Benzoylated Uronic Acid Building Blocks and Synthesis of N-Uronate Conjugates of Lamotrigine

Abstract
:
1. Introduction
2. Results and Discussion
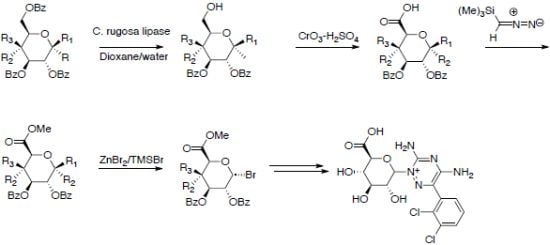
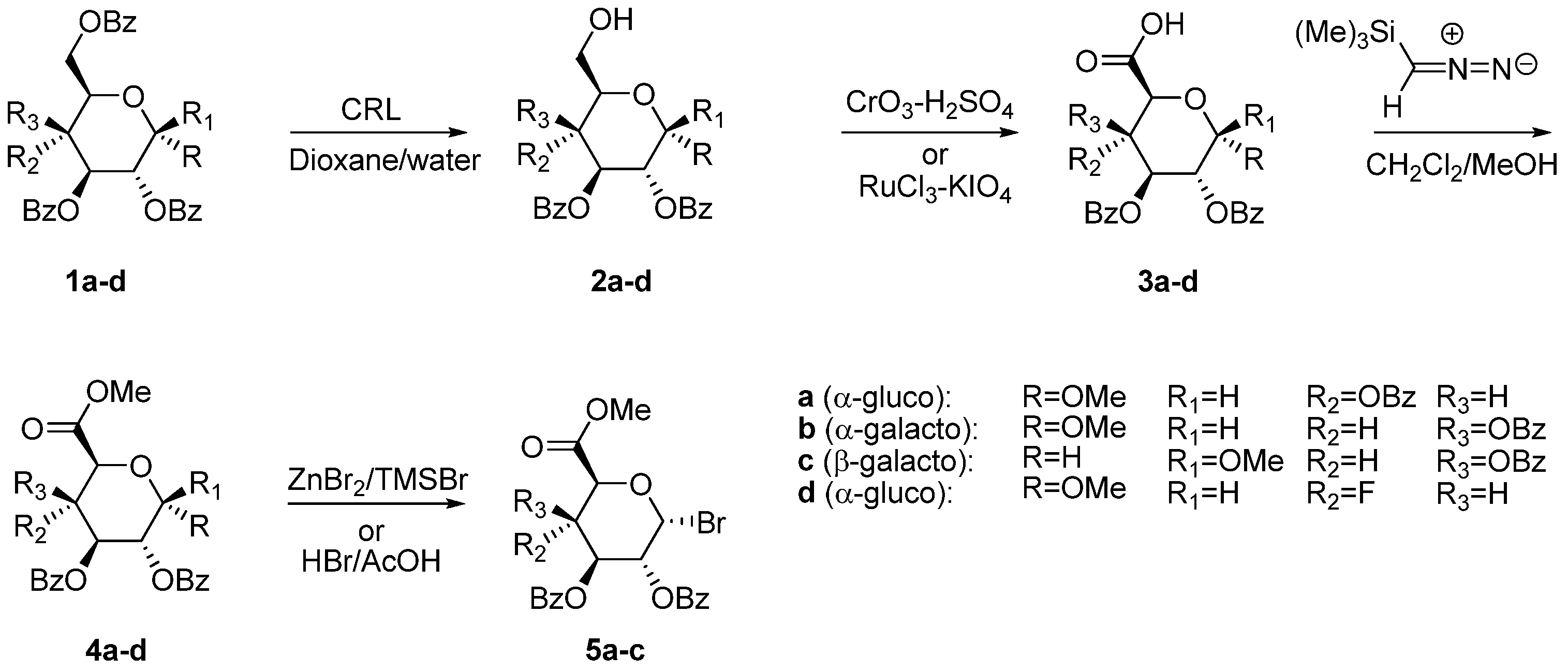
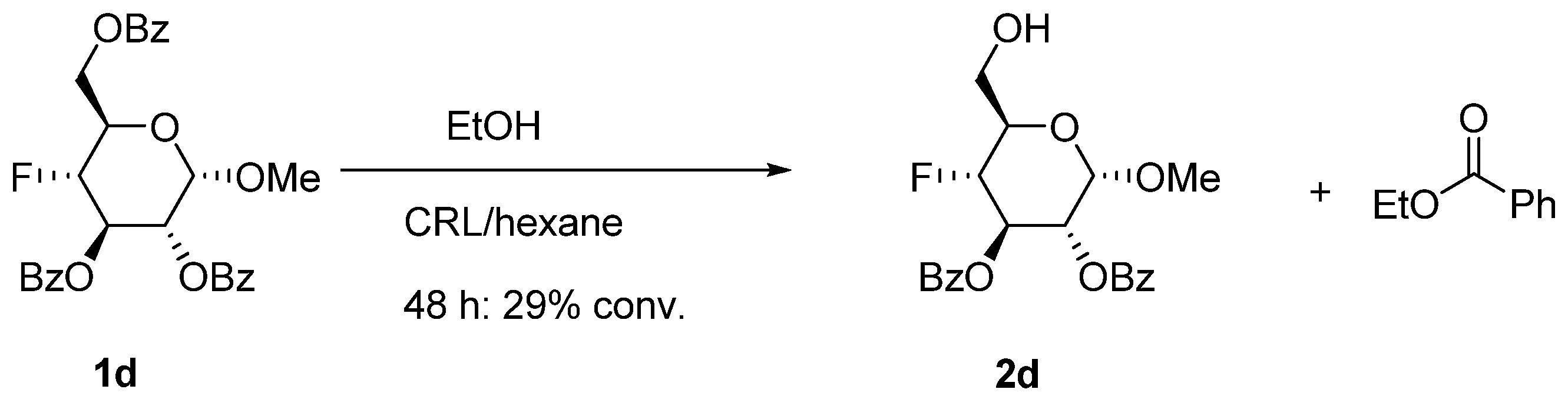
2.1. Uronic Acid Building Blocks
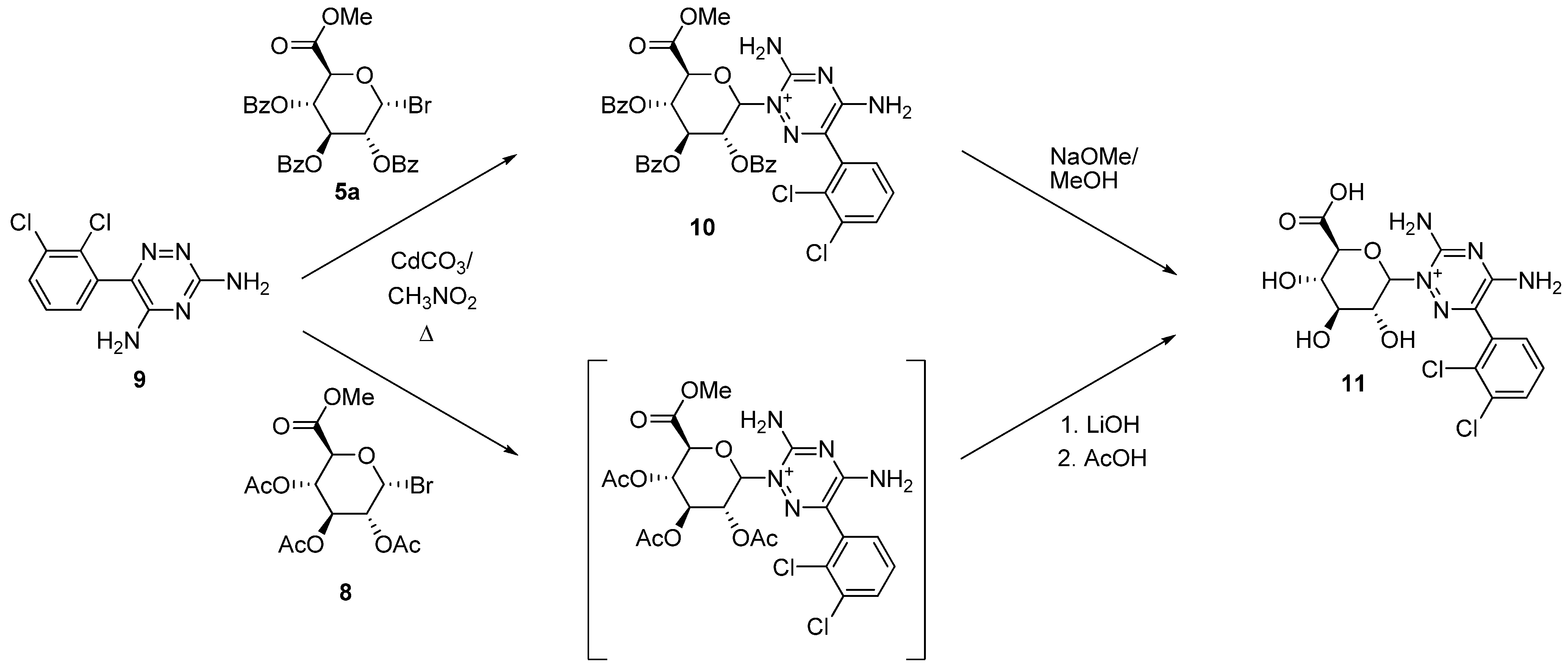
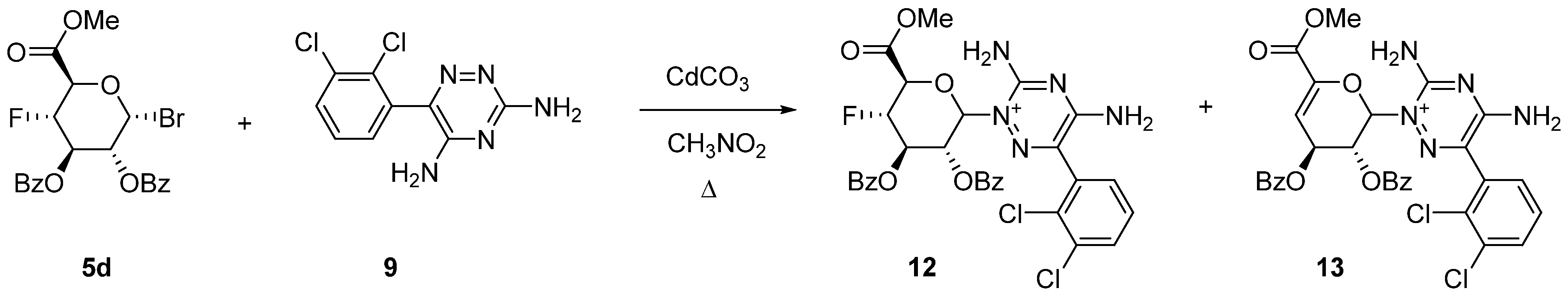
2.2. Uronic Acid Conjugates
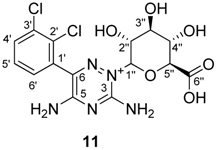
2.3. NMR Characterisation of Lamotrigin Glucuronide
3. Experimental
3.1. Chemicals and Equipment
3.2. NMR Spectroscopy
3.3. Methyl 2,3-di-O-Benzoyl-4-deoxy-4-fluoro-α-d-glucopyranoside (2d)
3.4. Oxidation to Uronic Acid Derivatives 3a–d
3.4.1. Methyl 2,3,4-tri-O-Benzoyl-α-d-glucopyranuronic Acid (3a)
3.4.2. Methyl 2,3,4-tri-O-Benzoyl-α-d-galactopyranuronic Acid (3b)
3.4.3. Methyl 2,3,4-tri-O-Benzoyl-β-d-galactopyranuronic Acid (3c)
3.4.4. Methyl 2,3-di-O-Benzoyl-4-fluoro-α-d-glucopyranuronic Acid (3d)
3.5. Methylation
3.5.1. Methyl (Methyl 2,3,4-tri-O-Benzoyl-α-d-glucoside)uronate (4a)
3.5.2. Methyl (Methyl 2,3,4-tri-O-Benzoyl-α-d-galactoside)uronate (4b) [38,42]
3.5.3. Methyl (Methyl 2,3,4-tri-O-Benzoyl-β-d-galactoside)uronate (4c)
3.5.4. Methyl (Methyl 2,3-di-O-Benzoyl-4-fluoro-α-d-glucoside)uronate (4d)
3.6. Bromination Using HBr in AcOH
3.6.1. Methyl (2,3,4-tri-O-Benzoyl-α-d-glycopyranosyl bromide)uronate (5a) [2,4,43]
3.6.2. Methyl (2,3,4-tri-O-Benzoyl-α-d-galactopyranosyl bromide)uronate (5b)
3.7. Bromination Using ZnBr2/TMSBr
3.7.1. Methyl (2,3,4-tri-O-Benzoyl-α-d-glycopyranosyl bromide)uronate (5a) Using Bromo-trimethylsilane/ZnBr2
3.7.2. Methyl (2,3-di-O-Benzoyl-4-deoxy-4-fluoro-α-d-glucosyl bromide)uronate (5d)
3.8. Bromo-2,3,4-tri-O-acetyl-α-d-glucopyranuronic Acid Methyl Ester (8) [30]
3.9. Lamotrigine Conjugates
3.9.1. Lamotrigine Conjugate 10
3.9.2. Lamotrigine N-2-glucuronide 11
3.9.3. Uronic Acid Conjugates 12 and 13
