, Y.; Oberley, L.W.; Li, Y. A simple method for clinical assay of superoxide dismutase. Clin. Chem. 1988, 34, 497–500. [Google Scholar]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar]
- Aebi, H. Catalase in vitro. Method. Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Carlberg, I.; Mannervik, B. Glutathione reductase. Method. Enzymol. 1985, 113, 484–490. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are commercially available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y.; Zhang, F.-G.; Meng, C.; Tian, S.-Y.; Wang, Y.-X.; Zhao, W.; Chen, J.; Zhang, X.-S.; Liang, Y.; Zhang, S.-D.; et al. Inhibition of Sevoflurane Postconditioning Against Cerebral Ischemia Reperfusion-Induced Oxidative Injury in Rats. Molecules 2012, 17, 341-354. https://doi.org/10.3390/molecules17010341
Zhang Y, Zhang F-G, Meng C, Tian S-Y, Wang Y-X, Zhao W, Chen J, Zhang X-S, Liang Y, Zhang S-D, et al. Inhibition of Sevoflurane Postconditioning Against Cerebral Ischemia Reperfusion-Induced Oxidative Injury in Rats. Molecules. 2012; 17(1):341-354. https://doi.org/10.3390/molecules17010341
Chicago/Turabian StyleZhang, Yan, Fu-Geng Zhang, Chun Meng, Shou-Yuan Tian, Ya-Xin Wang, Wei Zhao, Jun Chen, Xiu-Shan Zhang, Yu Liang, Shi-Dong Zhang, and et al. 2012. "Inhibition of Sevoflurane Postconditioning Against Cerebral Ischemia Reperfusion-Induced Oxidative Injury in Rats" Molecules 17, no. 1: 341-354. https://doi.org/10.3390/molecules17010341