3. Experimental
3.1. General
Unless specified, all reagents and starting materials were purchased from commercial sources and used as received. Solvents were purified following standard literature procedures. Analytical TLC was performed on silica gel60 F254 precoated on glass plates, with detection by fluorescence and/or by staining with 5% concentrated sulphuric acid in EtOH. 1H- (400 MHz) and 13C-NMR spectra (100 MHz) were recorded on a Bruker DRX-400 spectrometer at 25 °C. Chemical shifts (δ) for 1H and 13C spectra are expressed in ppm relative to internal Me4Si as standard. Signals were abbreviated as s, singlet; bs, broad singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Sugar signals were numbered as customary. ESI mass spectra and high resolution mass spectrometry (HRMS) were recorded using an Agilent Technologies 1100 Series instrument (ESI ionization). Optical rotations were measured in a 1.00 dm tube with an Optical Activity AA-10R polarimeter in methanol or chloroform.
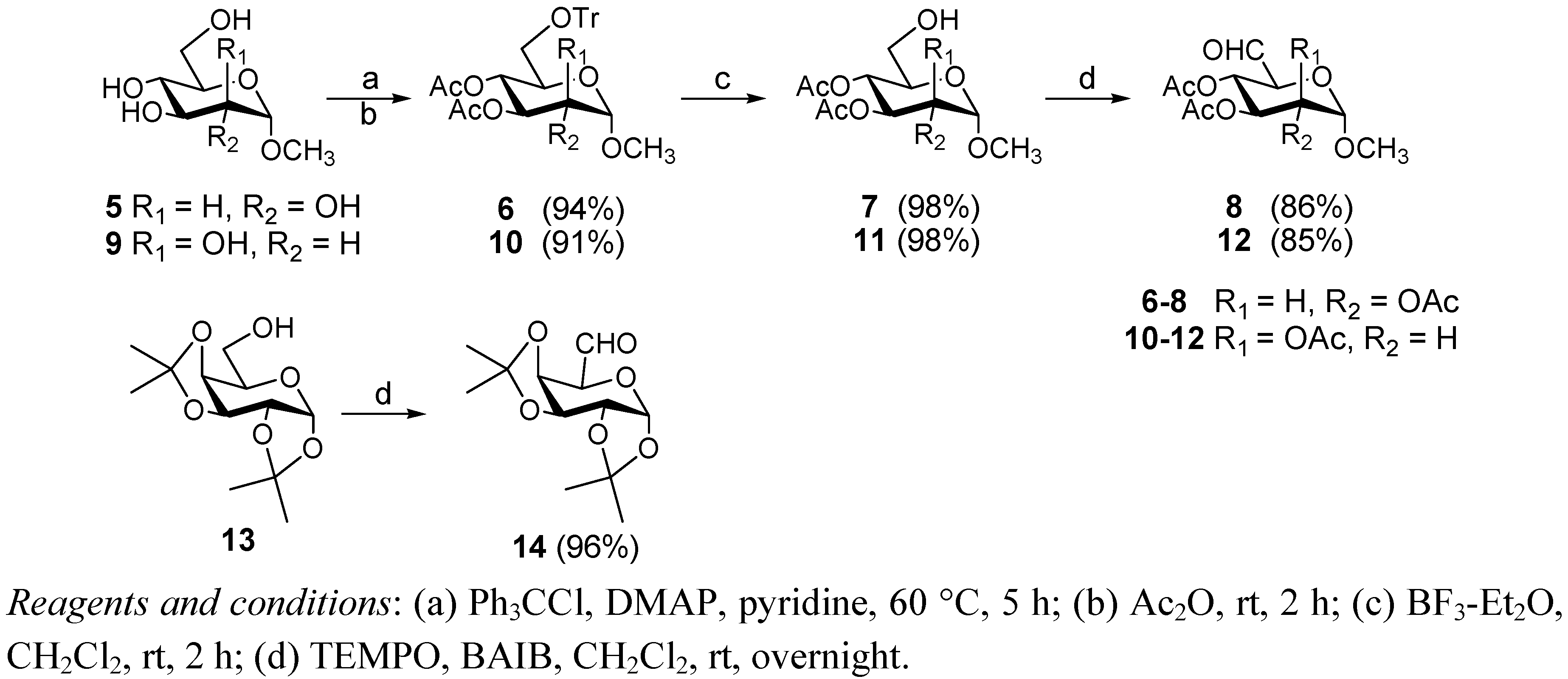
3.2. General Procedure for the Synthesis of Compounds 14a–e
Amine (compounds 15–19, 0.5 mmol) was added to a solution of compounds 14 (155 mg, 0.6 mmol) in dry methanol (2 mL) and acetic acid (1.5 mL) under stirring and under a nitrogen atmosphere at room temperature. After 10 min NaCNBH3 (40 mg, 0.6 mmol) was added. The solution was stirred at room temperature for 30 min. After completion of the reaction (TLC 3:1 petroleum ether b.p. 60–90 °C-EtOAc), the methanol was evaporated under reduced pressure, the reaction mixture was diluted with CH2Cl2 (5 mL) and washed with saturated NaHCO3 (3 × 5 mL) and with water (5 mL). The organic phase was dried over sodium sulphate and the solvent was evaporated under reduced pressure to give a crude mass which was purified by flash chromatography (4:1 petroleum ether b.p. 60–90 °C-EtOAc).