4. Conclusions
Acknowledgements
References and Notes
- Arsequell, G.; Sarries, N.; Valencia, G. Synthesis of glycosylated hydroxyproline building blocks. Tetrahedron Lett. 1995, 36, 7323–7326. [Google Scholar] [CrossRef]
- Coutant, C.; Jacquinet, J.C. 2-Deoxy-2-trichloroacetamido-d-glucopyranose derivatives in oligosaccharide synthesis: From hyaluronic acid to chondroitin 4-sulfate trisaccharides. J. Chem. Soc. Perkin Trans. 1 1995, 1573–1581. [Google Scholar] [CrossRef]
- Jacquinet, J.C.; Rochepeau-Jobron, L.; Combal, J.P. Multigram syntheses of the disaccharide repeating units of chondroitin 4- and 6-sulfates. Carbohydr. Res. 1998, 314, 283–288. [Google Scholar] [CrossRef]
- Bergman, K.; Hilborn, J.; Bowden, T. Selective Michael-type addition of a d-glucuronic acid derivative in the synthesis of model substances for uronic acid containing polysaccharides. Express Polym. Lett. 2008, 2, 553–559. [Google Scholar] [CrossRef]
- Leydet, A.; Jeantet-Segonds, C.; Barthelemy, P.; Boyer, B.; Roque, J.P. Polyanion inhibitors of human immunodeficiency virus. Part III. Polymerized anionic surfactants derived from d-glucose. Rec. Trav. Chim. Pays-Bas 1996, 115, 421–426. [Google Scholar] [CrossRef]
- Lergenmueller, M.; Lichtenthaler, F.W. Glucuronic acid-based ulosyl donors for introducing α-d-GlcA and δ-d-ManA units. Carbohydr. Res. 2007, 342, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Crich, D.; Sasaki, K. Reaction of thioacids with isocyanates and isothiocyanates: A convenient amide ligation process. Org. Lett. 2009, 11, 3514–3517. [Google Scholar] [CrossRef] [PubMed]
- Monneret, C.; Schmidt, F. Preparation of Derivatives of Etoposide and Analogs, and Pharmaceutical Compositions Containing Them. WO Patent WO/2003/035661, 2003. [Google Scholar]
- Wang, S.M.; Chern, J.W.; Yeh, M.Y.; Ng, J.C.; Tung, E.; Roffler, S.R. Specific activation of glucuronide prodrugs by antibody-targeted enzyme conjugates for cancer therapy. Cancer Res. 1992, 52, 4484–4491. [Google Scholar] [PubMed]