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-galactopyranose (14a). Compound 15 (127 mg, 0.5 mmol) was used to prepare compound 14a (205 mg, 82.3%) as a colorless syrup; [α]25D −75 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.72 (s, 1H), 7.46 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.15–7.13 (m, 2H), 6.73–6.68 (m, 3H), 5.56 (d, J = 5.2 Hz, 1H, H-1), 4.64 (dd, J1 = 2.4 Hz, J2 = 8.0 Hz, 1H, H-3), 4.34 (dd, J1 = 2.4 Hz, J2 = 5.2 Hz, 1H, H-2), 4.26 (dd, J1 = 2.0 Hz, J2 = 8.0 Hz, 1H, H-4), 4.04–4.00 (m, 1H, H-5), 3.40–3.37 (m, 2H, H-6), 1.48 (s, 3H, CH3), 1.44 (s, 3H, CH3), 1.37 (s, 3H, CH3), 1.33 (s, 3H, CH3); 13C-NMR(CDCl3): δ 162.1 (C=O), 148.7, 138.3, 138.2, 135.0, 129.7, 122.0, 113.2, 109.5, 109.1, 108.7, 96.4 (C-1), 71.6, 70.8, 70.6 and 65.7 (C-2, C-3, C-4, C-5), 43.9 (C-6), 26.0 (CH3), 26.0 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C24H27N2O6F3: 497.2[M+H]+, found: 497.2[M+H]+.
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(3-chloro-4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-galactopyranose (14b). Compound 16 (145 mg, 0.5 mmol) was used to prepare compound 14b (195 mg, 73.6%) as a colorless syrup; [α]25D −59 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.58 (s, 1H), 7.51 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.10 (dd, J1 = 1.6 Hz, J2 = 8.8 Hz, 1H), 6.78 (d, J = 2.4 Hz, 1H), 6.71 (d, J = 2.8 Hz, 1H), 6.63–6.60 (m, 1H), 5.56 (d, J = 5.2 Hz, 1H, H-1), 4.64 (dd, J1 = 2.4 Hz, J2 = 8.0 Hz, 1H, H-3), 4.35 (dd, J1 = 2.4 Hz, J2 = 5.2 Hz, 1H, H-2), 4.26 (dd, J1 = 2.0 Hz, J2 = 8.0 Hz, 1H, H-4), 4.02–3.99 (m, 1H, H-5), 3.38–3.35 (m, 2H, H-6), 1.48 (s, 3H, CH3), 1.47 (s, 3H, CH3), 1.37 (s, 3H, CH3), 1.34 (s, 3H, CH3); 13C-NMR (CDCl3): δ 161.7 (C=O), 150.0, 138.7, 135.5, 132.0, 129.2, 126.5, 122.3, 113.3, 112.2, 112.1, 109.6, 108.8, 96.4 (C-1), 71.6, 70.8, 70.6 and 65.6 (C-2, C-3, C-4, C-5), 43.8 (C-6), 26.0 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C24H26N2O6F3Cl: 531.2 [M+H]+, found: 531.1 [M+H]+.
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(3-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-galactopyranose (14c). Compound 17 (135 mg, 0.5 mmol) was used to prepare compound 14c(192 mg, 75.3%) as a colorless syrup; [α]25D −47 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.63 (s, 1H), 7.41 (d, J = 1.6 Hz, 1H), 7.18–7.14 (m, 1H), 6.67–6.59 (m, 4H), 5.54 (d, J = 4.8 Hz, 1H, H-1), 5.04 (m, 2H, CH2), 4.62 (dd, J1 = 1.6 Hz, J2 = 8.0 Hz, 1H, H-3), 4.33 (t, J = 2.4 Hz, 1H, H-2), 4.25 (d, J = 7.6 Hz, 1H, H-4), 3.98 (d, J = 4.8 Hz, 1H, H-5), 3.40–3.29 (m, 2H, H-6), 1.47 (s, 3H, CH3), 1.37 (s, 3H, CH3), 1.36 (s, 3H, CH3), 1.31 (s, 3H, CH3); 13C-NMR (CDCl3): δ 161.9 (C=O), 148.8, 136.7, 136.1, 134.8, 130.0, 121.5, 117.3, 113.1, 109.5, 108.7, 96.4 (C-1), 71.7, 70.8, 70.6 and 65.7 (C-2, C-3, C-4, C-5), 52.3 (CH2), 43.9 (C-6), 26.0 (CH3), 25.8 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C25H29N2O6F3: 511.2 [M+H]+, found: 511.2 [M+H]+.
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-galactopyranose (14d). Compound 18 (135 mg, 0.5 mmol) was used to prepare compound 14d (207 mg, 81.2%) as a colorless syrup; [α]25D −68 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.60 (s, 1H), 7.39 (dd, J1 = 2.0 Hz, J2 = 9.6 Hz, 1H), 7.15 (d, J = 8.4 Hz, 2H), 6.65–6.62 (m, 3H), 5.54 (d, J= 4.8 Hz, 1H, H-1), 5.00 (s, 2H, CH2), 4.62 (dd, J1 = 2.0 Hz, J2 = 8.0 Hz, 1H, H-3), 4.33–4.31 (m, 1H, H-2), 4.25–4.23 (m, 1H, H-4), 3.99 (t, J = 6.0 Hz, 1H, H-5), 3.41–3.30 (m, 2H, H-6), 1.47 (s, 3H, CH3), 1.36 (s, 3H, CH3), 1.33 (s, 3H, CH3), 1.30 (s, 3H, CH3); 13C-NMR (CDCl3): δ 162.0 (C=O), 148.4, 136.4, 136.3, 134.7, 134.6, 130.1, 123.5, 121.4, 113.6, 109.5, 108.7, 96.4 (C-1), 71.7, 70.8, 70.6 and 65.6 (C-2, C-3, C-4, C-5), 52.1 (CH2), 43.9 (C-6), 26.0 (CH3), 25.7 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C25H29N2O6F3: 511.2 [M+H]+, found: 511.2 [M+H]+.
1,2;3,4-di-O-Isopropylidene-6-deoxy-6-(2-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-galactopyranose (14e). Compound 19 (135 mg, 0.5 mmol) was used to prepare compound 14e (210 mg, 82.4%) as a colorless syrup; [α]25D −62 (c 0.10, CHCl3); 1H-NMR (CDCl3): δ 7.68 (s, 1H), 7.42 (dd, J1 = 2.0 Hz, J2 = 9.6 Hz, 1H), 7.25 (m, 1H), 7.14 (d, J = 7.2 Hz, 1H), 6.72–6.64 (m, 3H), 5.44 (d, J = 5.2 Hz, 1H, H-1), 5.20–4.95 (m, 2H, CH2), 4.57 (dd, J1 = 2.0 Hz, J2 = 8.0 Hz, 1H H-3), 4.27 (dd, J1 = 2.0 Hz, J2 = 5.2 Hz, 1H H-2), 4.21 (d, J = 8.8 Hz, 1H, H-4), 3.94 (t, J = 6.4 Hz, 1H, H-5), 3.50–3.43 (m, 1H, H-6), 3.36–3.31 (m, 1H, H-6), 1.47 (s, 3H, CH3), 1.34 (s, 3H, CH3), 1.26 (d, 6H, 2xCH3); 13C-NMR (CDCl3): δ 162.4 (C=O), 146.6, 136.1, 136.1, 135.0, 131.3, 130.5, 121.3, 119.3, 116.9, 111.6, 110.3, 109.3, 108.5, 96.4 (C-1), 71.4, 70.8, 70.6 and 65.1 (C-2, C-3, C-4, C-5), 49.1 (CH2), 43.6 (C-6), 26.0 (CH3), 25.6 (CH3), 24.9 (CH3), 24.4 (CH3); ESI-MS: Calcd for C25H29N2O6F3: 511.2 [M+H]+, found: 511.2 [M+H]+.