- Bonnaffe, D.; Lubineau, A.; Alais, J.; Gavard, O.; Dilhas, A.; Lortat, J.H. Compounds that Bind to the Interferon-Gamma, Preparation Method Thereof and Medicaments Containing Same. France Patent FR 2841250, 2003. [Google Scholar]
- Murphy, P.V. Monosaccharide Derivatives. WO Patent WO 2003018598, 2003. [Google Scholar]
- Rye, C.S.; Withers, S.G. Elucidation of the mechanism of polysaccharide cleavage by chondroitin AC lyase from Flavobacterium heparinum. J. Am. Chem. Soc. 2002, 124, 9756–9767. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Kumar, V.; Xue, J.; Locke, R.D.; Matta, K.L. The first chemical synthesis of F-4-GlcAβ(1→3)GlcNAc-UDP with the potential of novel substrate and enzyme inhibitor for hyaluronic acid synthases (HASs). Tetrahedron Lett. 2009, 50, 5920–5922. [Google Scholar] [CrossRef]
- Williams, L.; Nguyen, T.; Li, Y.; Porter, T.N.; Raushel, F.M. Uronate isomerase: A nonhydrolytic member of the amidohydrolase superfamily with an ambivalent requirement for a divalent metal ion. Biochemistry 2006, 45, 7453–7462. [Google Scholar] [CrossRef] [PubMed]
- Nieman, C.E.; Wong, A.W.; He, S.; Clarke, L.; Hopwood, J.J.; Withers, S.G. Family 39 α-l-iduronidases and β-d-xylosidases react through similar glycosyl-enzyme intermediates: Identification of the human iduronidase nucleophile. Biochemistry 2003, 42, 8054–8065. [Google Scholar] [CrossRef] [PubMed]
- Arewang, C.J.; Lahmann, M.; Oscarson, S.; Tiden, A.K. Synthesis of urine drug metabolites: Glucuronic acid glycosides of phenol intermediates. Carbohydr. Res. 2007, 342, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Rukhman, I.; Yudovich, L.; Nisnevich, G.; Gutman, A.L. Selective synthesis of both isomers of morphine 6-β-d-glucuronide and their analogs. Tetrahedron 2001, 57, 1083–1092. [Google Scholar] [CrossRef]