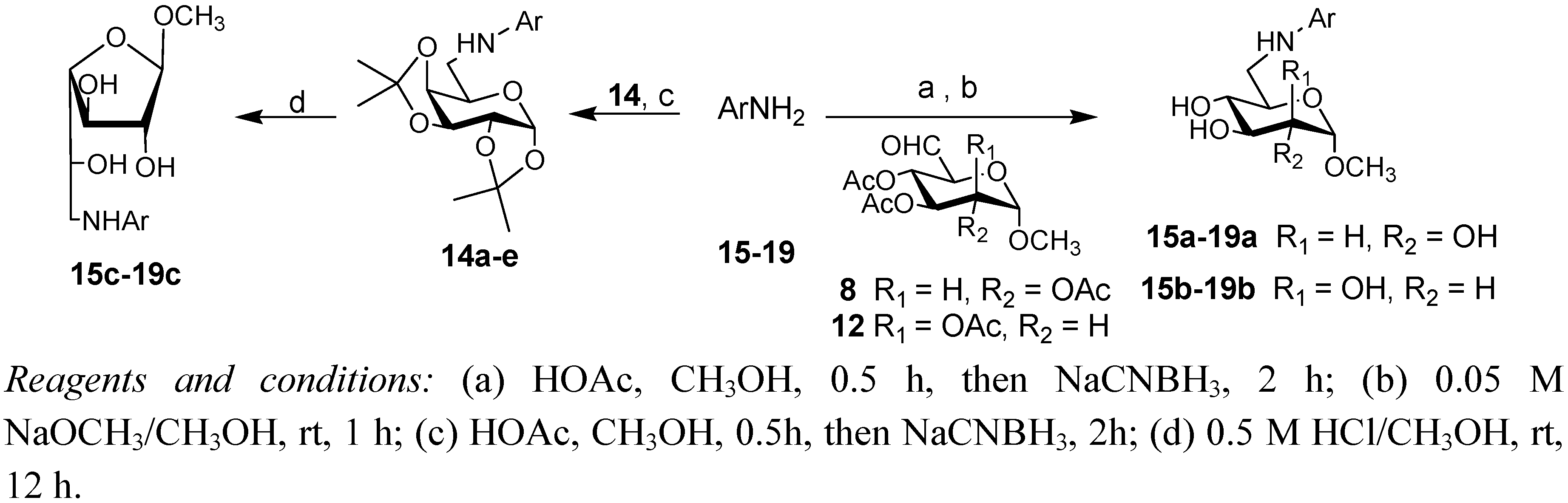
3.3. General Procedure for the Synthesis of Compounds 15a–19a and 15b–19b
Amine (compound 15–19, 0.5 mmol) was added to a solution of compounds 8 or 12 (0.6 mmol) in dry methanol (2 ml) and acetic acid (1.5 mL) under stirring and under nitrogen atmosphere at room temperature. After 10 min NaCNBH3 (40 mg, 0.6 mmol) was added. The solution was stirred at room temperature for 30 min. After completion of the reaction (TLC 3:1 petroleum ether b.p. 60–90 °C-EtOAc), the methanol was evaporated under reduced pressure, the reaction mixture was diluted with CH2Cl2 (5 mL) and washed with saturated NaHCO3 (3 × 5 mL) and with water (5 mL). The organic phase was dried over sodium sulphate and the solvent was evaporated under reduced pressure to give a crude mass which was dissolved in 0.05 M NaOMe/MeOH (5 mL) and stirred at ambient temperature for 30 min. The stirring reaction mixture was neutralized with prewashed Amberlite IR 120-H+. The resin was filtered off and the solvent was evaporated under reduced pressure. The crude product was purified by flash chromatography (7:1 CHCl3-MeOH).