- Bedford, C.T. Glucoronic acid conjugates. J. Chromatogr. B 1998, 717, 313–326. [Google Scholar] [CrossRef]
- Dickins, M.; Chen, C. Lamotrigine. Chemistry, Biotransformation and Pharmacokinetics. In Antiepileptic Drugs, 5th ed.; Levy, R.H., Mattson, R.H., Meldrum, B.S., Perucca, E., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2002; pp. 370–379. [Google Scholar]
- Rowland, A.; Elliot, D.J.; Williams, J.A.; Mackenzie, P.I.; Dickinson, R.G.; Miners, J.O. In vitro characterization of Lamotrigine N2-glucuronidation and the Lamotrigine-valproic acid interaction. Drug Metab. Dispos. 2006, 34, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Doig, M.V.; Clare, R.A. Use of thermospray liquid chromatography-mass spectrometry to aid in the identification of urinary metabolites of a novel antiepileptic drug, Lamotrigine. J. Chromatogr. A 1991, 554, 181–189. [Google Scholar] [CrossRef]
- Oehman, I.; Beck, O.; Vitols, S.; Tomson, T. Plasma concentrations of lamotrigine and its 2-N-glucuronide metabolite during pregnancy in women with epilepsy. Epilepsia 2008, 49, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Esmurziev, A.; Simic, N.; Sundby, E.; Hoff, B.H. (1)H and (13)C NMR data of methyl tetra-O-benzoyl-d-pyranosides in acetone-d(6). Magn. Reson. Chem. 2009, 47, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Esmurziev, A.; Simic, N.; Sundby, E.; Hoff, B.H. Synthesis and structure elucidation of benzoylated deoxyfluoropyranosides. Carbohydr. Res. 2010, 29, 348–367. [Google Scholar] [CrossRef]
- Carlsen, P.H.J.; Katsuki, T.; Martin, V.S.; Sharpless, K.B. A greatly improved procedure for ruthenium tetroxide catalyzed oxidations of organic compounds. J. Org. Chem. 1981, 46, 3936–3938. [Google Scholar] [CrossRef]
- Takahashi, H.; Shida, T.; Hitomi, Y.; Iwai, Y.; Miyama, N.; Nishiyama, K.; Sawada, D.; Ikegami, S. Divergent synthesis of l-sugars and l-imino-sugars from l-sugars. Chem. Eur. J. 2006, 12, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Gerber-Lemaire, S.; Vogel, P. Anomeric effects in pyranosides and related acetals. Carbohydr. Chem. 2009, 35, 13–32. [Google Scholar]
- Kuszmann, J. Introduction to carbohydrates. Org. Chem. Sugars 2006, 25–52. [Google Scholar]
- Higashi, K.; Nakayama, K.; Shioya, E.; Kusama, T. Direct transformation of O-glycoside into glycosyl bromide with the combination of trimethylsilyl bromide and zinc bromide. Chem. Pharm. Bull. 1991, 39, 2502–2504. [Google Scholar] [CrossRef]
- Caldwell, W.S.; Greene, J.M.; Byrd, G.D.; Chang, K.M.; Uhrig, M.S.; DeBethizy, J.D.; Crooks, P.A.; Bhatti, B.S.; Riggs, R.M. Characterization of the glucuronide conjugate of cotinine: A previously unidentified major metabolite of nicotine in smokers’ urine. Chem. Res. Toxicol. 1992, 5, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Vashishtha, S.C.; Hawes, E.M.; McKay, G.; McCann, D.J. Synthesis and identification of the quaternary ammonium-linked glucuronide of 1-phenylimidazole in human liver microsomes and investigation of the human UDP-glucuronosyltransferases involved. Drug Metab. Dispos. 2000, 28, 1009–1013. [Google Scholar] [PubMed]
- Upadhyaya, P.; McIntee, E.J.; Hecht, S.S. Preparation of pyridine-N-glucuronides of tobacco-specific nitrosamines. Chem. Res. Toxicol. 2001, 14, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, S.; Conrow, R.B. Steroid conjugates. VI. Improved Koenigs--Knorr synthesis of aryl glucuronides using cadmium carbonate, a new and effective catalyst. J. Org. Chem. 1971, 36, 863–870. [Google Scholar] [CrossRef]
- Foulon, L.; Serradeil-Legal, C.; Valette, G. Nouveaux Derives Pyridiniques D’Indolin-2-one, leur Preparation et leur Application en Therapeutique. France Patent FR 2875499, 2006. [Google Scholar]