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-glucopyranoside (15a). Compound 15 (127 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 15a (143 mg, 66.5%) as a colorless syrup; [α]25D −63 (c 0.10, MeOH); 1H-NMR (MeOH-d4) δ 7.97 (s, 1H), 7.66 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H), 7.01 (d, J = 8.8 Hz, 2H), 6.76 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 9.2 Hz, 1H), 4.62 (d, J = 3.6 Hz, 1H, H-1), 3.71–3.63 (m, 1H), 3.59–3.55 (m, 2H), 3.38–3.35 (m, 1H), 3.28 (s, 3H), 3.22–3.17 (m, 2H); 13C-NMR (MeOH-d4) δ 164.5 (C=O), 151.0, 140.6, 140.5, 137.3, 130.3, 128.1, 122.2, 113.6, 101.2 (C-1), 75.1, 73.6, 73.5 and 71.4 (C-2, C-3, C-4, C-5), 55.5 (OCH3), 45.7 (C-6); HRMS: Calcd for C19H21N2O6F3 [M+H]+ 431.1431, found 431.1425.
Methyl 6-deoxy-6-(3-chloro-4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-glucopyranoside (16a). Compound 16 (145 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 16a (152 mg, 65.5%) as a colorless syrup; [α]25D −34 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.92 (s, 1H), 7.70 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.08 (d, J = 8.8 Hz, 2H), 6.84 (m, 1H), 6.68 (m, 2H), 4.62 (d, J = 3.6 Hz, 1H, H-1), 3.63–3.60 (m, 1H), 3.59–3.51 (m, 2H), 3.38–3.34 (m, 1H), 3.29 (s, 3H, OCH3), 3.26–3.17 (m, 3H); 13C-NMR (MeOH-d4): δ 164.1 (C=O), 152.5, 141.2, 137.8, 132.7, 130.2, 126.7, 122.5, 113.8, 112.8, 101.2 (C-1), 75.1, 73.6, 73.3 and 71.6 (C-2, C-3, C-4, C-5), 55.6 (OCH3), 45.5 (C-6); HRMS: Calcd for C19H21N2O6F3Cl: 465.1041 [M+H]+, found: 465.1038 [M+H]+.
Methyl 6-deoxy-6-(3-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-glucopyranoside (17a). Compound 17 (135 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 17a (150 mg, 67.6%) as a colorless syrup; [α]25D −45 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.06 (s, 1H), 7.55 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H), 7.02 (t, J = 8.0 Hz, 1H), 6.61–6.58 (m, 3H), 6.49 (d, J = 7.6 Hz, 1H), 5.03 (s, 2H, CH2), 4.57 (d, J = 2.8 Hz, 1H, H-1), 3.62–3.48 (m, 3H), 3.35–3.12 (m, 1H), 3.16 (s, 3H, OCH3), 3.19–3.06 (m, 2H); 13C-NMR (MeOH-d4): δ 161.2 (C=O), 147.8, 136.6, 136.6, 135.0, 134.1, 127.8, 123.3, 120.6, 118.9, 114.6, 111.3, 111.0, 108.7, 108.4, 98.2 (C-1), 72.2, 70.7, 70.7 and 68.19 (C-2, C-3, C-4, C-5), 52.5 (OCH3), 50.9 (CH2), 43.0 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1583 [M+H]+.
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-glucopyranoside (18a). Compound 18 (135 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 18a (155 mg, 69.8%) as a colorless syrup; [α]25D −54 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.03 (s, 1H), 7.54 (dd, J1 = 2.8 Hz, J2 =9.6 Hz, 1H), 7.07 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 4.4 Hz, 2H), 6.56 (d, J = 9.6 Hz, 1H), 4.98 (s, 2H, CH2), 4.57 (d, J = 3.6 Hz, 1H, H-1), 3.63–3.58 (m, 1H), 3.55–3.49 (m, 2H), 3.34–3.31 (m, 1H), 3.24–3.23 (m, 1H), 3.18 (s, 3H, OCH3), 3.15–3.06 (m, 2H); 13C-NMR (MeOH-d4): δ 162.8 (C=O), 148.9, 137.7, 137.7, 135.5, 129.3, 123.5, 120.3, 113.1, 99.8 (C-1), 73.7, 72.2, 72.2 and 69.6 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 52.1 (CH2), 44.5 (C-6); HRMS: Calcd for C20H23N2O6F3: 467.1400 [M+Na]+, found: 465.1391 [M+Na]+.
Methyl 6-deoxy-6-(2-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-glucopyranoside (19a). Compound 19 (135 mg, 0.5 mmol) and 8 (190 mg, 0.6 mmol) were used to prepare compound 19a (147 mg, 66.2%) as a colorless syrup; [α]25D −57 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.90 (s, 1H), 7.55 (dd, J1 = 1.6 Hz, J2 =7.6 Hz, 1H), 7.15 (m, 2H), 6.70 (d, J = 8.0 Hz, 1H), 6.61 (m, 2H), 5.06 (m, 2H, CH2), 4.51 (d, J = 3.6 Hz, 1H, H-1), 3.67–3.59 (m, 1H), 3.52–3.47 (m, 2H), 3.37–3.31 (m, 1H), 3.22 (s, 1H), 3.16–3.10 (m, 2H), 3.02 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 162.9 (C=O), 146.6, 137.0, 137.0, 135.6, 131.0, 130.0, 127.3, 120.3, 119.5, 116.6, 111.1, 110.6, 99.8 (C-1), 73.7, 72.2, 72.1 and 69.0 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 48.2 (CH2), 44.2 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1586 [M+H]+.
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-mannopyranoside (15b). Compound 15 (127 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 15b (140 mg, 65.1%) as a colorless syrup; [α]25D +82 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.98 (s, 1H), 7.67 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H), 7.08 (d, J = 8.8 Hz, 2H), 6.76 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 9.2 Hz, 1H), 4.61 (d, J= 1.2 Hz, 1H, H-1), 3.76 (s, 1H), 3.64–3.59 (m, 3H), 3.54–3.51 (m, 1H), 3.32–3.28 (m, 1H), 3.26 (s, 3H, OCH3), 13C-NMR (MeOH-d4): δ 164.5 (C=O), 151.0, 140.6, 140.5, 140.5, 137.3, 130.2, 128.1, 126.2, 123.6, 122.2, 113.0, 111.5, 111.1, 102.8(C-1), 72.6, 72.1, 72.1 and 69.9 (C-2, C-3, C-4, C-5), 55.2 (OCH3), 45.5 (C-6); HRMS: Calcd for C19H21N2O6F3: 431.1431 [M+H]+, found: 431.1425 [M+H]+.
Methyl 6-deoxy-6-(3-chloro-4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-α-D-mannopyranoside (16b). Compound 16 (145 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 16b (145 mg, 62.5%) as a colorless syrup; [α]25D −59 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.93 (s, 1H), 7.71 (dd, J1 = 2.4 Hz, J2 = 9.6 Hz, 1H), 7.09 (d, J = 2.2 Hz, 2H), 6.84 (dd, J1 = 0.4 Hz, J2 = 1.6 Hz, 1H), 6.71–6.66 (m, 2H), 4.60 (d, J = 1.6 Hz, 1H, H-1), 3.76 (s, 1H), 3.64–3.57 (m, 3H), 3.52–3.48 (m, 1H), 3.30–3.26 (m, 2H), 3.28 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 162.7 (C=O), 151.1, 139.8, 139.7, 136.4, 131.3, 128.8, 125.3, 124.7, 122.1, 121.1, 112.3, 111.4, 111.4, 110.2, 109.9, 101.4 (C-1), 71.2, 70.9, 70.7 and 68.4 (C-2, C-3, C-4, C-5), 53.9 (OCH3), 43.8 (C-6); HRMS: Calcd for C19H21N2O6F3Cl: 465.1041 [M+H]+, found: 465.1039 [M+H]+.
Methyl 6-deoxy-6-(3-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-mannopyranoside (17b). Compound 17 (135 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 17b (154 mg, 69.4%) as a colorless syrup; [α]25D −83 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.08 (s, 1H), 7.60 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.06 (dd, J1 = 7.6 Hz, J2 = 8.0 Hz, 1H), 6.63–6.60 (m, 3H), 6.52 (d, J = 7.2 Hz, 1H), 5.07 (s, 2H, CH2), 4.57 (d, J = 1.6 Hz, 1H, H-1), 3.74 (t, 1H), 3.62–3.56 (m, 3H), 3.49–3.46 (m, 1H), 3.27–3.25 (m, 1H), 3.17 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 162.7 (C=O), 149.3, 138.1, 138.1, 136.5,135.7, 129.3, 124.8, 120.4, 116.0, 112.7, 112.3, 110.2, 109.9, 101.3 (C-1), 71.2, 70.7, 70.3 and 68.6 (C-2, C-3, C-4, C-5), 53.7 (OCH3), 52.4 (CH2), 44.2 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1585 [M+H]+.