- Honda, T.; Kato, M.; Inoue, M.; Shimamoto, T.; Shima, K.; Nakanishi, T.; Yoshida, T.; Noguchi, T. Synthesis and antitumor activity of quaternary ellipticine glycosides, a series of novel and highly active antitumor agents. J. Med. Chem. 1988, 31, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Pilgrim, W.; Murphy, P.V. SnCl4- and TiCl4-Catalyzed anomerization of acylated O- and S-glycosides: Analysis of factors that lead to higher α:β anomer ratios and reaction rates. J. Org. Chem. 2010, 75, 6747–6755. [Google Scholar] [CrossRef] [PubMed]
- Adorjan, I.; Jaeaeskelaeinen, A.S.; Vuorinen, T. Synthesis and characterization of the hexenuronic acid model methyl 4-deoxy-β-l-threo-hex-4-enopyranosiduronic acid. Carbohydr. Res. 2006, 341, 2439–2443. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.L.; Horner, M.W.; Hough, L.; Richardson, A.C. Selective benzoylation of methyl(methyl α-d-galactopyranoside)uronate. Carbohyd. Res. 1971, 17, 213–215. [Google Scholar] [CrossRef]
- Timoshchuk, V.A.; Kulinkovich, L.N. Synthesis and conformational analysis of 4,5-unsaturated uronic acid nucleosides. Zh. Obshch. Khim. 1988, 58, 1651–1658. [Google Scholar]
- Kulinkovich, L.N.; Timoshchuk, V.A. Synthesis of unsaturated nucleosides of uronic acids. Zh. Obshch. Khim. 1983, 53, 1652–1655. [Google Scholar]
- Esmurziev, A.; Sundby, E.; Hoff, B.H. Regioselective C-6 hydrolysis of methyl O-benzoyl-pyranosides catalyzed by Candida rugosa lipase. Eur. J. Org. Chem. 2009, 1592–1597. [Google Scholar] [CrossRef]
- Morell, S.; Link, K.P. Derivatives of d-galacturonic acid. I. Esterification and acylation of d-galacturonic acid. J. Biol. Chem. 1935, 108, 763–771. [Google Scholar]
- Zorbach, W.W.; Valiaveedan, G.D. Methyl (cortison-21-yl 2,3,4-tri-O-acetyl-β-d-glucosid)uronate. J. Org. Chem. 1964, 29, 2462–2463. [Google Scholar] [CrossRef]
Sample Availability: Samples of the lamotrigine N-2-glucuronide 11 are now commercially available from Chiron AS. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δ, 1H mult. (J in Hz) | δ, 13C | IP-COSY | NOESY | HMBC a |
|---|---|---|---|---|---|
| 3 | - | 155.3 | - | - | - |
| 5 | - | 156.0 | - | - | - |
| 6 | - | 140.8 | - | - | - |
| 1’ | - | 130.8 | - | - | - |
| 2’ | - | 132.1 | - | - | - |
| 3’ | - | 134.1 | - | - | - |
| 4’ | 7.79 m | 133.9 | 5′ and/or 6′ | 5′ and/or 6’ | 2′, 6′, 3′ (w) |
| 5’ | 7.50 m | 129.5 | b 4′ | b 4′, 5-NHH (w), 5-NHH (w) | 1′, 3′ |
| 6’ | 7.51 m | 130.6 | 2′, 4′, 6 (w) | ||
| 1” | 5.53 br s | 88.9 | 2″ | 3″, 5″, 3-NH2 (w) | 3, 2″, 3″ (w) |
| 2” | 4.13 br t (9) c | 70.0 | 1″, 3″ | 4″, 3″ (w) | 1″, 3″ |
| 3” | 3.71 t (9.4) c | 76.7 | 2″, 4″ | 1″, 5″, 2″ (w), 4″ (w) | 2″, 4″, 1″ (w) |
| 4” | 3.59 t (9.8) c | 71.7 | 3″, 5″ | 2″, 5″, 3″ (w) | 3″, 5″, 6″ |
| 5” | 4.02 d (10.1) | 79.2 | 4” | 1″, 3″, 4” | 4″, 6″, 1″ (w), 3″ (w) |
| 6” | - | 175.1 | - | - | - |
| 3-NH2 | 8.21 br s | - | na | 1″ (w) | na |
| 5-NHH | 8.00 br s | - | na | 5-NHH, 5′ and/or 6′ | 5, 6 |
| 5-NHH | 8.74 br s | - | na | 5-NHH, 5′ and/or 6′ | 5 (w), 6 (w) |
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Esmurziev, A.M.; Reimers, A.; Andreassen, T.; Simic, N.; Sundby, E.; Hoff, B.H. Benzoylated Uronic Acid Building Blocks and Synthesis of N-Uronate Conjugates of Lamotrigine. Molecules 2012, 17, 820-835. https://doi.org/10.3390/molecules17010820
Esmurziev AM, Reimers A, Andreassen T, Simic N, Sundby E, Hoff BH. Benzoylated Uronic Acid Building Blocks and Synthesis of N-Uronate Conjugates of Lamotrigine. Molecules. 2012; 17(1):820-835. https://doi.org/10.3390/molecules17010820
Chicago/Turabian StyleEsmurziev, Aslan M., Arne Reimers, Trygve Andreassen, Nebojsa Simic, Eirik Sundby, and Bård Helge Hoff. 2012. "Benzoylated Uronic Acid Building Blocks and Synthesis of N-Uronate Conjugates of Lamotrigine" Molecules 17, no. 1: 820-835. https://doi.org/10.3390/molecules17010820