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-mannopyranoside (18b). Compound 18 (135 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 18b (150 mg, 67.6%) as a colorless syrup; [α]25D −67 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.06 (s, 1H), 7.58 (dd, J1 = 2.8 Hz, J2 = 2.4 Hz, 1H), 7.11 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 6.58 (d, J = 9.6 Hz, 1H), 5.01 (s, 2H, CH2), 4.57 (d, J= 1.6 Hz, 1H, H-1), 3.73 (t, 1H), 3.61–3.55 (m, 3H), 3.51–3.46 (m, 1H), 3.27–3.25 (m, 3H), 3.19 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 164.2 (C=O), 150.4, 139.1, 139.1, 136.9,130.7, 126.2, 124.9, 123.5, 121.7, 114.4, 111.6, 111.2, 102.7 (C-1), 72.6, 72.1, 71.8 and 70.1 (C-2, C-3, C-4, C-5), 55.1 (OCH3), 53.5 (CH2), 45.7 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1589 [M+H]+.
Methyl 6-deoxy-6-(2-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-α-D-mannopyranoside (19b). Compound 19 (135 mg, 0.5 mmol) and 12 (190 mg, 0.6 mmol) were used to prepare compound 19b (151 mg, 68.0%) as a colorless syrup; [α]25D −72 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.95 (s, 1H), 7.58 (d, J = 9.6 Hz 1H), 7.23–7.17 (m, 2H), 6.72–6.70 (m, 2H), 6.65 (t, J = 7.6 Hz, 1H), 5.19–5.09 (m, 2H, CH2), 4.62 (s, 1H, H-1), 3.75 (s, 1H), 3.69–3.60 (m, 3H), 3.46–3.42 (m, 1H), 3.34–3.27 (m, 2H), 3.14 (s, 3H, OCH3); 13C-NMR (MeOH-d4): δ 163.0 (C=O), 146.7, 137.1, 137.0, 136.9, 135.7, 130.9, 130.1, 124.6, 121.9, 120.2, 119.5, 116.5, 110.9, 110.9, 110.6, 101.5(C-1), 71.3, 71.0, 70.0 and 68.5 (C-2, C-3, C-4, C-5), 53.8 (OCH3), 43.8 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1587 [M+H]+, found: 445.1581 [M+H]+.
3.4. General Procedure for the Synthesis of Compounds 15c–19c
Compound 14a–e (0.2 mmol) was added to a solution of 0.5 M HCl/MeOH (5 mL) under stirring and under nitrogen atmosphere at room temperature. The progress of reaction was monitored by TLC (6:1CHCl3-MeOH). After completion of the reaction the solvent was evaporated under reduced pressure, the residue was purified by flash chromatography (7:1CHCl3-MeOH).
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-β-D-galactofuranoside (15c). Compound 14a (100 mg, 0.2 mmol) was used to prepare compound 15c (45 mg, 52.3%) as a colorless syrup; [α]25D −48 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.93 (s, 1H), 7.62 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.8 Hz, 2H), 6.58 (d, J = 9.6 Hz, 1H), 4.69 (s, 1H, H-1), 3.85–3.77 (m, 4H), 3.34–3.13 (m, 5H); 13C-NMR (MeOH-d4): δ 163.3 (C=O), 149.7, 139.4, 136.1, 129.0, 127.0, 121.0, 114.8, 112.4, 110.3, 109.9, 109.3 (C-1), 83.9, 82.0, 77.6 and 68.8 (C-2, C-3, C-4, C-5), 54.3 (OCH3), 46.3 (C-6); HRMS: Calcd for C19H21N2O6F3: 431.1424 [M+H]+, found: 431.1420 [M+H]+.
Methyl 6-deoxy-6-(3-chloro-4-(5-trifluoromethyl-2(1H)-pyridone-1-yl)-anilino)-β-D-galactofuranoside (16c). Compound 14b (106 mg, 0.2 mmol) was used to prepare compound 16c (42 mg, 45.2%) as a yellow syrup; [α]25D −64 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.95 (s, 1H), 7.69 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 6.79 (t, J = 2.4 Hz, 1H), 6.66–6.63 (m, 2H), 4.73 (d, J = 1.6 Hz, 1H, H-1), 3.96–3.95 (m, 1H), 3.87–3.85 (m, 2H), 3.83–3.78 (m, 1H), 3.38–3.35 (m, 1H), 3.32 (s, 3H, OCH3),3.25–3.23 (m, 1H), 3.22–3.16 (m, 1H); 13C-NMR (MeOH-d4): δ 162.7 (C=O), 150.9, 139.7, 136.4, 131.3, 128.9, 125.2, 112.0, 111.2, 109.1, 83.6, 81.7, 77.3 and 68.5 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 45.8 (C-6); HRMS: Calcd for C19H20N2O6F3Cl: 465.1035 [M+H] +, found: 465.1038 [M+H]+.
Methyl 6-deoxy-6-(3-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-β-D-galactofuranoside (17c). Compound 14c (100 mg, 0.2 mmol) was used to prepare compound 17c (52 mg, 58.4%) as a yellow syrup; [α]25D −34 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.07 (s, 1H), 7.58 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.05–7.01 (m, 1H), 6.60–6.55 (m, 3H), 6.48 (d, J = 7.6 Hz, 1H), 5.04 (s, 2H, CH2), 4.71 (d, J= 1.2 Hz, 1H, H-1), 3.95–3.93 (m, 1H), 3.87–3.83 (m, 2H), 3.80–3.79 (m, 1H), 3.30 (s, 3H, OCH3), 3.24 (m, 1H), 3.15–3.10 (m, 1H); 13C-NMR (MeOH-d4): δ 164.1(C=O), 150.7, 139.5, 138.0, 137.1, 130.7, 121.8, 117.2, 113.6, 113.4, 111.7, 110.5, 85.3, 83.2, 78.9 and 70.0 (C-2, C-3, C-4, C-5), 55.3 (OCH3), 53.8 (CH2), 47.7 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1581 [M+H]+, found: 445.1587 [M+H]+.
Methyl 6-deoxy-6-(4-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-β-D-galactofuranoside (18c). Compound 14d (100 mg, 0.2 mmol) was used to prepare compound 18c (47 mg, 52.8%) as a yellow syrup; [α]25D −75 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 8.06 (s, 1H), 7.56 (dd, J1 = 2.8 Hz, J2 = 9.6 Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H), 6.61–6.54 (m, 3H), 4.98 (s, 2H, CH2), 4.70 (d, J = 2.0 Hz, 1H, H-1), 3.94–3.91 (m, 1H), 3.85–3.82 (m, 2H), 3.80–3.76 (m, 1H), 3.29 (s, 3H, OCH3),3.25–3.23 (m, 3H), 3.16–3.11 (m, 1H); 13C-NMR (MeOH-d4): δ 162.8 (C=O), 148.9, 137.8, 135.5, 129.4, 123.4, 120.3, 112.6, 109.1, 83.7, 81.9, 77.4 and 68.6 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 52.1 (CH2), 46.2 (C-6); HRMS: Calcd for C20H23N2O6F3: 467.1400 [M+Na]+, found: 465.1380 [M+Na]+.
Methyl 6-deoxy-6-(2-(5-trifluoromethyl-2(1H)-pyridone-1-yl-methylene)-anilino)-β-D-galactofuranoside (19c). Compound 14e (100 mg, 0.2 mmol) was used to prepare compound 19c (49 mg, 55.1%) as a yellow syrup; [α]25D −72 (c 0.10, MeOH); 1H-NMR (MeOH-d4): δ 7.97 (s, 1H), 7.57 (dd, J1 = 2.8 Hz, J2 = 7.2 Hz, 1H), 7.16–7.10 (m, 2H), 6.68 (d, J = 8.0 Hz, 1H), 6.63–6.59 (m, 2H), 5.09 (s, 2H, CH2), 4.73 (s, 1H, H-1), 3.97–3.96 (m, 1H), 3.88–3.84 (m, 3H), 3.36–3.17 (m, 5H); 13C-NMR (MeOH-d4): δ 163.0 (C=O), 146.5, 137.3, 137.2, 135.7, 130.4, 129.9, 120.3, 119.5, 116.5, 110.8, 109.1, 84.0, 81.8, 77.5 and 68.5 (C-2, C-3, C-4, C-5), 54.0 (OCH3), 48.5 (CH2), 46.2 (C-6); HRMS: Calcd for C20H23N2O6F3: 445.1581 [M+H]+, found: 445.1583 [M+H]+.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
















